
Computational Simulation of HIV Protease Inhibitors to the Main Protease (Mpro) of SARS-CoV-2: Implications for COVID-19 Drugs Design

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
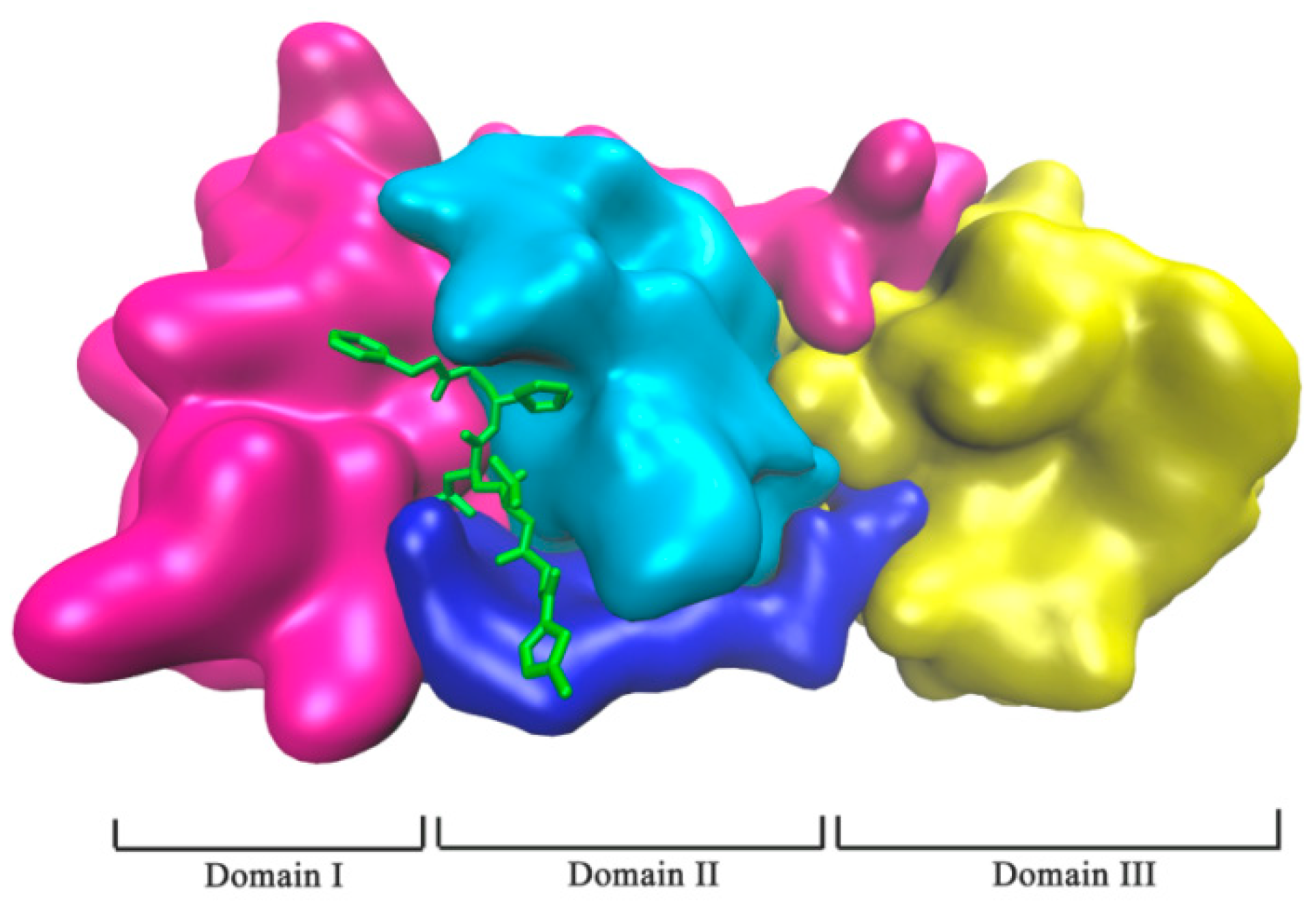
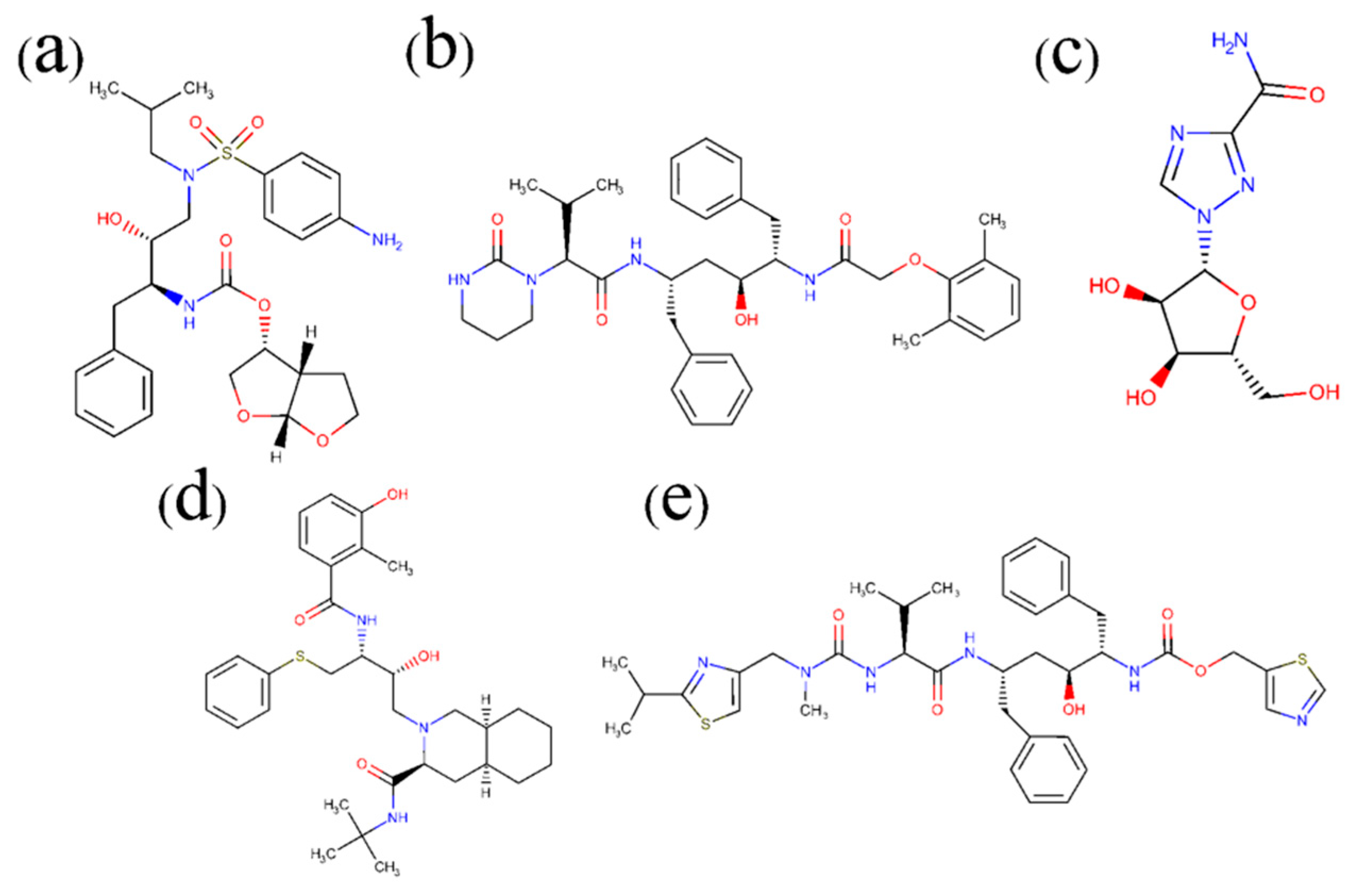
2.1. Complex Formation by Docking
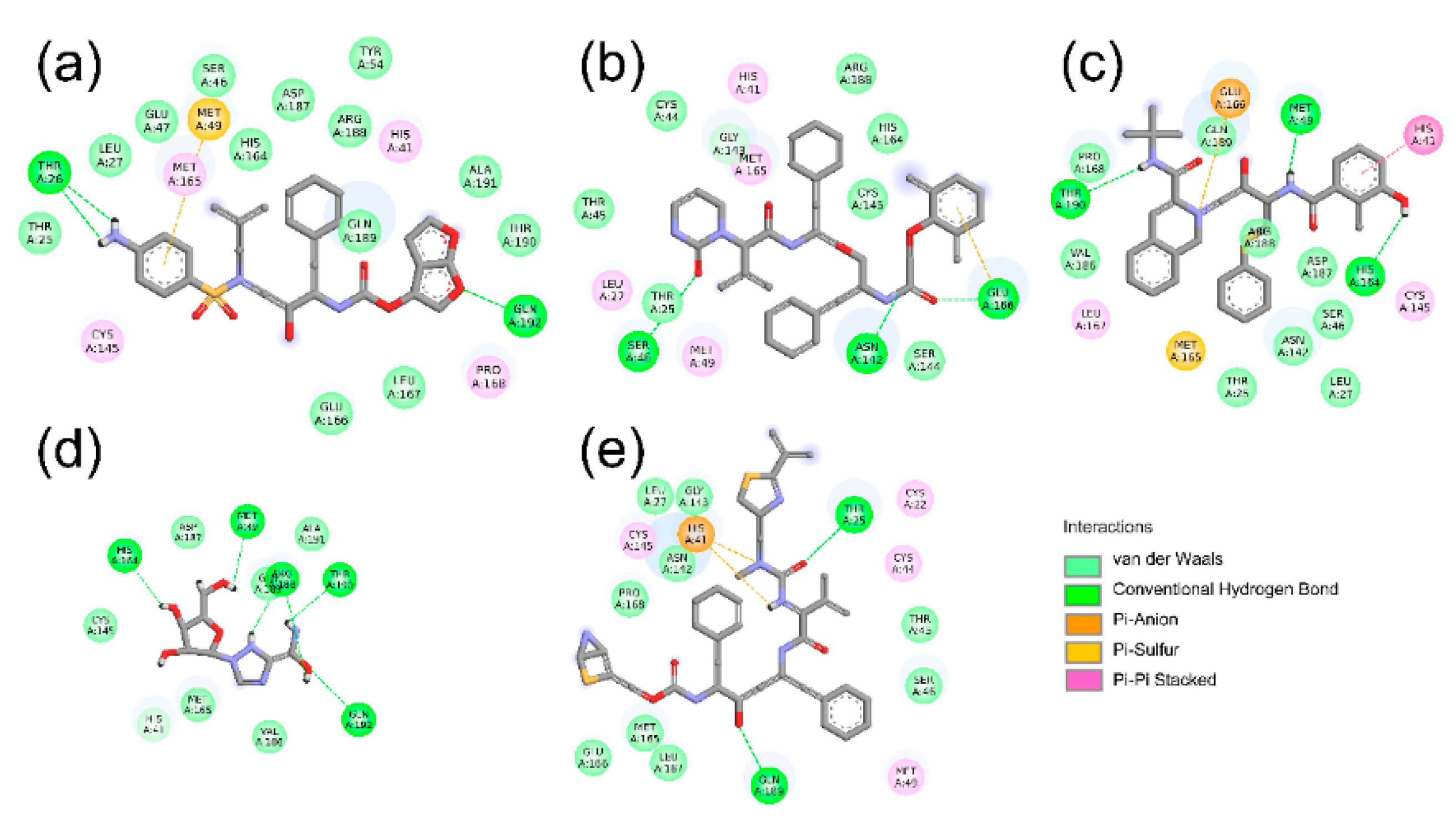
2.2. Hydrogen Bond and Salt Bridge Interactions
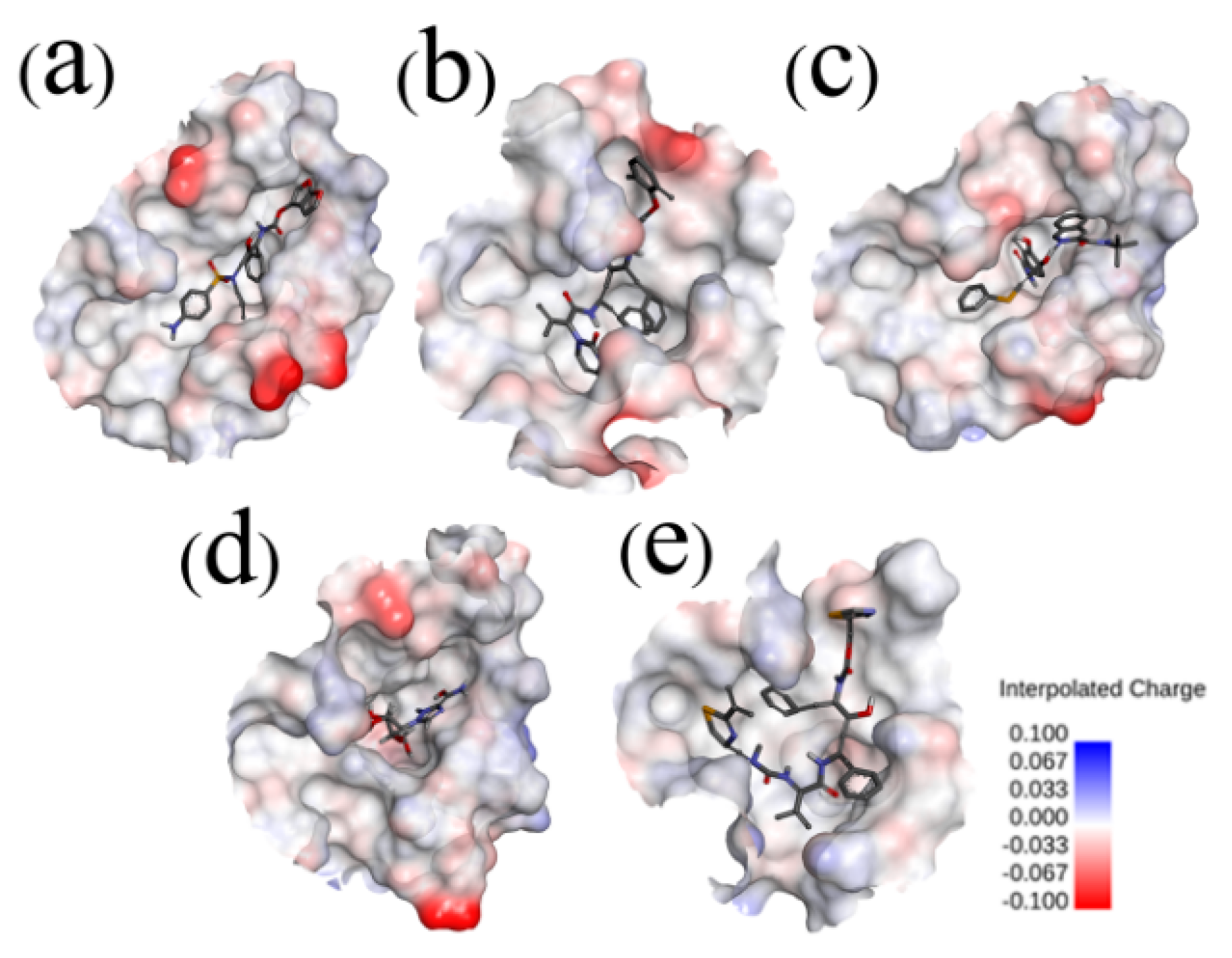
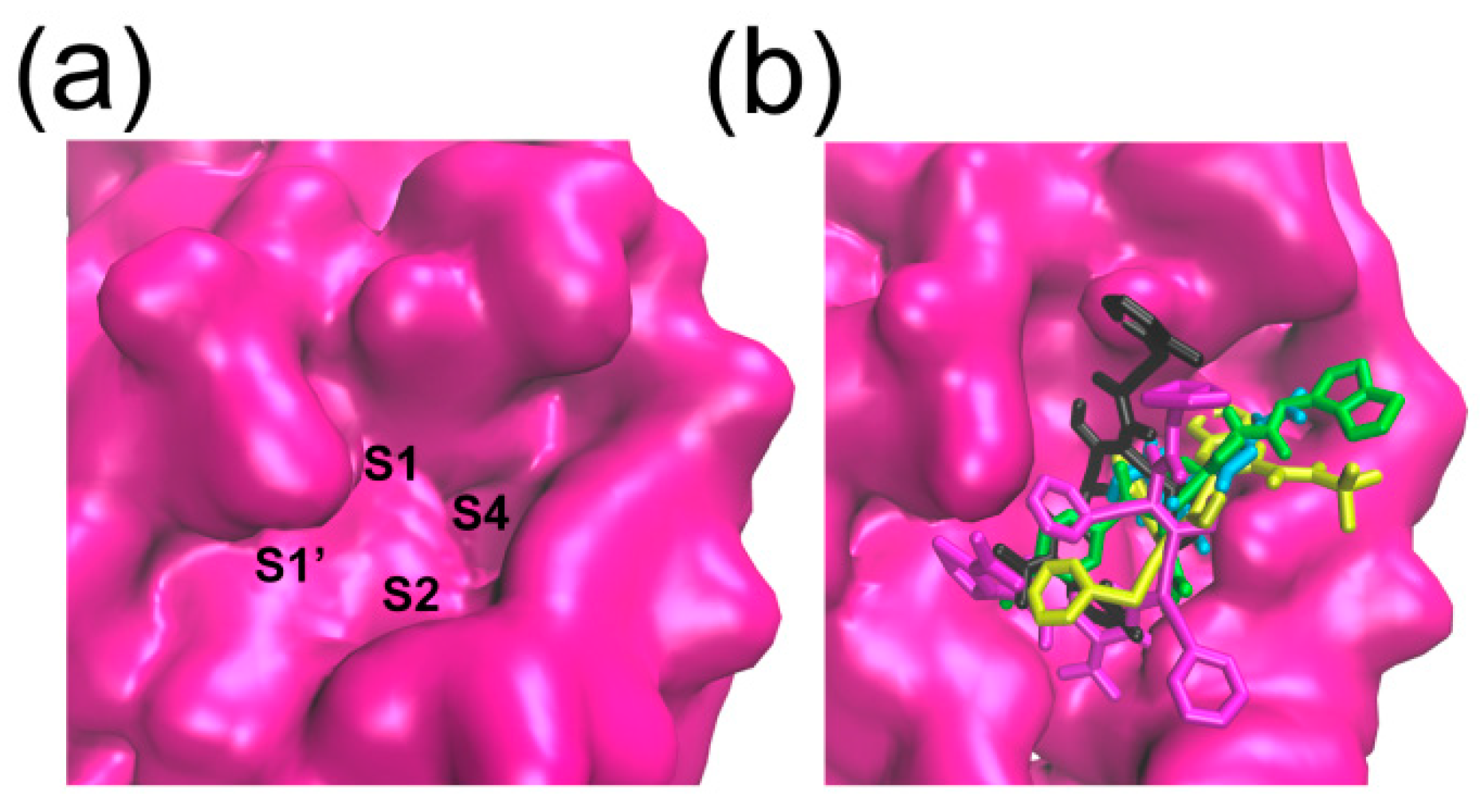
2.3. Binding Pocket Analysis
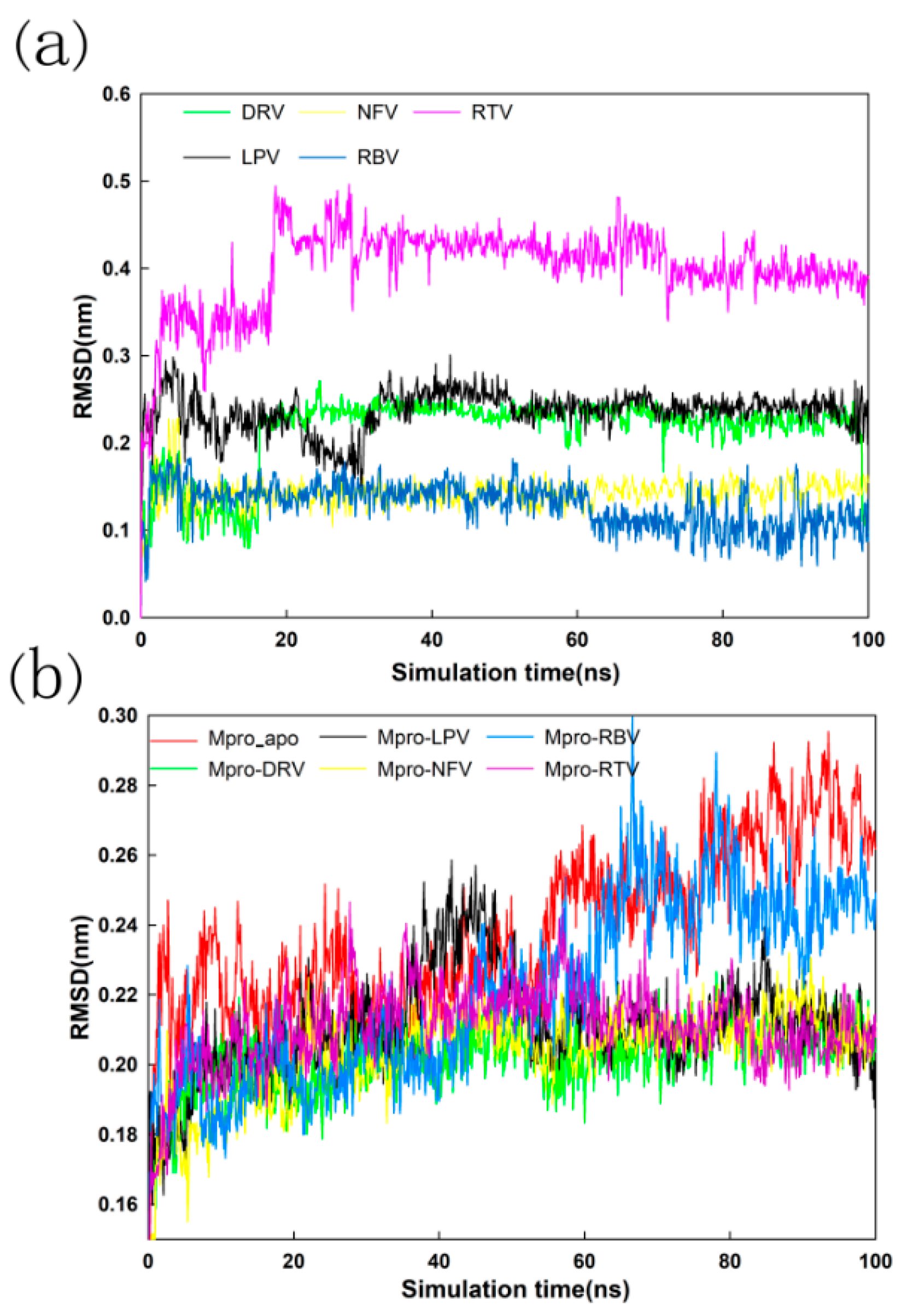
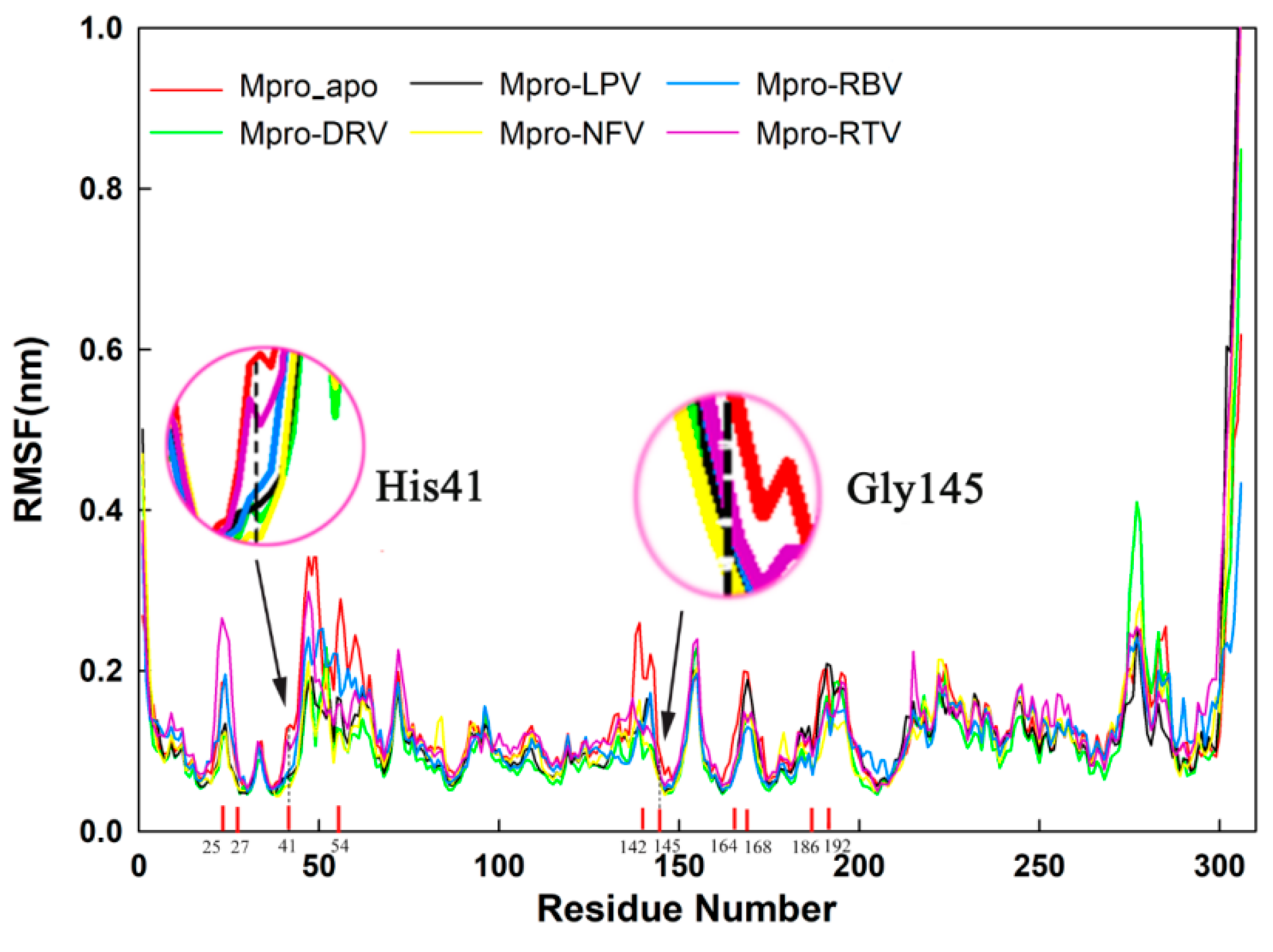
2.4. Stability Analysis of Docked Complexes
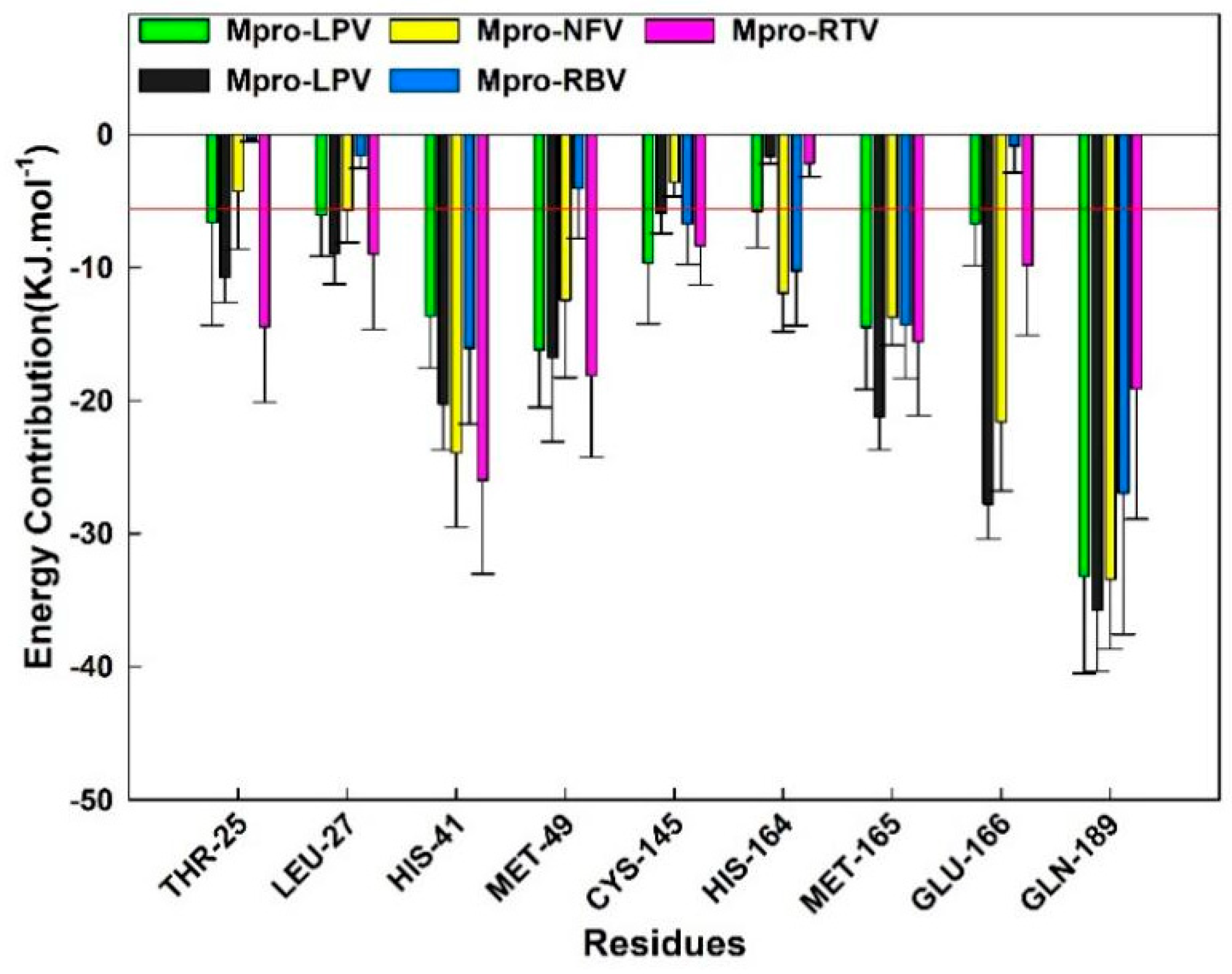
2.5. MM–GBSA and Energy Decomposition
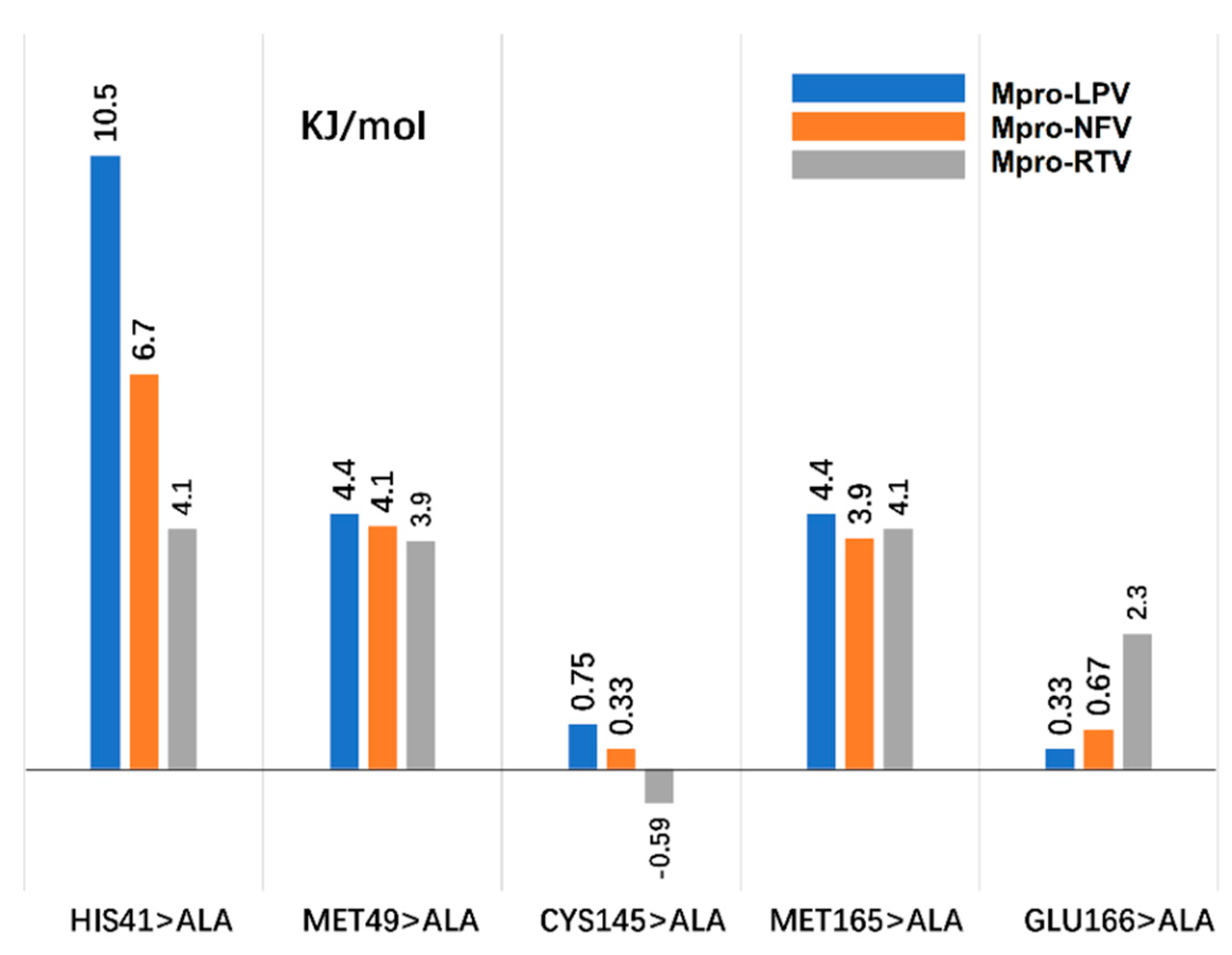
2.6. Computational Alanine Scanning (CAS)
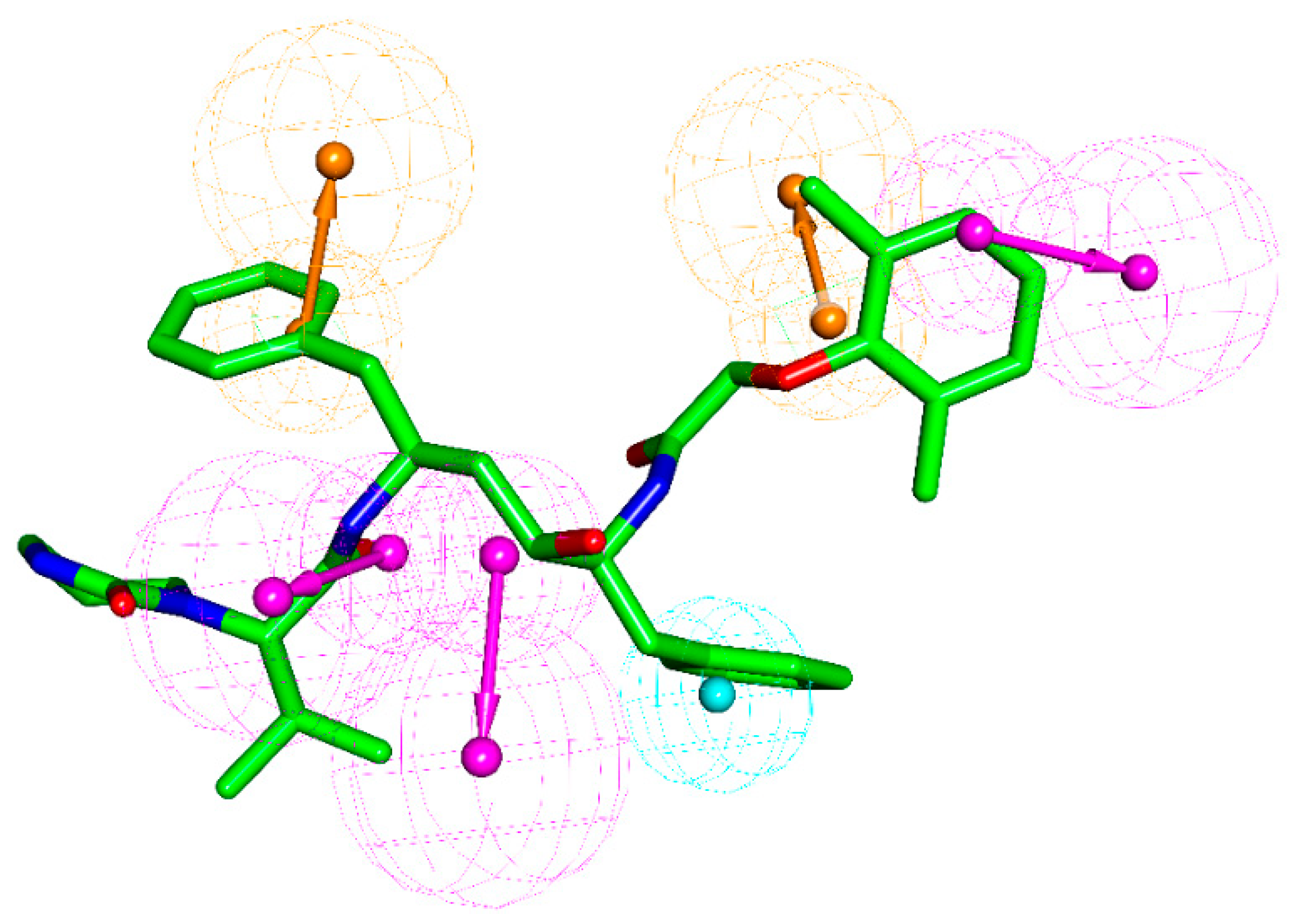
2.7. Pharmacophore Model Analysis
3. Materials and Methods
3.1. Structure Preparation
3.2. Docking and MD Simulations
3.3. Binding Energy Calculation by MM–GBSA
3.4. Computational Alanine Scanning (CAS)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Joshi, R.S.; Jagdale, S.S.; Bansode, S.B.; Shankar, S.S.; Tellis, M.B.; Pandya, V.K.; Chugh, A.; Giri, A.P.; Kulkarni, M.J.J.J.o.B.S. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 2020, 39, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Song, W.; Huang, H.; Sun, Q. Pharmacological therapeutics targeting RNA-dependent RNA polymerase, proteinase and spike protein: From mechanistic studies to clinical trials for COVID-19. J. Clin. Med. 2020, 9, 1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, I.-L.; Mahindroo, N.; Liang, P.-H.; Peng, Y.-H.; Kuo, C.-J.; Tsai, K.-C.; Hsieh, H.-P.; Chao, Y.-S.; Wu, S.-Y. Structure-based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J. Med. Chem. 2006, 49, 5154–5161. [Google Scholar] [CrossRef]
- Du, Q.; Wang, S.; Wei, D.; Sirois, S.; Chou, K.-C.J.A.B. Molecular modeling and chemical modification for finding peptide inhibitor against severe acute respiratory syndrome coronavirus main proteinase. Anal. Biochem. 2005, 337, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.J.N. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchan, A. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 13, 3213–3224. [Google Scholar] [CrossRef]
- Mills, A.M.; Nelson, M.; Jayaweera, D.; Ruxrungtham, K.; Cassetti, I.; Girard, P.-M.; Workman, C.; Dierynck, I.; Sekar, V.; Abeele, C.V.; et al. Once-daily darunavir/ritonavir vs. lopinavir/ritonavir in treatment-naive, HIV-1-infected patients: 96-week analysis. AIDS 2009, 23, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Yang, R.; Yoshinaka, Y.; Amari, S.; Nakano, T.; Cinatl, J.; Rabenau, H.; Doerr, H.W.; Hunsmann, G.; Otaka, A.; et al. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004, 318, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Boffito, M.; Arnaudo, I.; Raiteri, R.; Bonora, S.; Sinicco, A.; Garbo, A.D.; Reynolds, H.E.; Hoggard, P.G.; Back, D.J.; Perri, G.D. Clinical use of lopinavir/ritonavir in a salvage therapy setting: Pharmacokinetics and pharmacodynamics. AIDS 2002, 16, 2081–2083. [Google Scholar] [CrossRef] [PubMed]
- Bolcato, G.; Bissaro, M.; Pavan, M.; Sturlese, M.; Moro, S. Targeting the coronavirus SARS-CoV-2: Computational insights into the mechanism of action of the protease inhibitors lopinavir, ritonavir and nelfinavir. Sci. Rep. 2020, 10, 20927. [Google Scholar] [CrossRef]
- Cardoso, W.B.; Mendanha, S.A. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. J. Mol. Struct. 2021, 1225, 129143. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, A.; Mohammad, T.; Anwar, S.; AlAjmi, M.F.; Hussain, A.; Rehman, M.T.; Islam, A.; Hassan, M.I. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: Possible implication in COVID-19 therapy. Biosci. Rep. 2020, 40, BSR20201256. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Zhou, H.X. Profiling SARS-CoV-2 Main Protease (M(PRO)) Binding to Repurposed Drugs Using Molecular Dynamics Simulations in Classical and Neural Network-Trained Force Fields. ACS Comb. Sci. 2020, 22, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Madruga, J.V.; Berger, D.; McMurchie, M.; Suter, F.; Banhegyi, D.; Ruxrungtham, K.; Norris, D.; Lefebvre, E.; de Béthune, M.-P.; Tomaka, F.; et al. Efficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV-infected patients in TITAN: A randomised controlled phase III trial. Lancet 2007, 370, 49–58. [Google Scholar] [CrossRef]
- Kraus, M.; Müller-Ide, H.; Rückrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk. Res. 2014, 38, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Chandwani, A.; Shuter, J. Lopinavir/ritonavir in the treatment of HIV-1 infection: A review. Ther. Clin. Risk Manag. 2008, 4, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Yao, T.-T.; Qian, J.-D.; Zhu, W.-Y.; Wang, Y.; Wang, G.-Q. A systematic review of lopinavir therapy for SARS coronavirus and MERS coronavirus—A possible reference for coronavirus disease-19 treatment option. J. Med. Virol. 2020, 92, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, S.; Singh, M.; Ravichandiran, V.; Murty, U.S.N.; Srivastava, H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, N.; Matsuyama, S.; Hoshino, T.; Yamamoto, N. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. BioRxiv 2020, 026476. [Google Scholar] [CrossRef]
- Yang, Z.W.; Zhao, Y.Z.; Zang, Y.J.; Wang, H.; Zhu, X.; Meng, L.J.; Yuan, X.H.; Zhang, L.; Zhang, S.L. Rapid Structure-Based Screening Informs Potential Agents for Coronavirus Disease (COVID-19) Outbreak. Chin. Phys. Lett. 2020, 37, 058701. [Google Scholar] [CrossRef]
- Luan, B.; Huynh, T.; Cheng, X.; Lan, G.; Wang, H.R. Targeting Proteases for Treating COVID-19. J. Proteome Res. 2020, 19, 4316–4326. [Google Scholar] [CrossRef]
- Arshad, U.; Pertinez, H.; Box, H.; Tatham, L.; Rajoli, R.K.R.; Curley, P.; Neary, M.; Sharp, J.; Liptrott, N.J.; Valentijn, A.; et al. Prioritization of Anti-SARS-Cov-2 Drug Repurposing Opportunities Based on Plasma and Target Site Concentrations Derived from their Established Human Pharmacokinetics. Clin. Pharmacol. Ther. 2020, 108, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.; Sun, H.Y.; Wang, J.M.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T.J. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lin, S.J.; Ren, J.Y.; Liu, T.; Wang, Y.M.; Li, C.M.; Xu, W.W.; He, Y.W.; Zheng, W.H.; Zhao, J.; et al. Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor H (CFH) Antibody Ab42 and CFH Polypeptide. Int. J. Mol. Sci. 2019, 20, 2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, B.K.; Faheem; Sekhar, K.V.G.C.; Ojha, R.; Prajapati, V.K.; Pai, A.; Murugesan, S. Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases. J. Biomol. Struct. Dyn. 2020, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Vandelli, A.; Monti, M.; Milanetti, E.; Armaos, A.; Rupert, J.; Zacco, E.; Bechara, E.; Ponti, R.D.; Tartaglia, G.G. Structural analysis of SARS-CoV-2 genome and predictions of the human interactome. Nucleic Acids Res. 2020, 48, 11270–11283. [Google Scholar] [CrossRef]
- Balakrishnan, V.; Lakshminarayanan, K. Screening of FDA Approved Drugs Against SARS-CoV-2 Main Protease: Coronavirus Disease. Int. J. Pept. Res. Ther. 2020, 65, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Accelrys. Discovery Studio 4.5. Available online: http://accelrys.com (accessed on 12 November 2021).
- Yang, Z.W.; Wu, X.M.; Yang, G.; Zu, Y.G.; Zhou, L.J. Understanding the chiral recognitions between neuraminidases and inhibitors: Studies with DFT, docking, and MD methods. Int. J. Quantum Chem. 2012, 112, 909–921. [Google Scholar] [CrossRef]
- Ren, J.; Yuan, X.; Li, J.; Lin, S.; Yang, B.; Chen, C.; Zhao, J.; Zheng, W.; Liao, H.; Yang, Z. Assessing the performance of the g_mmpbsa tools to simulate the inhibition of oseltamivir to influenza virus neuraminidase by molecular mechanics Poisson–Boltzmann surface area methods. J. Chin. Chem. Soc. 2020, 67, 46–53. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, Y.; Hao, D.; Wang, H.; Li, S.; Jia, L.; Yuan, X.; Zhang, L.; Meng, L.; Zhang, S. Computational identification of potential chemoprophylactic agents according to dynamic behavior of peroxisome proliferator-activated receptor gamma. RSC Adv. 2021, 11, 147–159. [Google Scholar] [CrossRef]
- Luo, Q.; Zhang, C.; Miao, L.; Zhang, D.; Bai, Y.; Hou, C.; Liu, J.; Yan, F.; Mu, Y.; Luo, G. Triple mutated antibody scFv2F3 with high GPx activity: Insights from MD, docking, MDFE, and MM-PBSA simulation. Amino Acids 2013, 44, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, G.; Zhou, L. Mutation effects of neuraminidases and their docking with ligands: A molecular dynamics and free energy calculation study. J. Comput. Aided Mol. Des. 2013, 27, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Hao, D.; Wang, H.; Yang, Z.; Liu, H.; Zhang, S. Structure-based methoxyflavone derivatives with potent inhibitory activity against various influenza neuraminidases. J. Biomol. Struct. Dyn. 2020, 38, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Pullman, B., Ed.; Springer: Dordrecht, The Netherlands, 1981; Volume 14, pp. 331–342. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.-H.; Wang, Y.-C.; Jin, W.-J.; Zhao, B.-B.; Chen, C.-F.; Yang, J.; Wang, J.-F.; Guo, Y.-Y.; Liu, J.-J.; Zhang, D. Structure-based high-throughput epitope analysis of hexon proteins in B and C species human adenoviruses (HAdVs). PLoS ONE 2012, 7, e32938. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Alberto, A.; Ribas-Aparicio, R.M.; Aparicio-Ozores, G.; Castelan-Vega, J.A. Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors. Comput. Biol. Chem. 2020, 88, 107325. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Yu, W.; Wu, X.; Ren, J.; Zhang, X.; Wang, Y.; Li, C.; Xu, W.; Li, J.; Li, G.; Zheng, W.; et al. Mechanistic Insights to the Binding of Antibody CR3022 Against RBD from SARS-CoV and HCoV-19/SARS-CoV-2: A Computational Study. Comb. Chem. High Throughput Screen. 2021, 24, 1069–1082. [Google Scholar] [CrossRef]
- Sargolzaei, M.; Afshar, M.; Jorabchi, M. Binding of 1-substituted carbazolyl-3, 4-dihydro-β-carbolines with DNA: Molecular dynamics simulation and MM-GBSA analysis. Mol. Biol. 2016, 50, 313–319. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham , T.E., III; DeBolt, S.; Fergusond, D.; Seibele, G.; Kollmanc, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Martins, S.A.; Perez, M.A.S.; Moreira, I.S.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Computational Alanine Scanning Mutagenesis: MM-PBSA vs TI. J. Chem. Theory Comput. 2013, 9, 1311–1319. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Donor and Acceptor | Category |
|---|---|---|
| SARS-CoV-2 Mpro-DRV | GLN192:H–DRV:O26 | Hydrogen Bond |
| MET165:HA–DRV:O10 | Hydrogen Bond | |
| DRV:H39–THR26:O | Hydrogen Bond | |
| DRV:H40–THR26:O | Hydrogen Bond | |
| HIS41:Pi-Orbitals–DRV:Alkyl | Hydrophobic | |
| DRV:Pi-Orbitals–CYS145:Alkyl | Hydrophobic | |
| DRV:Pi-Orbitals–PRO168:Alkyl | Hydrophobic | |
| DRV:C15–MET49:Alkyl | Hydrophobic | |
| SARS-CoV-2 Mpro-LPV | GLU166:OE1–LPV:C42 | Electrostatic |
| SER46:HG–LPV:O12 | Hydrogen Bond | |
| GLU166:H–LPV:O38 | Hydrogen Bond | |
| LPV:H78–ASN142:OD1 | Hydrogen Bond | |
| GLY143:HA2–LPV:O20 | Hydrogen Bond | |
| HIS41:Pi-Orbitals–LPV:Alkyl | Hydrophobic | |
| SARS-CoV-2 Mpro-NFV | NFV:N7–GLU166:OE1 | Electrostatic |
| NFV:H56–THR190:O | Hydrogen Bond | |
| NFV:H71–MET49:SD | Hydrogen Bond | |
| NFV:H77–HIS164:O | Hydrogen Bond | |
| HIS41:Pi-Orbitals–NFV:Pi-Orbitals | Hydrophobic | |
| NFV:C33–CYS145:Alkyl | Hydrophobic | |
| HIS41:Pi-Orbitals–NFV:C33 | Hydrophobic | |
| NFV:Pi-Orbitals–LEU167:Alkyl | Hydrophobic | |
| NFV:Pi-Orbitals–MET49:Alkyl | Hydrophobic | |
| SARS-CoV-2 Mpro-RBV | GLN192:HE21–RBV:O5 | Hydrogen Bond |
| RBV:H18–THR190:O | Hydrogen Bond | |
| RBV:H25–HIS164:O | Hydrogen Bond | |
| RBV:H29–MET49:SD | Hydrogen Bond | |
| RBV:H6–ARG188:O | Hydrogen Bond | |
| RBV:H7–ARG188:O | Hydrogen Bond | |
| RBV:H14–ARG188:O | Hydrogen Bond | |
| SARS-CoV-2 Mpro-RTV | RTV:N39–HIS41:Pi-Orbitals | Electrostatic |
| THR25:HG1–RTV:O41 | Hydrogen Bond | |
| GLN189:HE21–RTV:O22 | Hydrogen Bond | |
| RTV:H77–HIS41:Pi-Orbitals | Hydrogen Bond | |
| RTV:C50–HIS41:Pi-Orbitals | Hydrophobic | |
| RTV:Pi-Orbitals–MET49:Alkyl | Hydrophobic |
| Complex | His41 (nm) | Gly145 (nm) |
|---|---|---|
| Mpro_apo | 0.128 ± 0.009 | 0.090 ± 0.007 |
| Darunavir | 0.070 ± 0.010 | 0.056 ± 0.007 |
| Lopinavir | 0.067 ± 0.012 | 0.063 ± 0.010 |
| Nelfinavir | 0.057 ± 0.008 | 0.057 ± 0.012 |
| Ribavirin | 0.072 ± 0.009 | 0.067 ± 0.009 |
| Ritonavire | 0.113 ± 0.013 | 0.066 ± 0.011 |
| Complex | ΔEvdw | ΔEele | ΔGGB | ΔGSA | TΔS a | ΔGS b |
|---|---|---|---|---|---|---|
| Darunavir (DRV) | −276.98 ± 17.49 | −337.44 ± 36.65 | 558.19 ± 35.31 | −27.03 ± 1.34 | −97.48 ± 14.94 | 14.22 ± 49.00 |
| Lopinavir (LPV) | −260.66 ± 12.51 | −75.65 ± 15.94 | 134.10 ± 14.64 | −30.63 ± 1.42 | −116.12 ± 16.42 | −116.72 ± 32.90 |
| Nelfinavir (NFV) | −318.70 ± 35.94 | −382.42 ± 56.90 | 542.03 ± 62.51 | −26.61 ± 2.85 | −88.83 ± 25.89 | −96.86 ± 63.66 |
| Ribavirin (RBV) | −122.93 ± 15.10 | −75.65 ± 32.34 | 172.46 ± 27.15 | −16.28 ± 1.30 | −88.07 ± 21.02 | 45.69 ± 49.64 |
| Ritonavire (RTV) | −311.54 ± 42.89 | −13.97 ± 35.69 | 151.96 ± 33.18 | −31.34 ± 2.22 | −101.34 ± 29.29 | −103.55 ± 55.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.; Wu, X.; Zhao, Y.; Chen, C.; Yang, Z.; Zhang, X.; Ren, J.; Wang, Y.; Wu, C.; Li, C.; et al. Computational Simulation of HIV Protease Inhibitors to the Main Protease (Mpro) of SARS-CoV-2: Implications for COVID-19 Drugs Design. Molecules 2021, 26, 7385. https://doi.org/10.3390/molecules26237385
Yu W, Wu X, Zhao Y, Chen C, Yang Z, Zhang X, Ren J, Wang Y, Wu C, Li C, et al. Computational Simulation of HIV Protease Inhibitors to the Main Protease (Mpro) of SARS-CoV-2: Implications for COVID-19 Drugs Design. Molecules. 2021; 26(23):7385. https://doi.org/10.3390/molecules26237385
Chicago/Turabian StyleYu, Wei, Xiaomin Wu, Yizhen Zhao, Chun Chen, Zhiwei Yang, Xiaochun Zhang, Jiayi Ren, Yueming Wang, Changwen Wu, Chengming Li, and et al. 2021. "Computational Simulation of HIV Protease Inhibitors to the Main Protease (Mpro) of SARS-CoV-2: Implications for COVID-19 Drugs Design" Molecules 26, no. 23: 7385. https://doi.org/10.3390/molecules26237385