
An Outline of the Latest Crystallographic Studies on Inhibitor-Enzyme Complexes for the Design and Development of New Therapeutics against Tuberculosis

 , ,
, ,  , , and
, , and 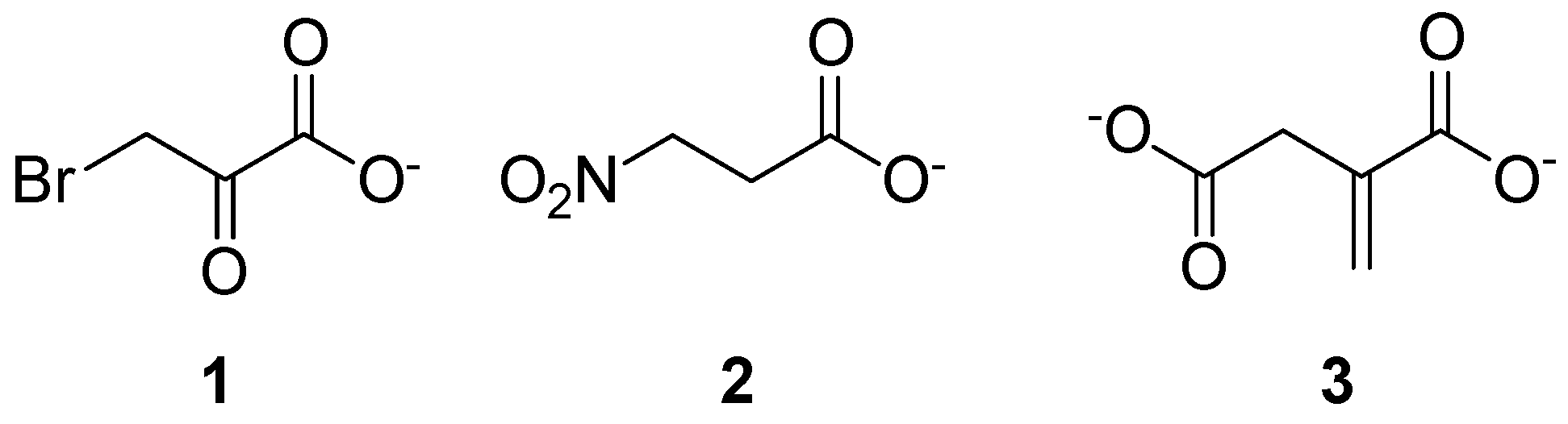{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
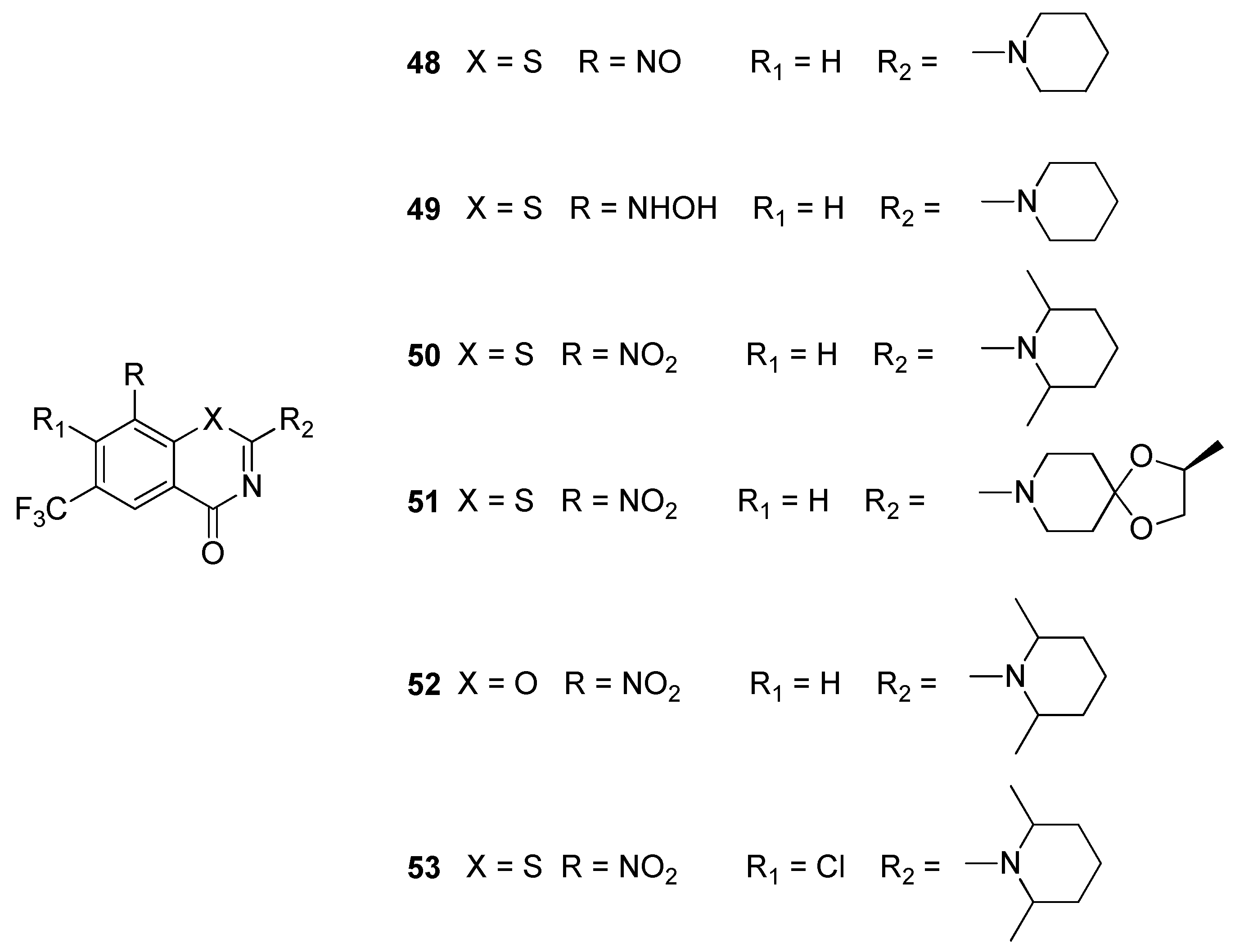{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
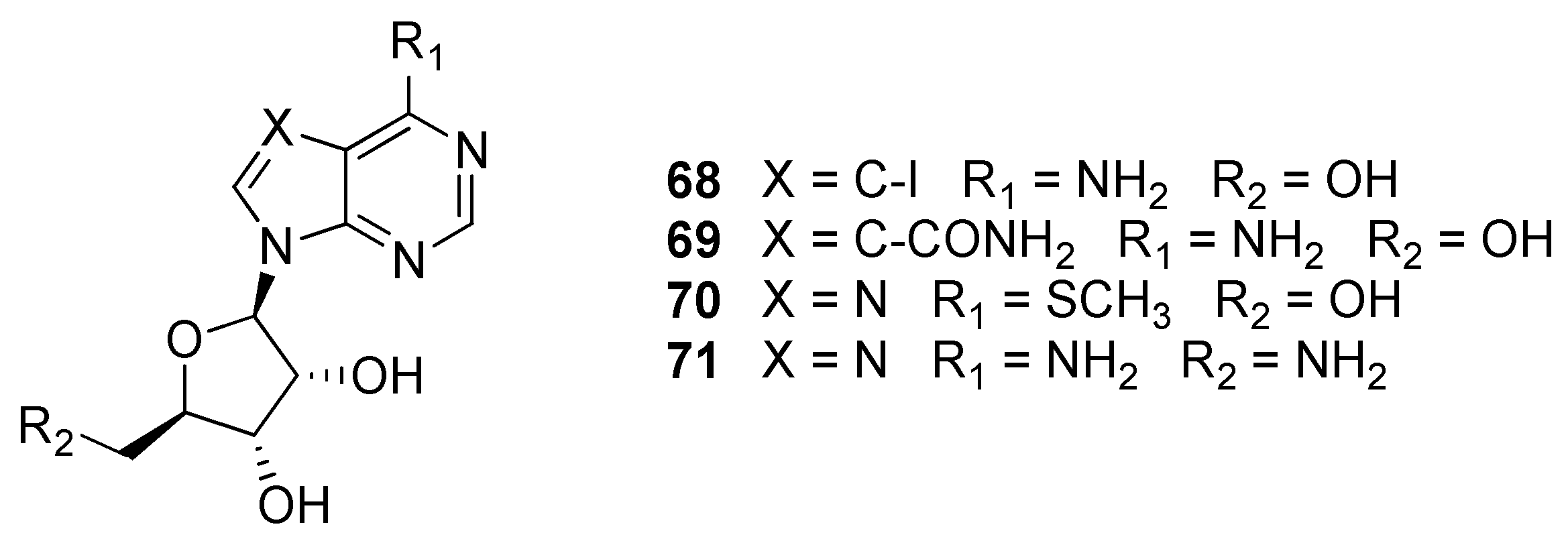{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
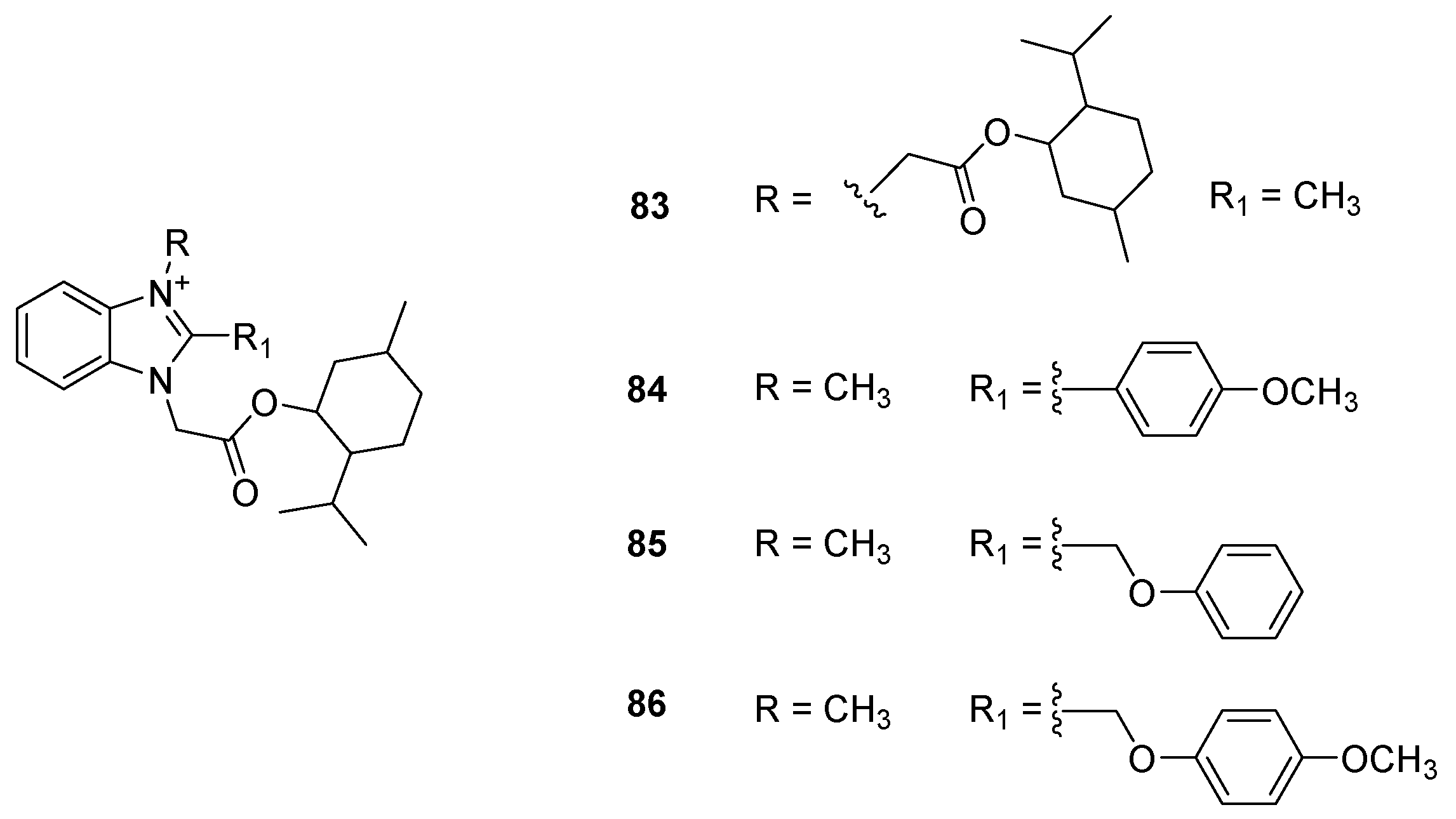{kind=link}
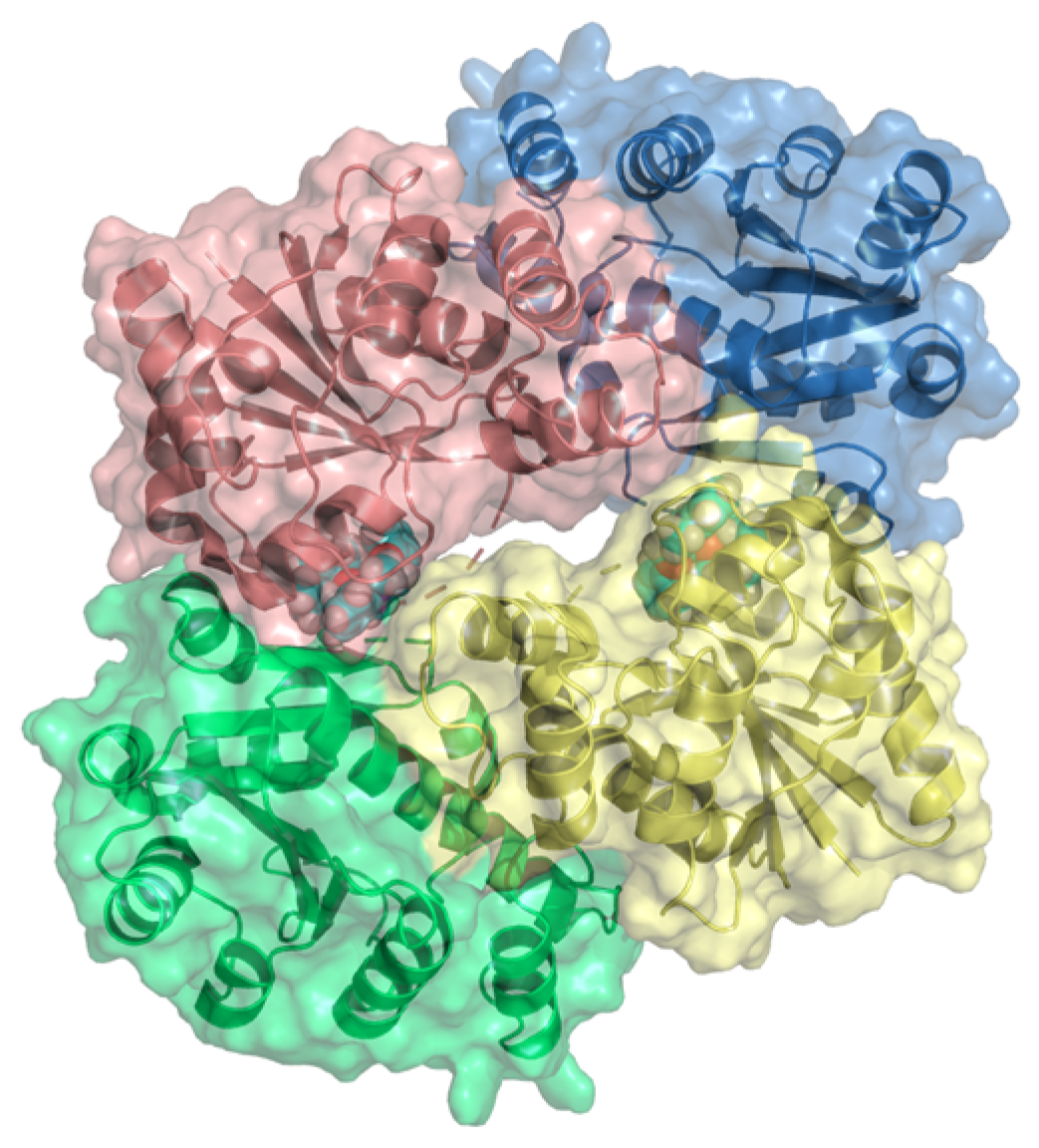{kind=link}
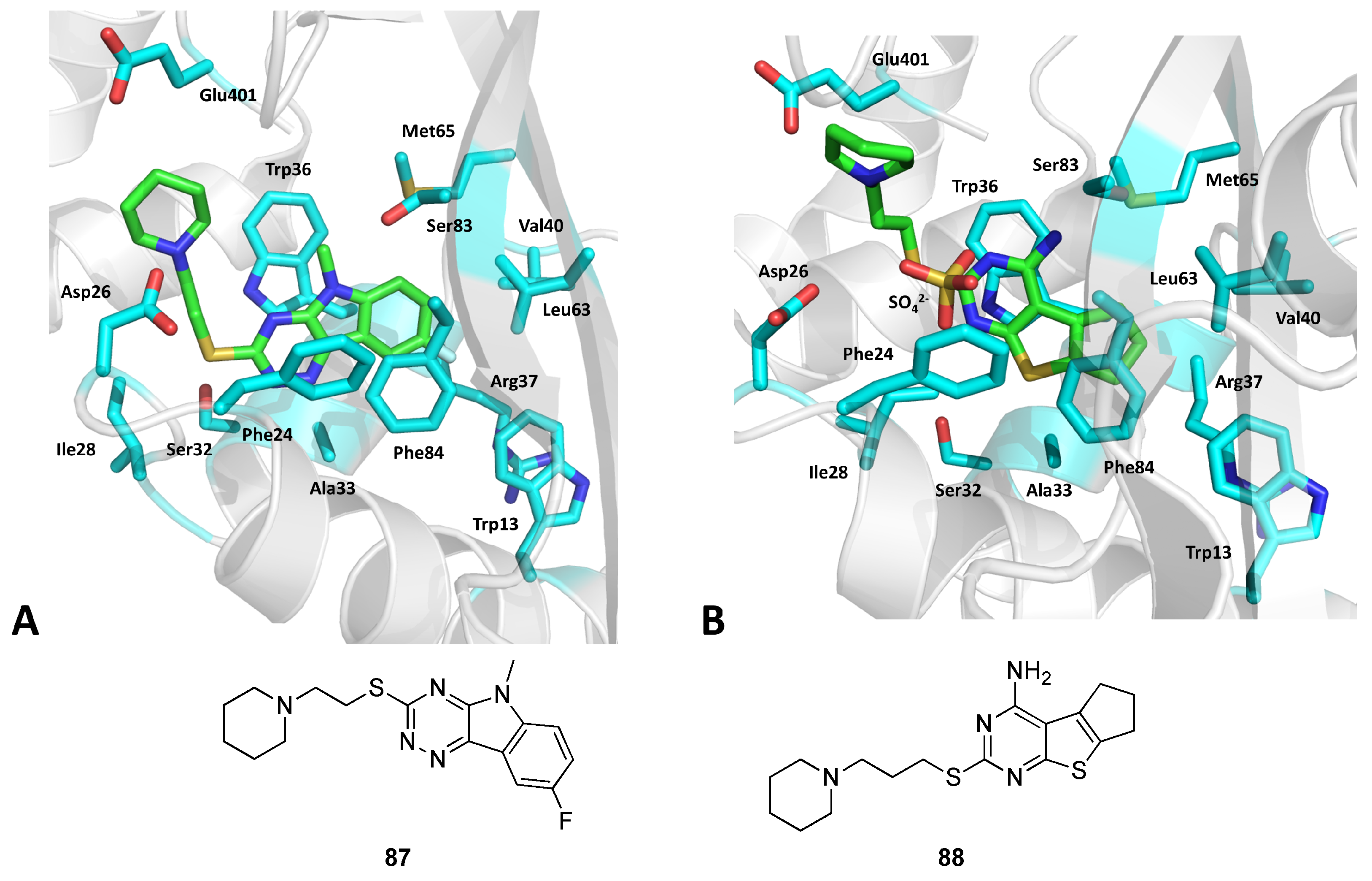{kind=link}
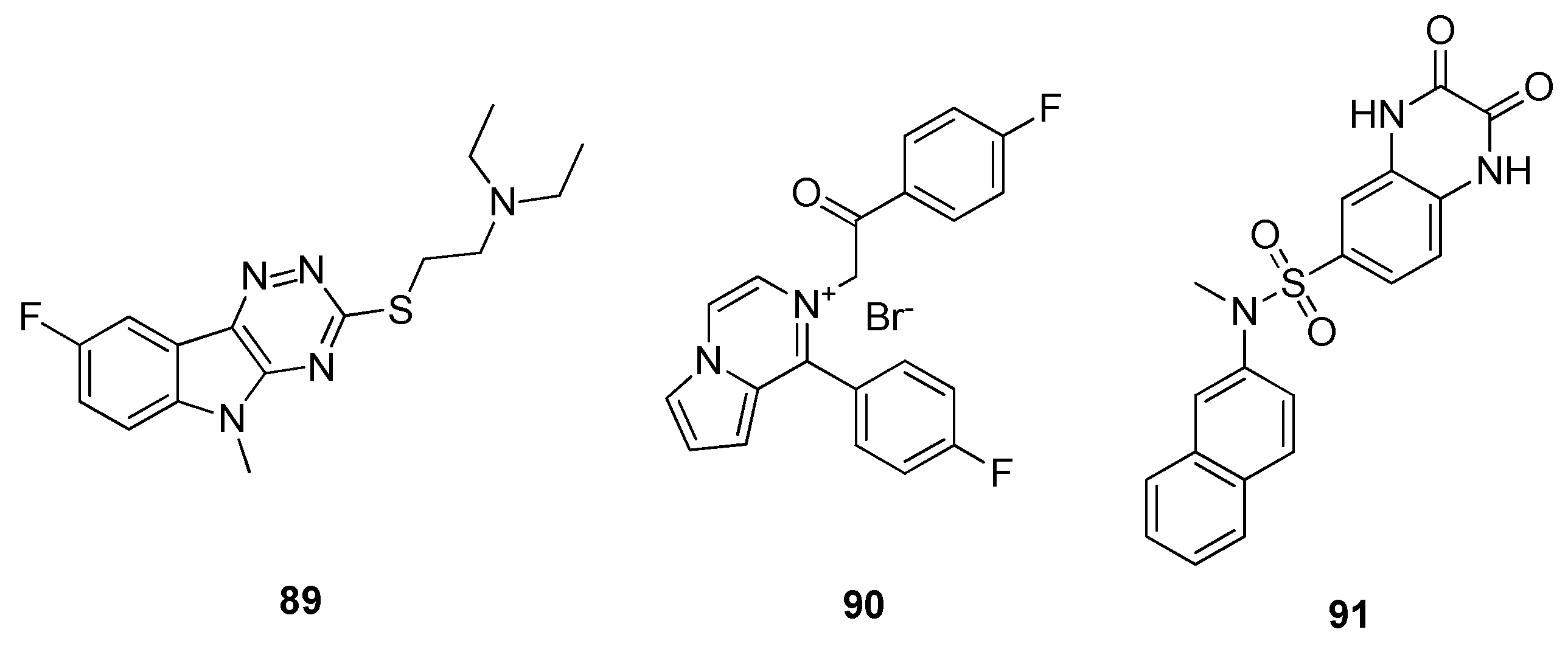{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mtb Enzymatic Targets
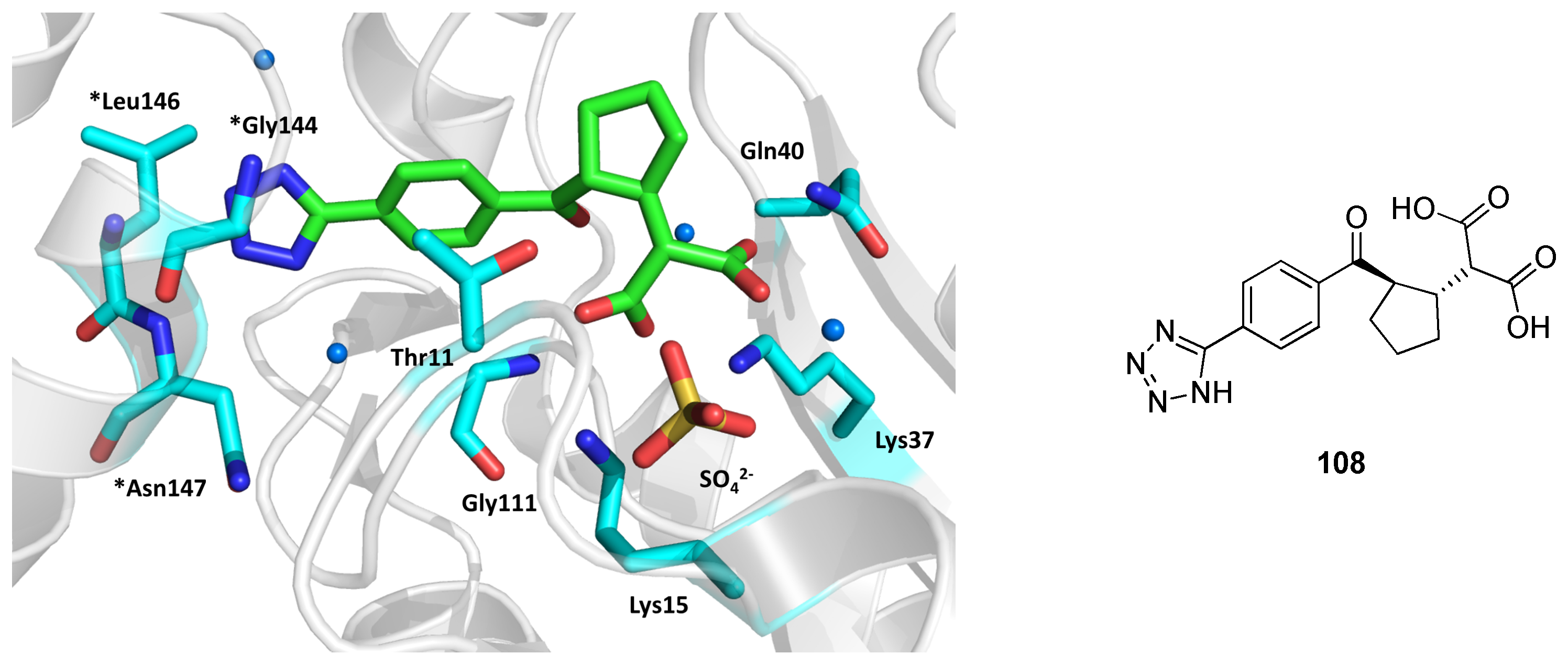
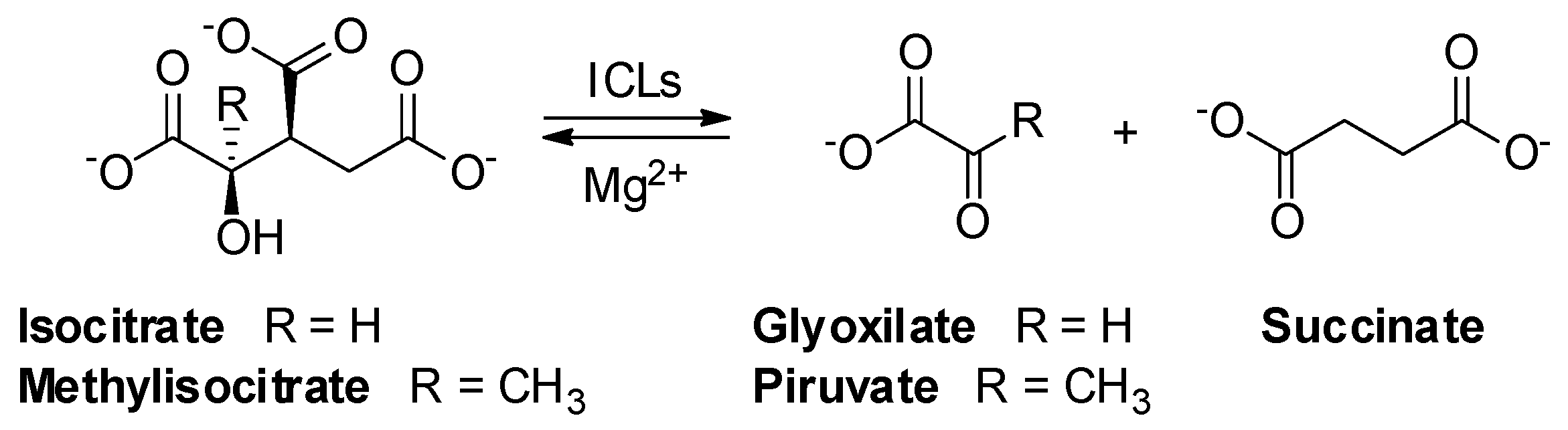
2.1. Isocitrate Lyase 1 (ICL1)
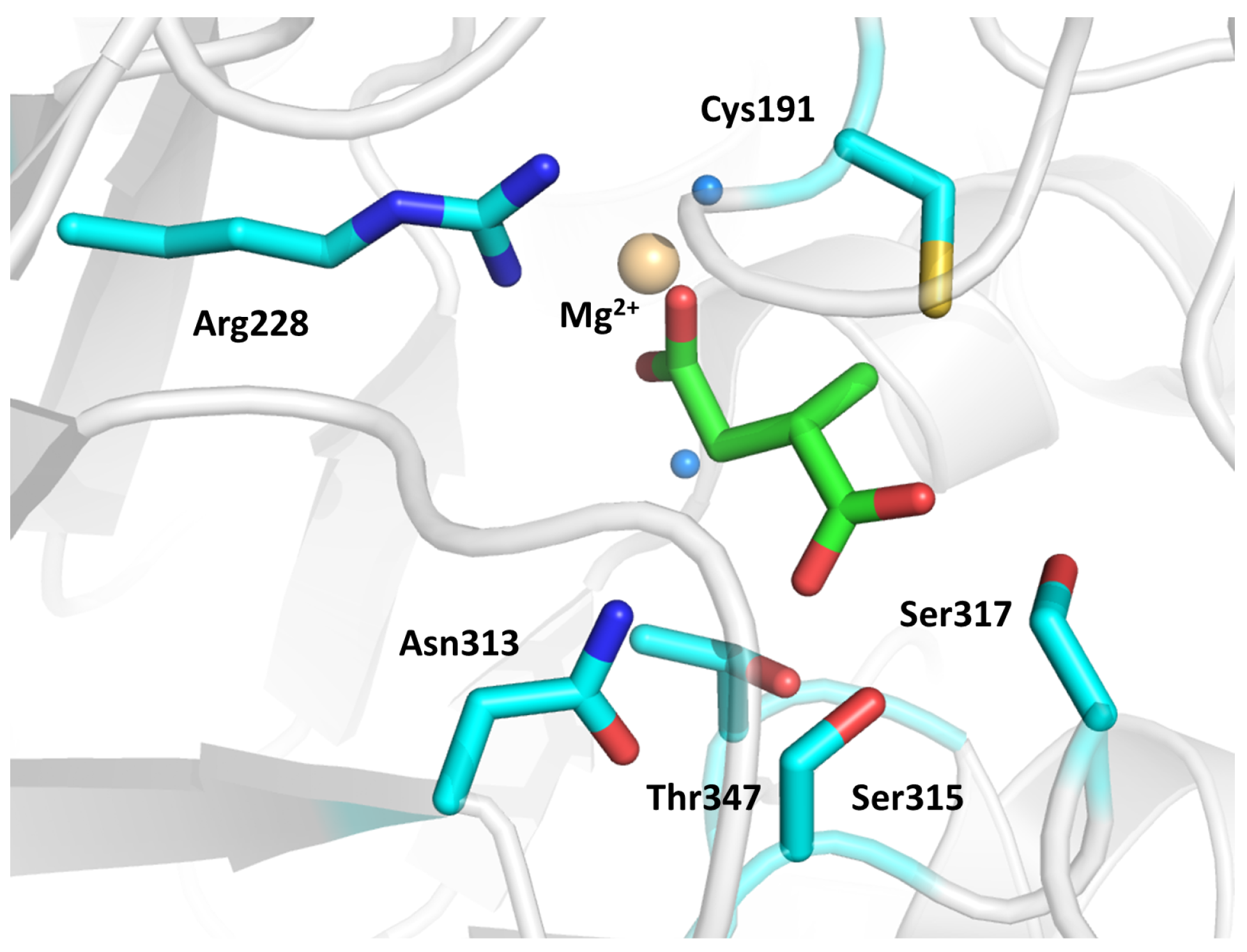
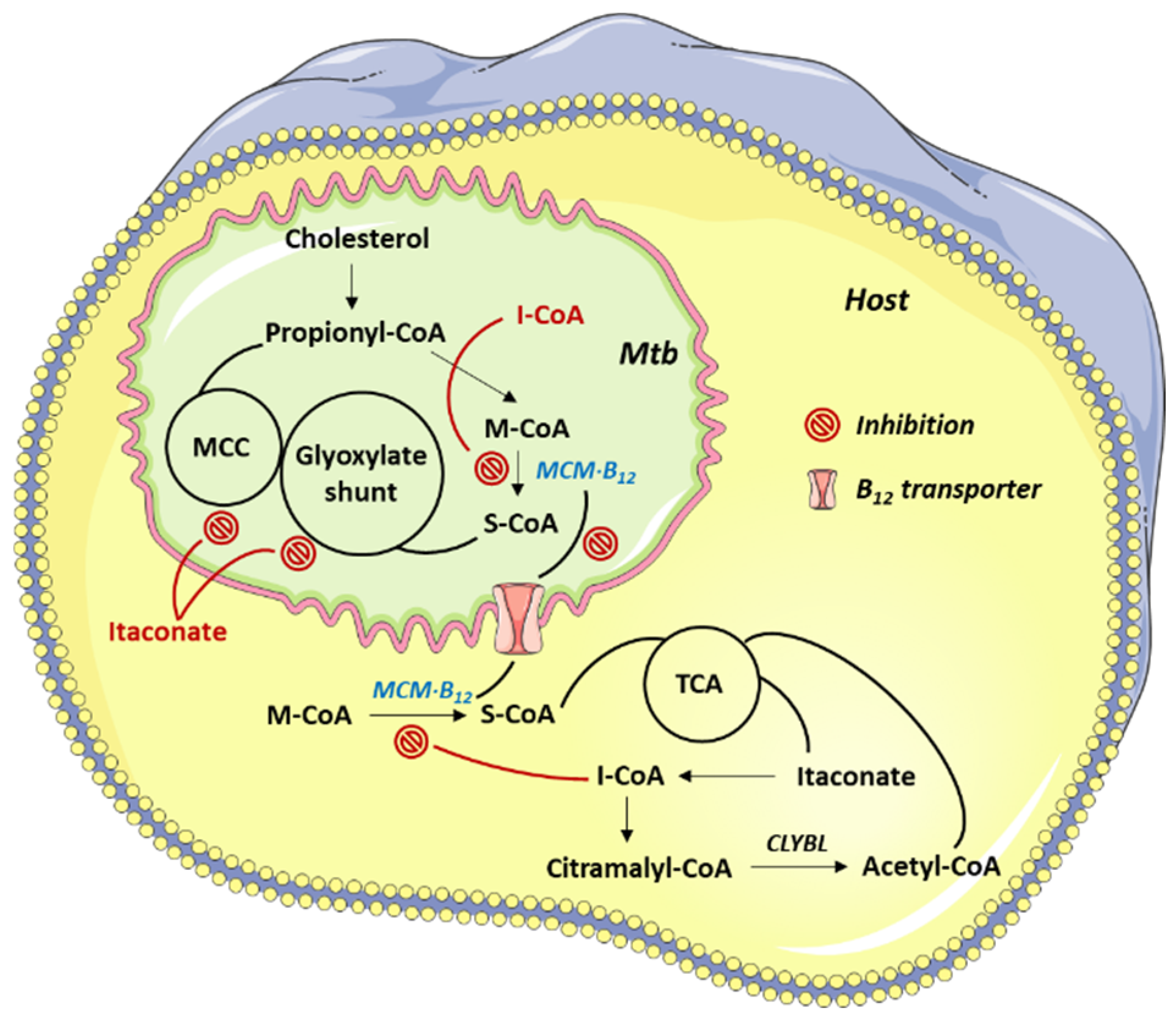
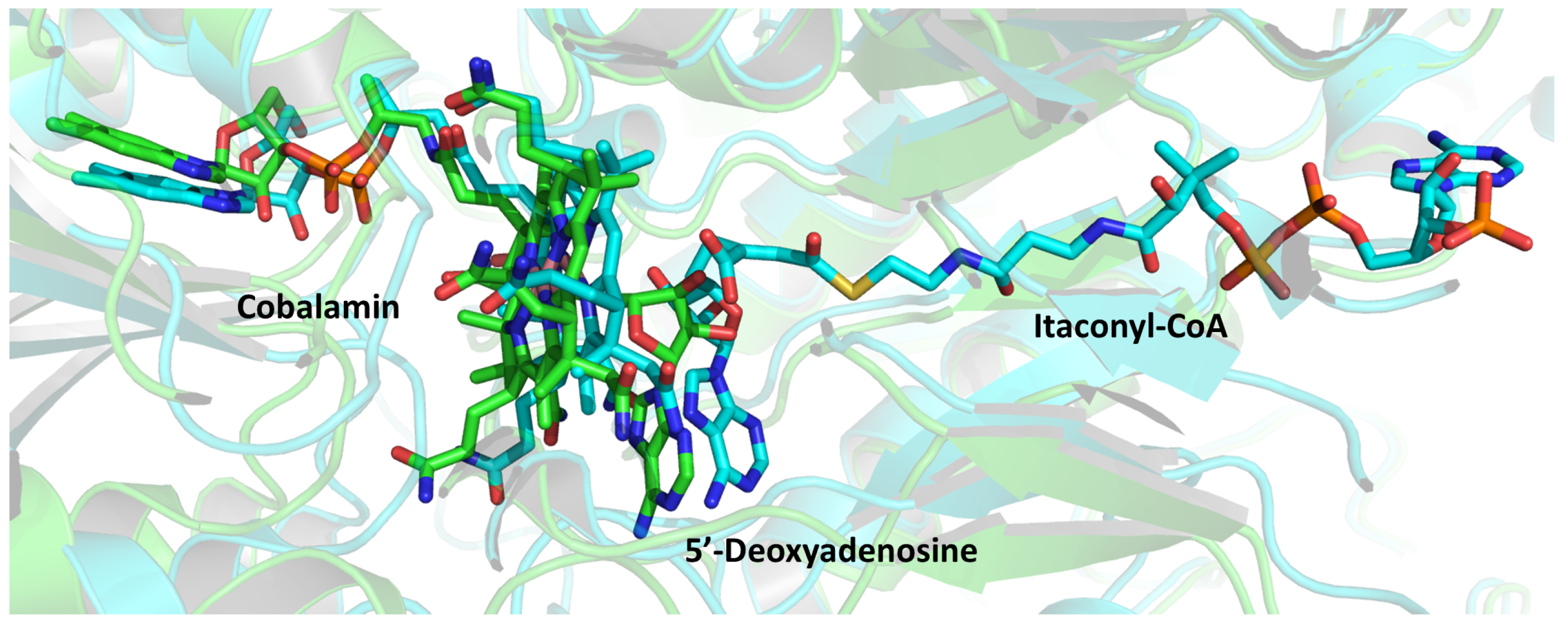
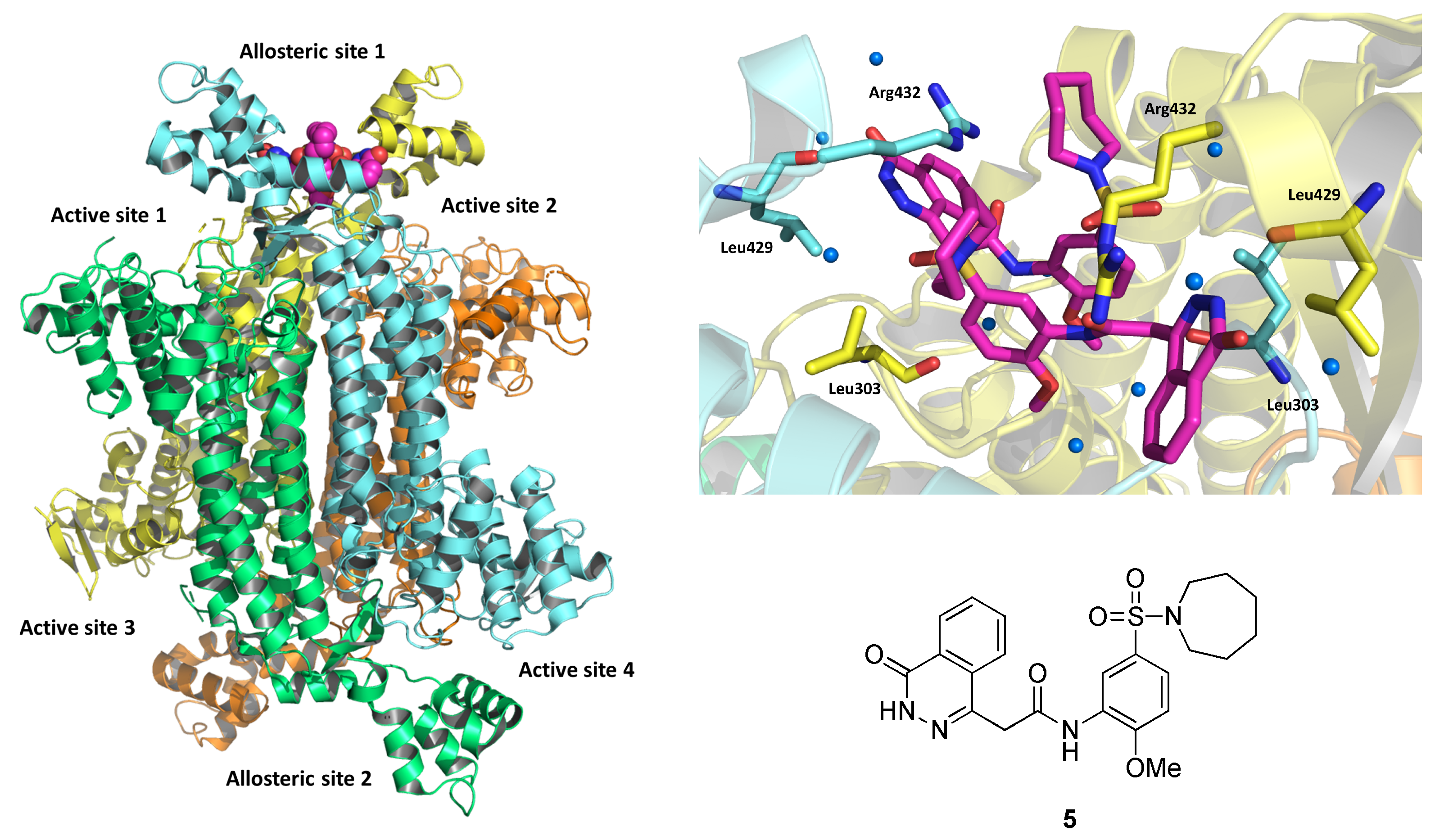
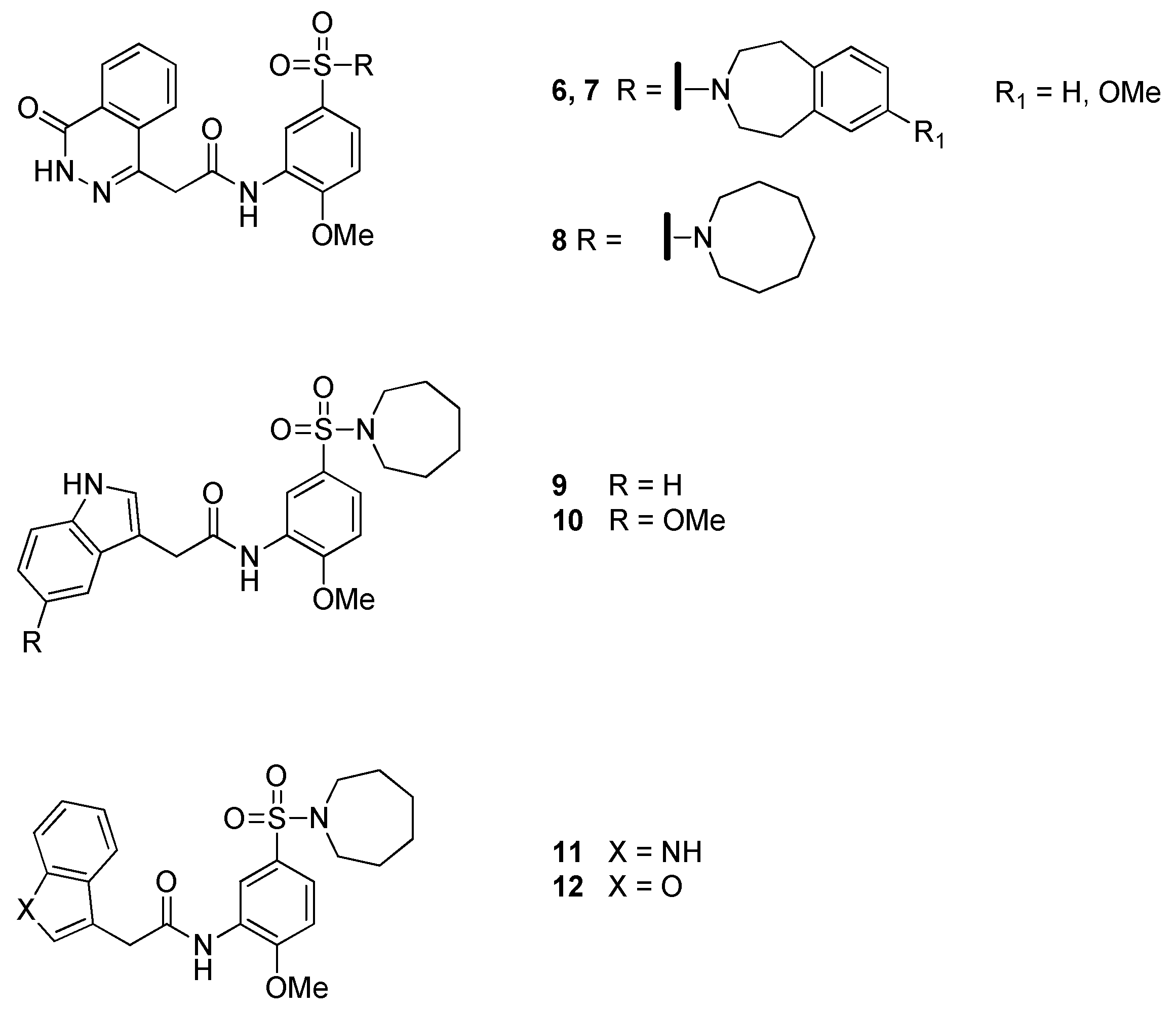
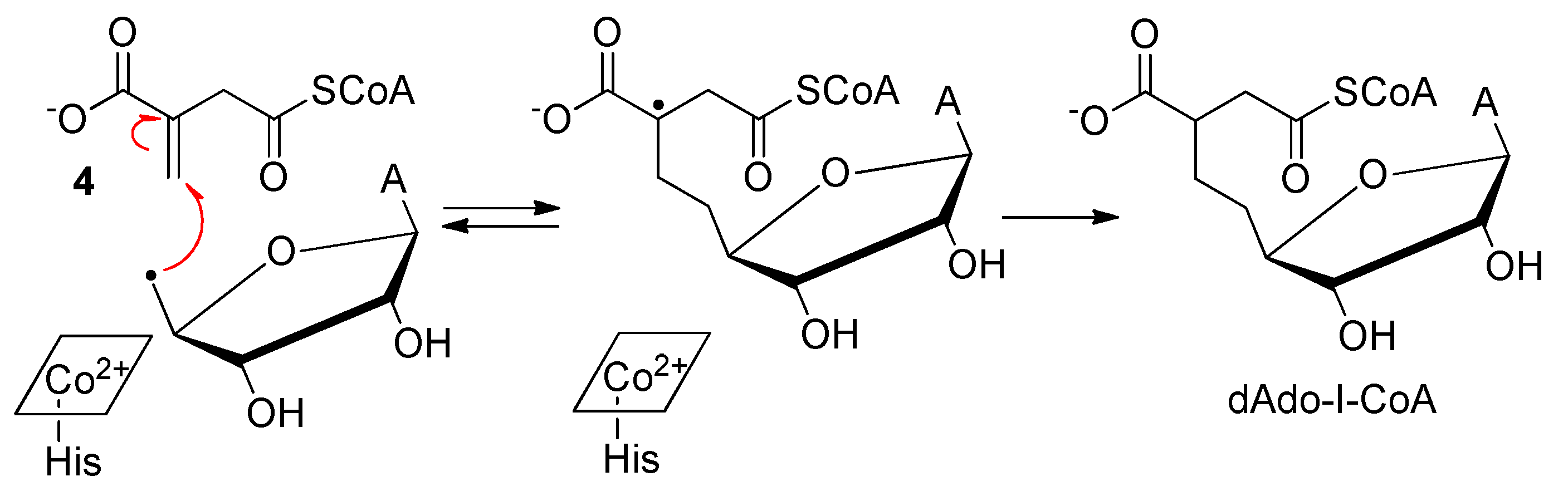
2.2. Methylmalonyl-CoA Mutase (MCM)
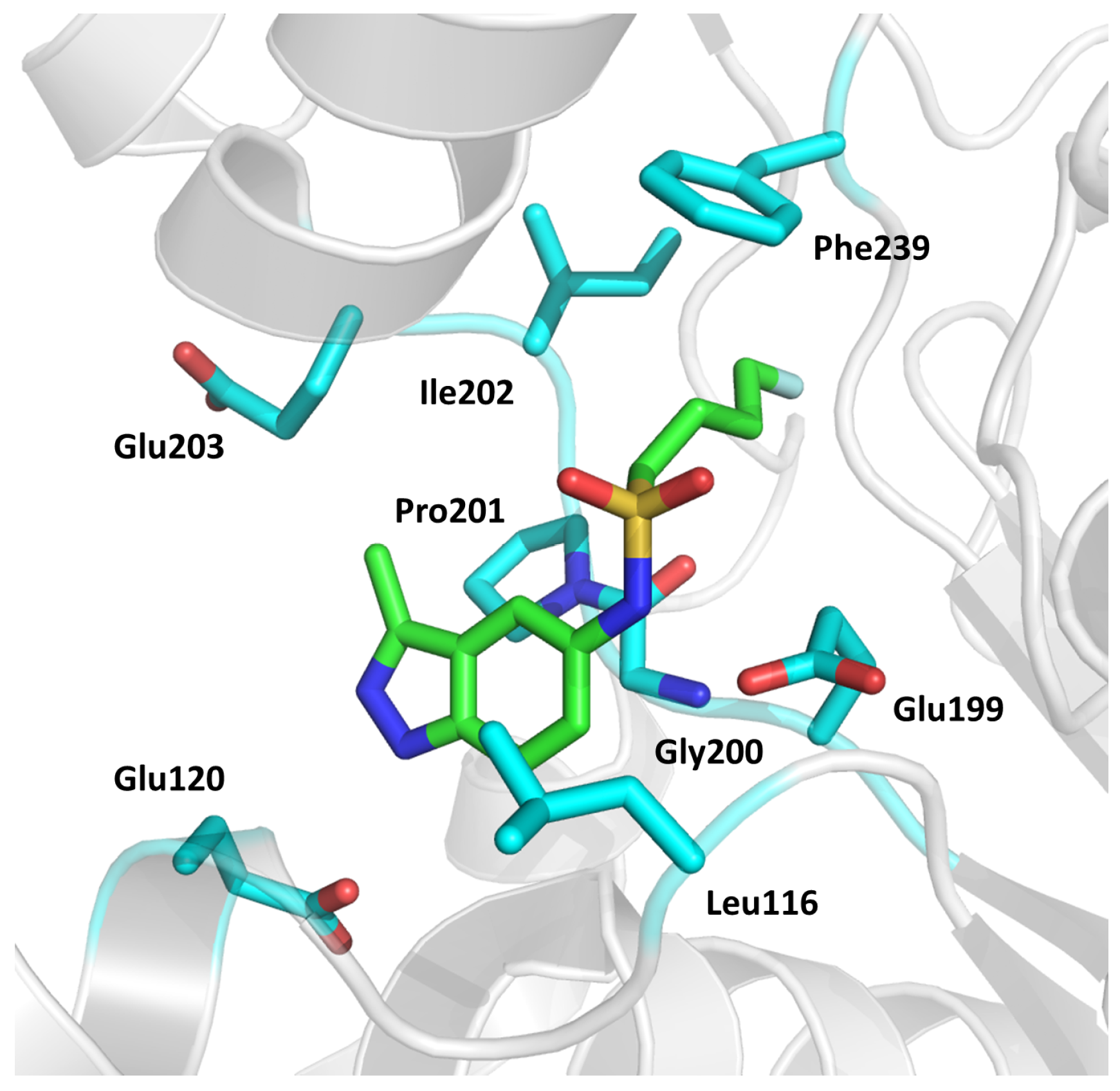
2.3. Fumarate Hydratase
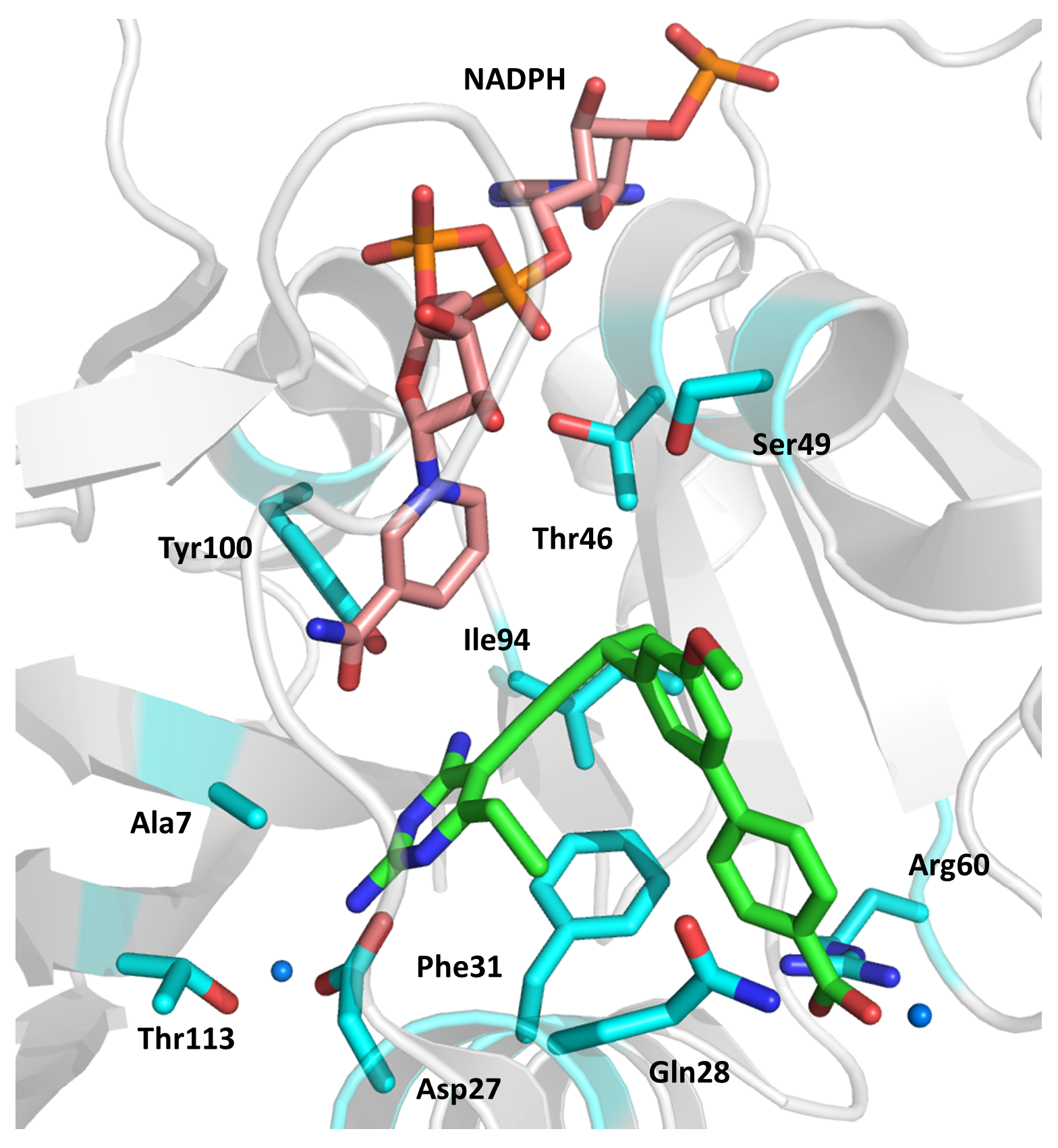
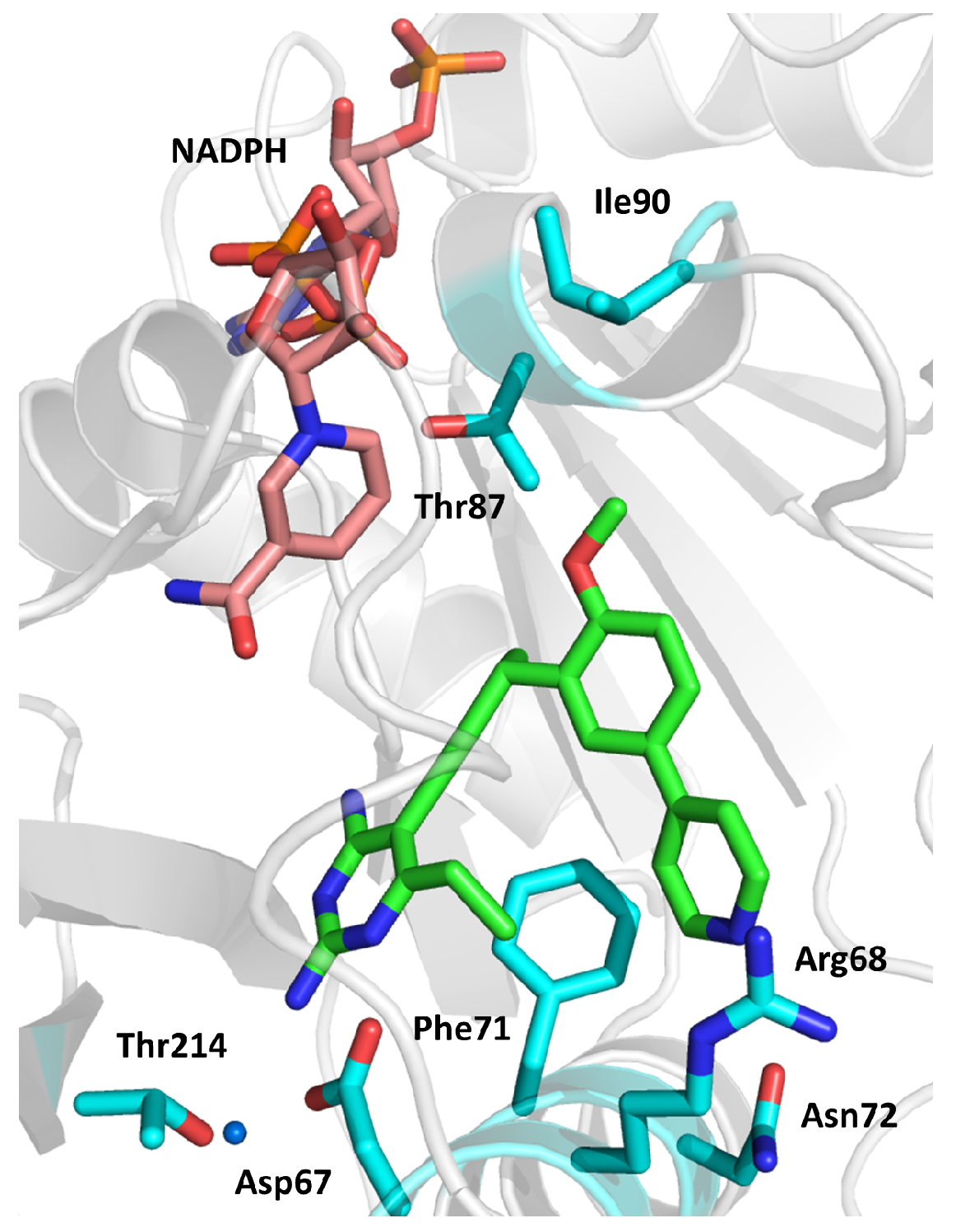
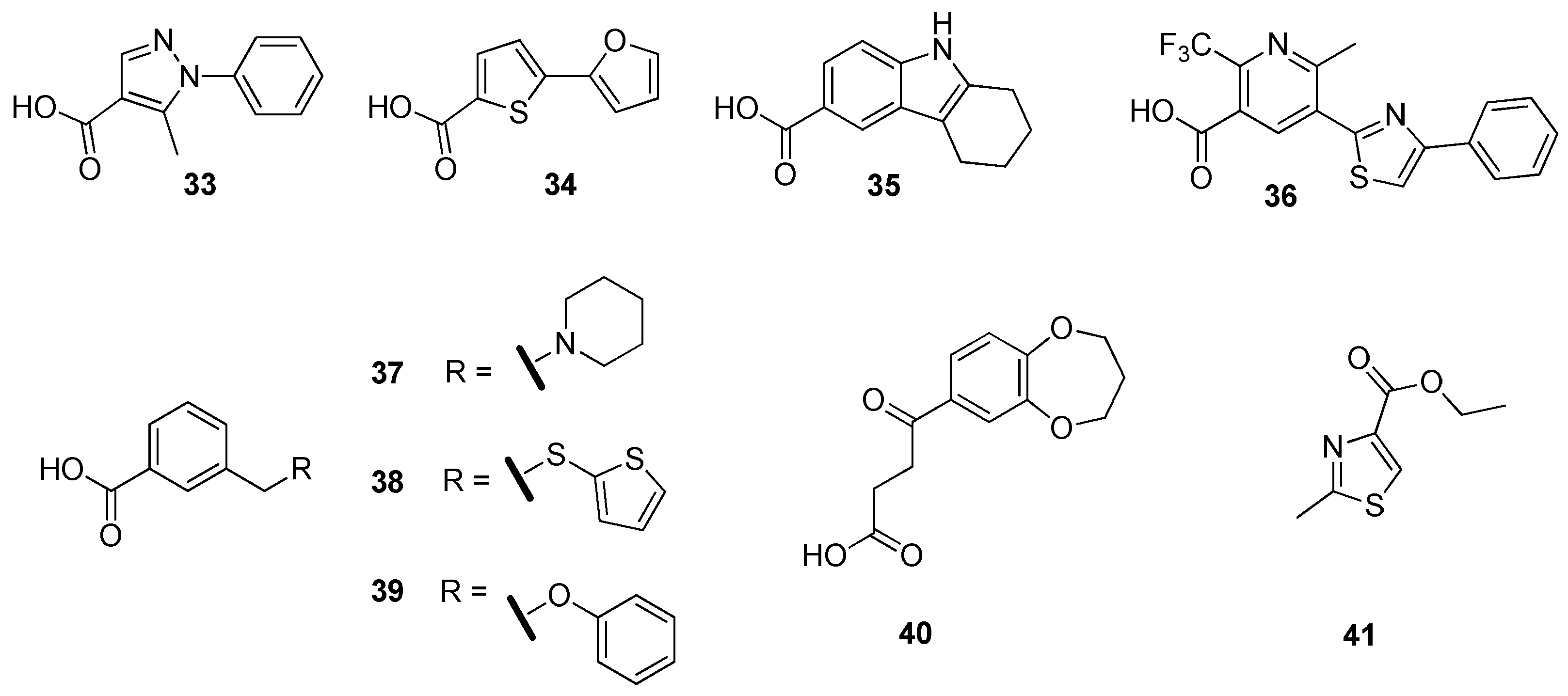
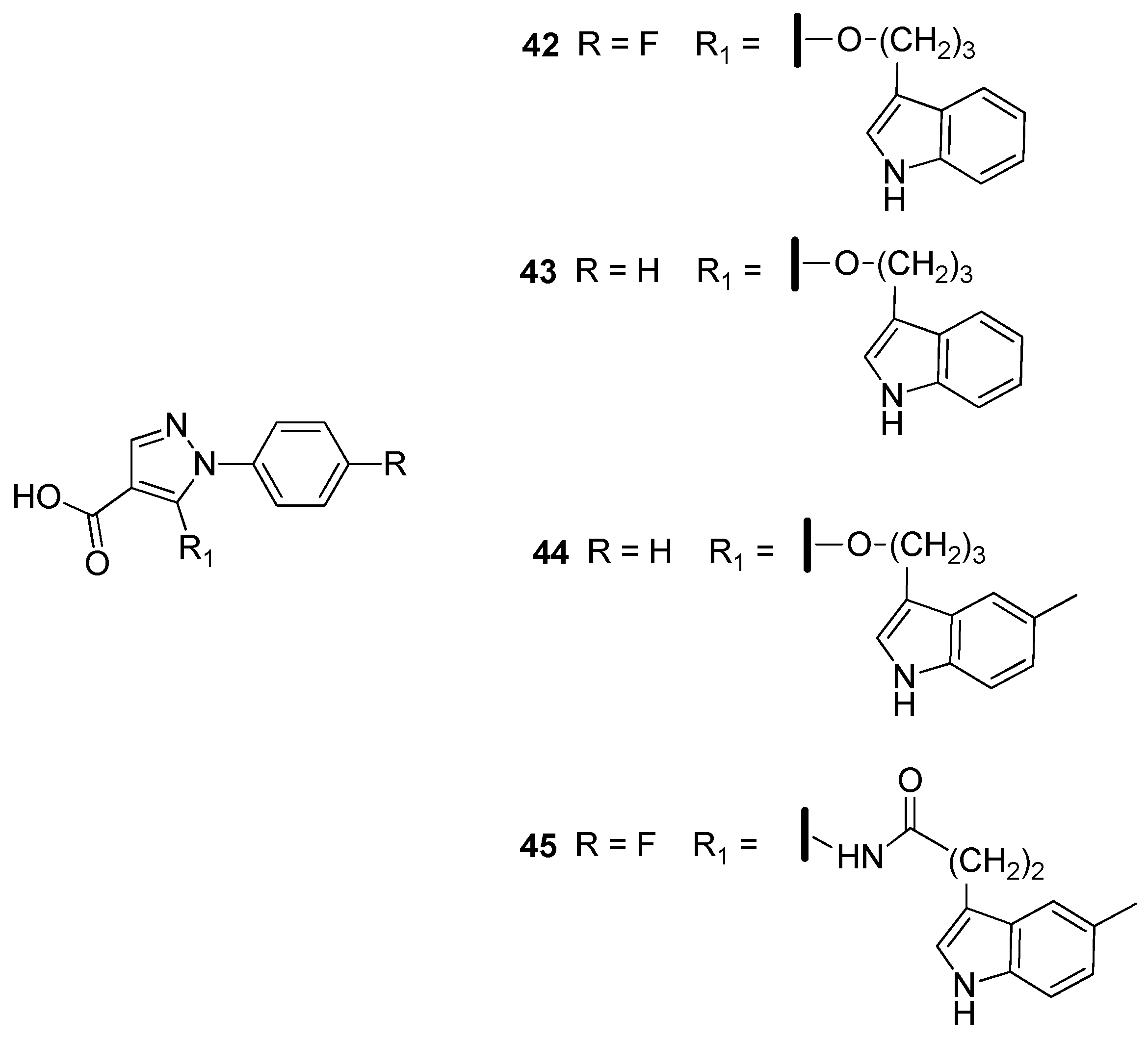
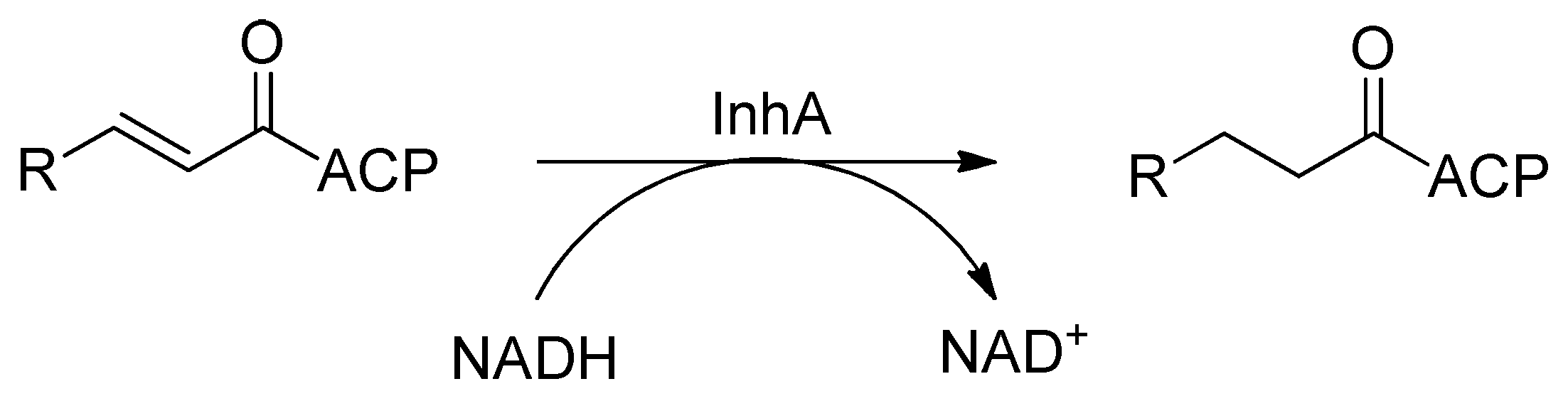
2.4. Enoyl-Acyl Carrier Protein Reductase (InhA)
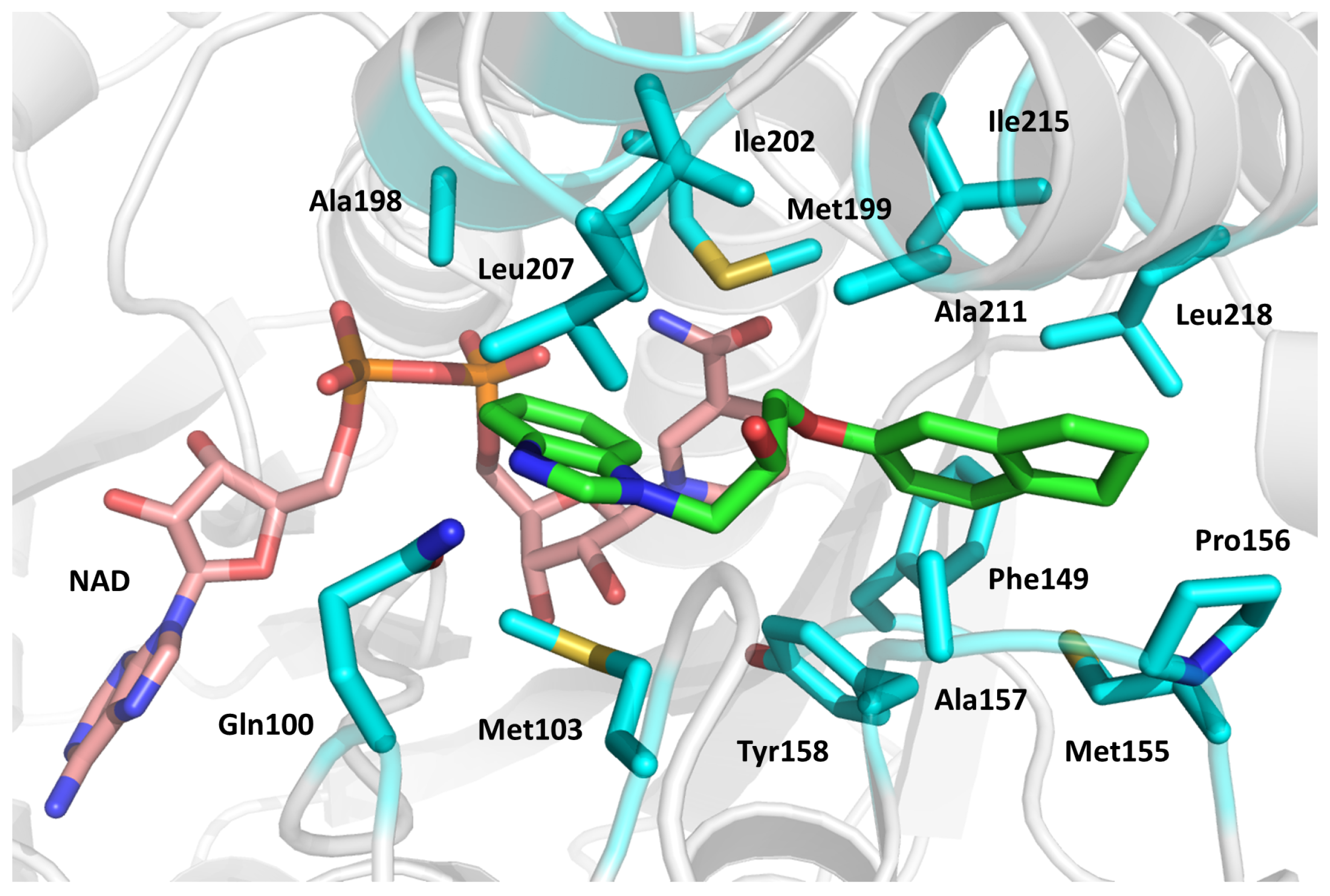
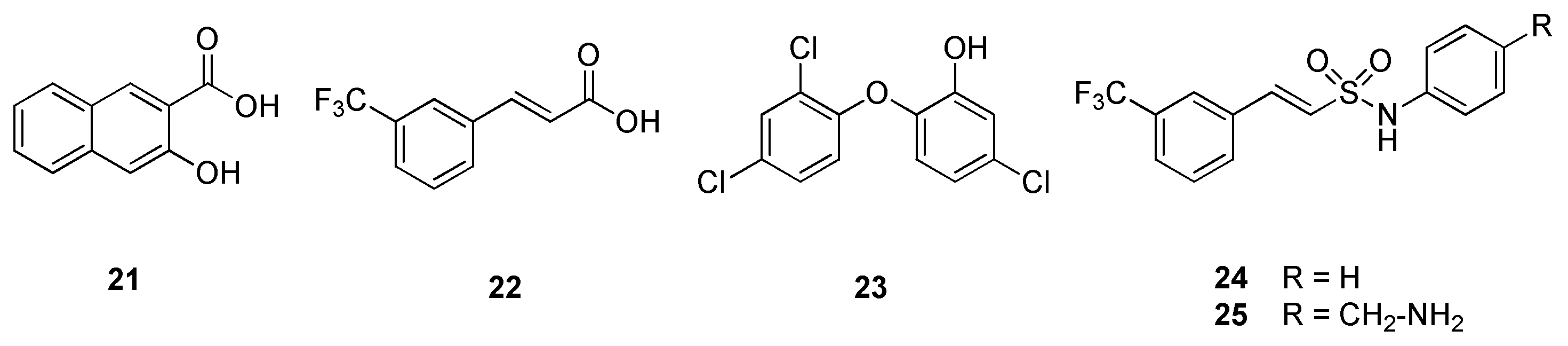
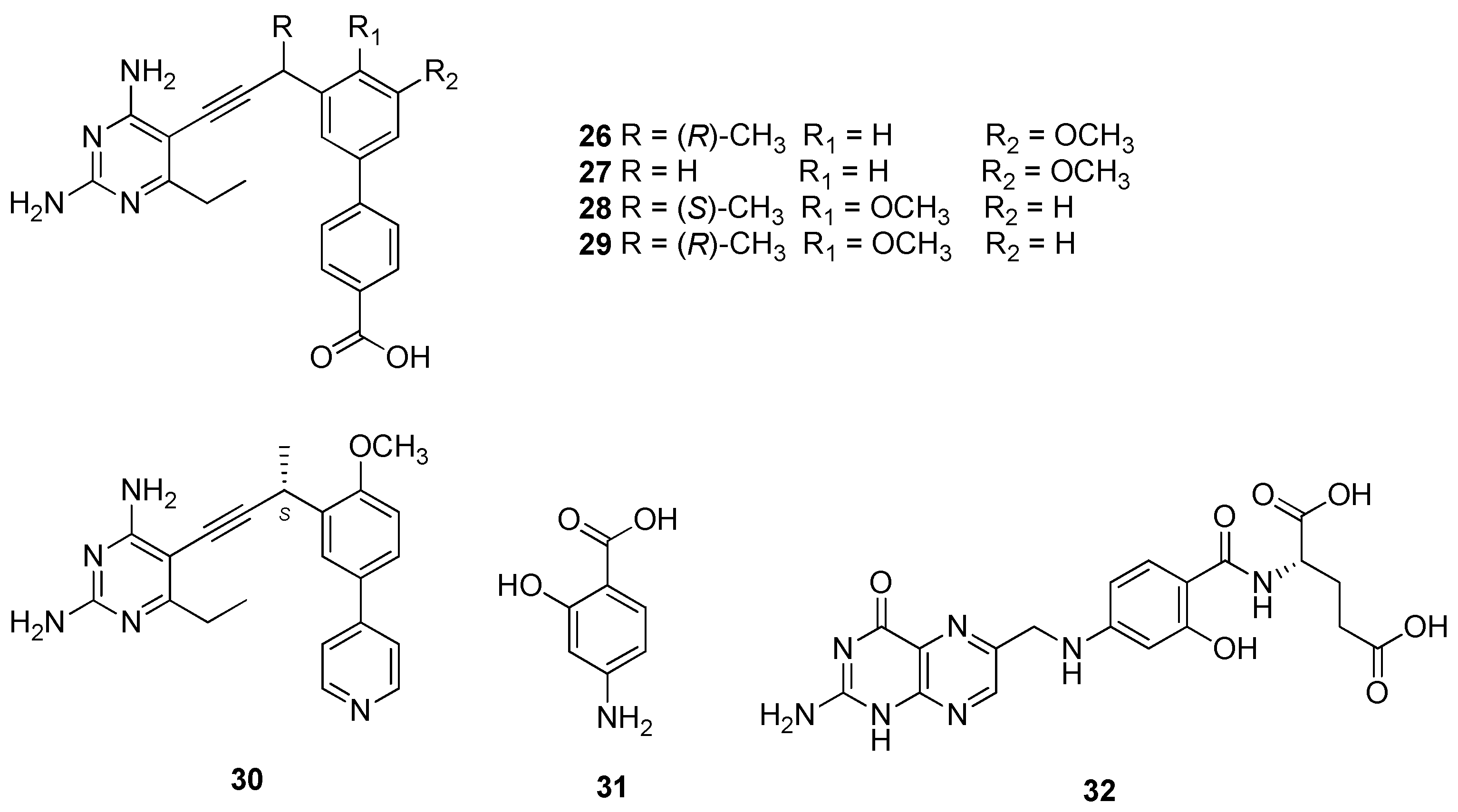
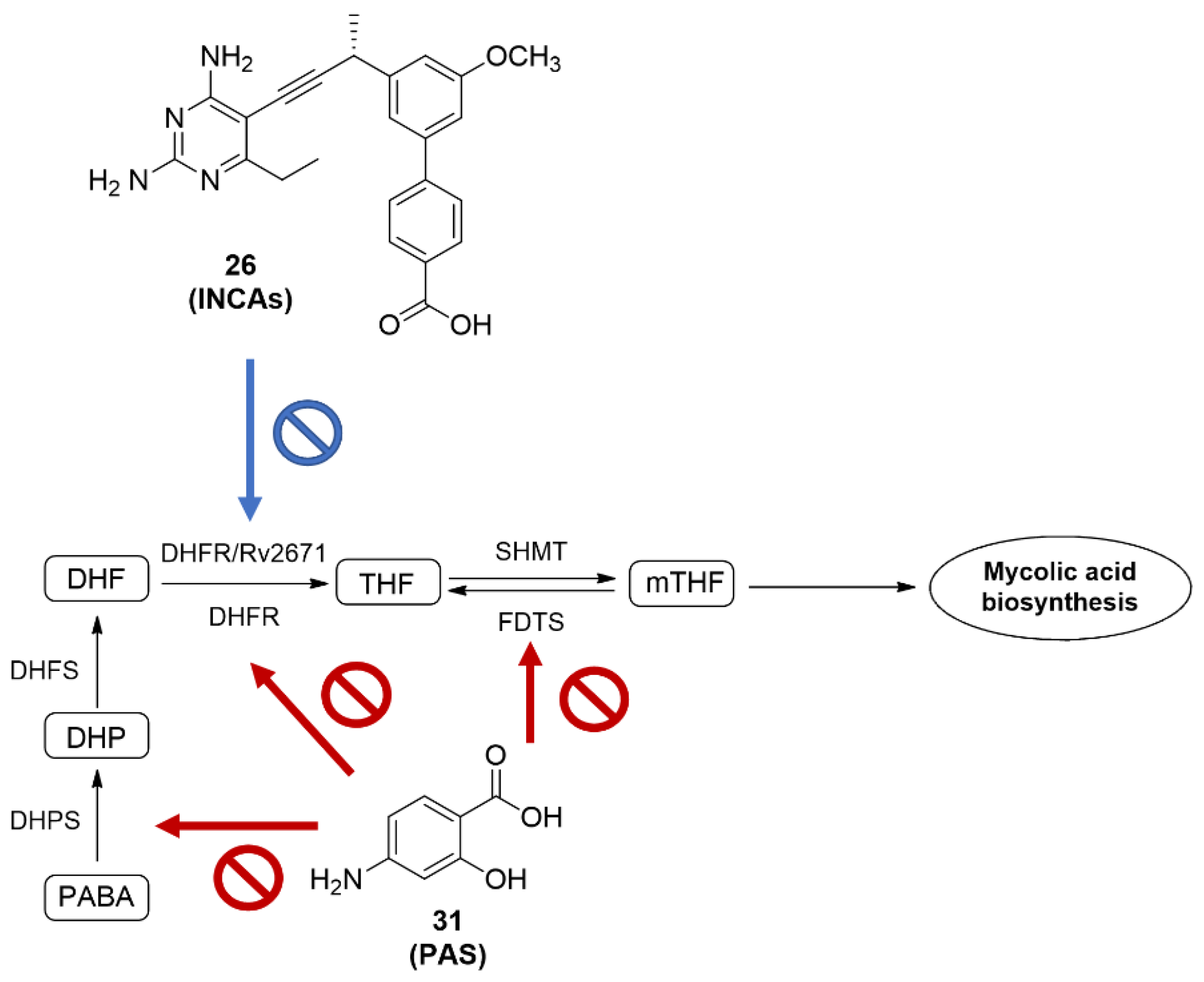
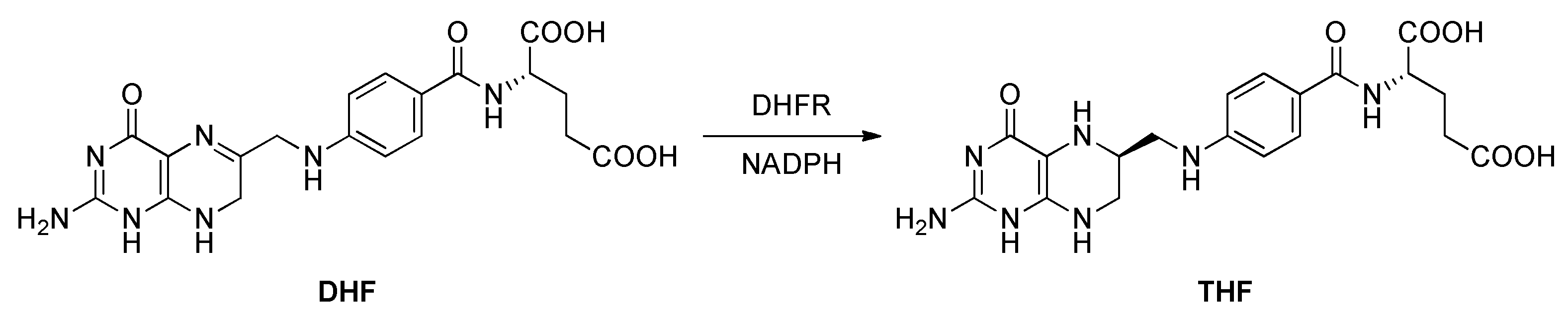
2.5. Dihydrofolate Reductase (DHFR)
- (1)
- The active site entrance, featuring a positive charge;
- (2)
- The slightly apolar central region of the active site;
- (3)
- The dipyrimidine binding site, characterized by strong negative charges;
- (4)
- The glycerol-binding site, a specific region found in Mtb DHFR and not in the human homolog.
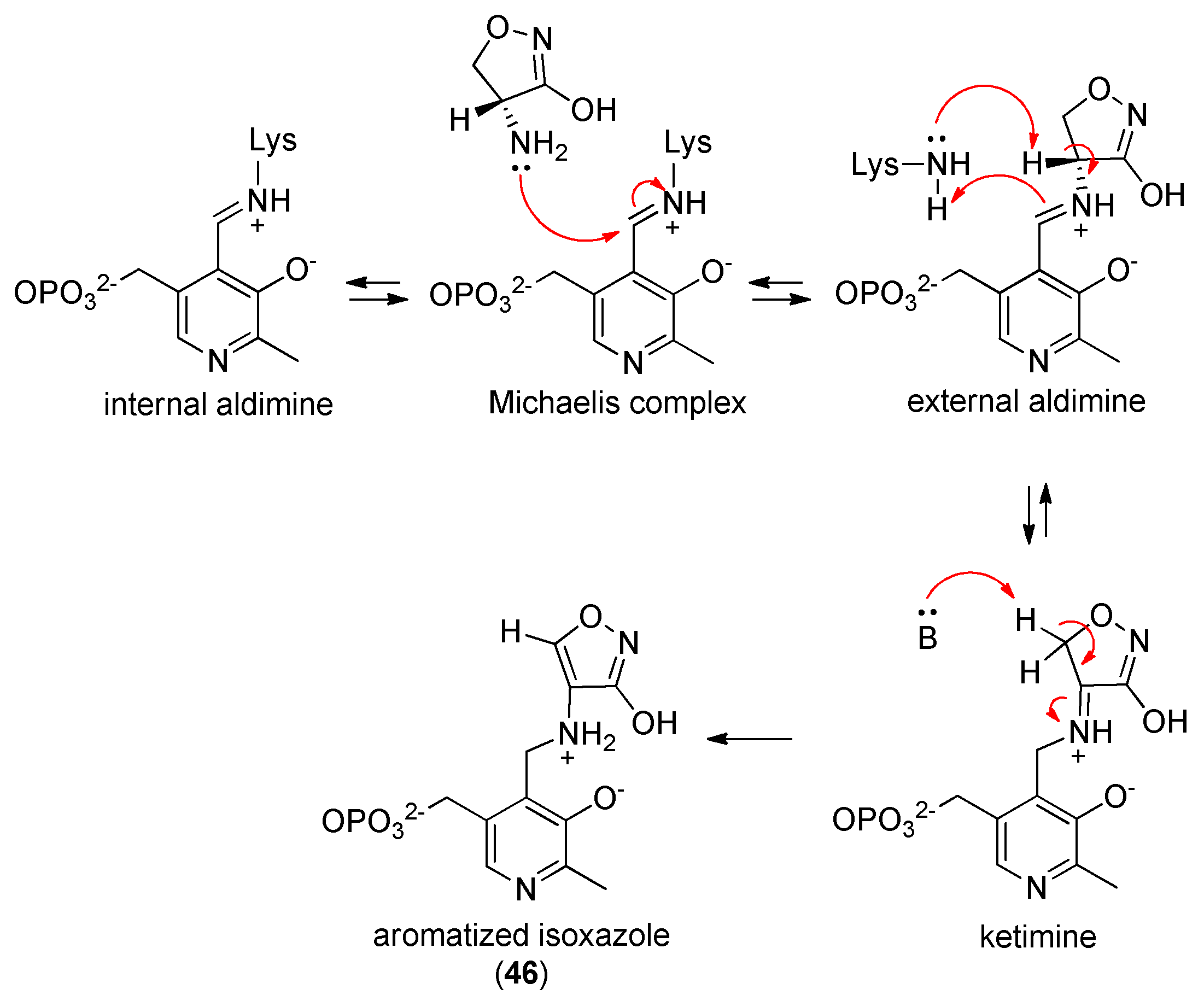
2.6. Alanine Racemase (Alr)
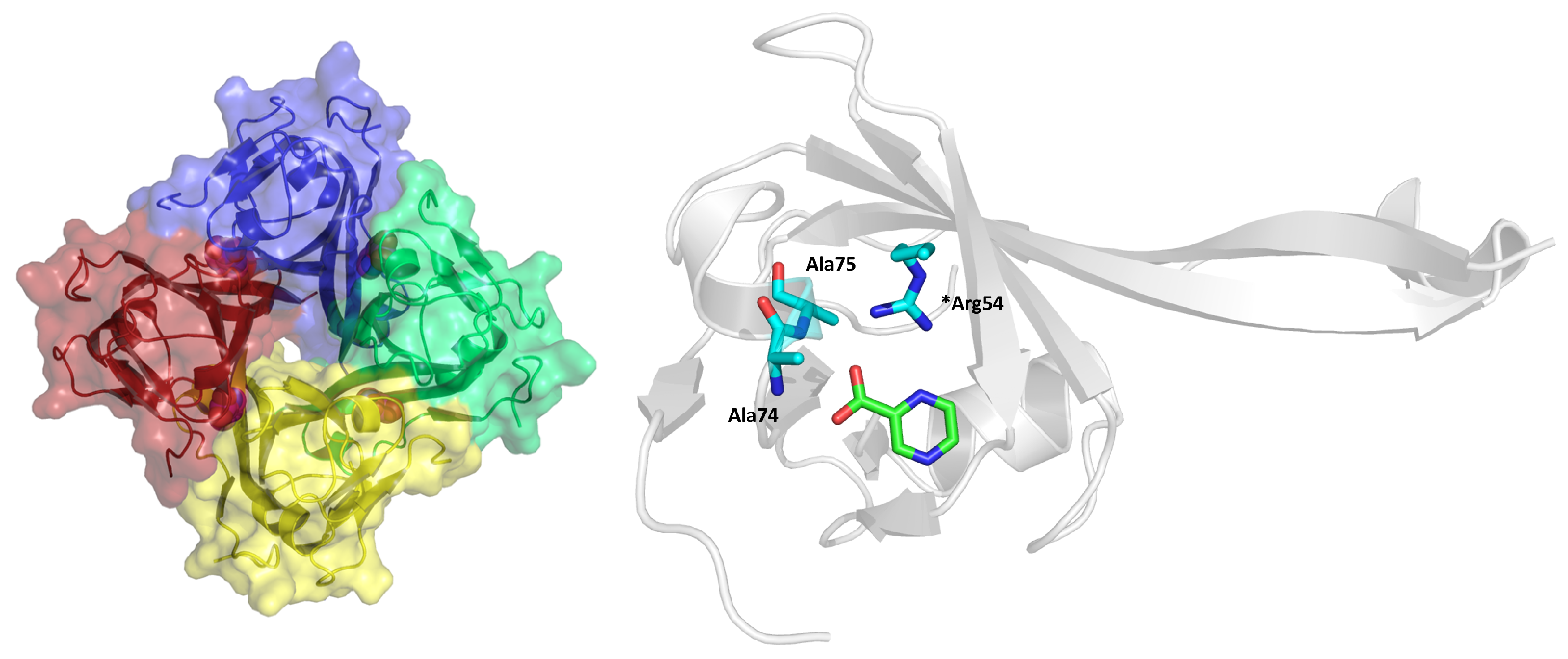
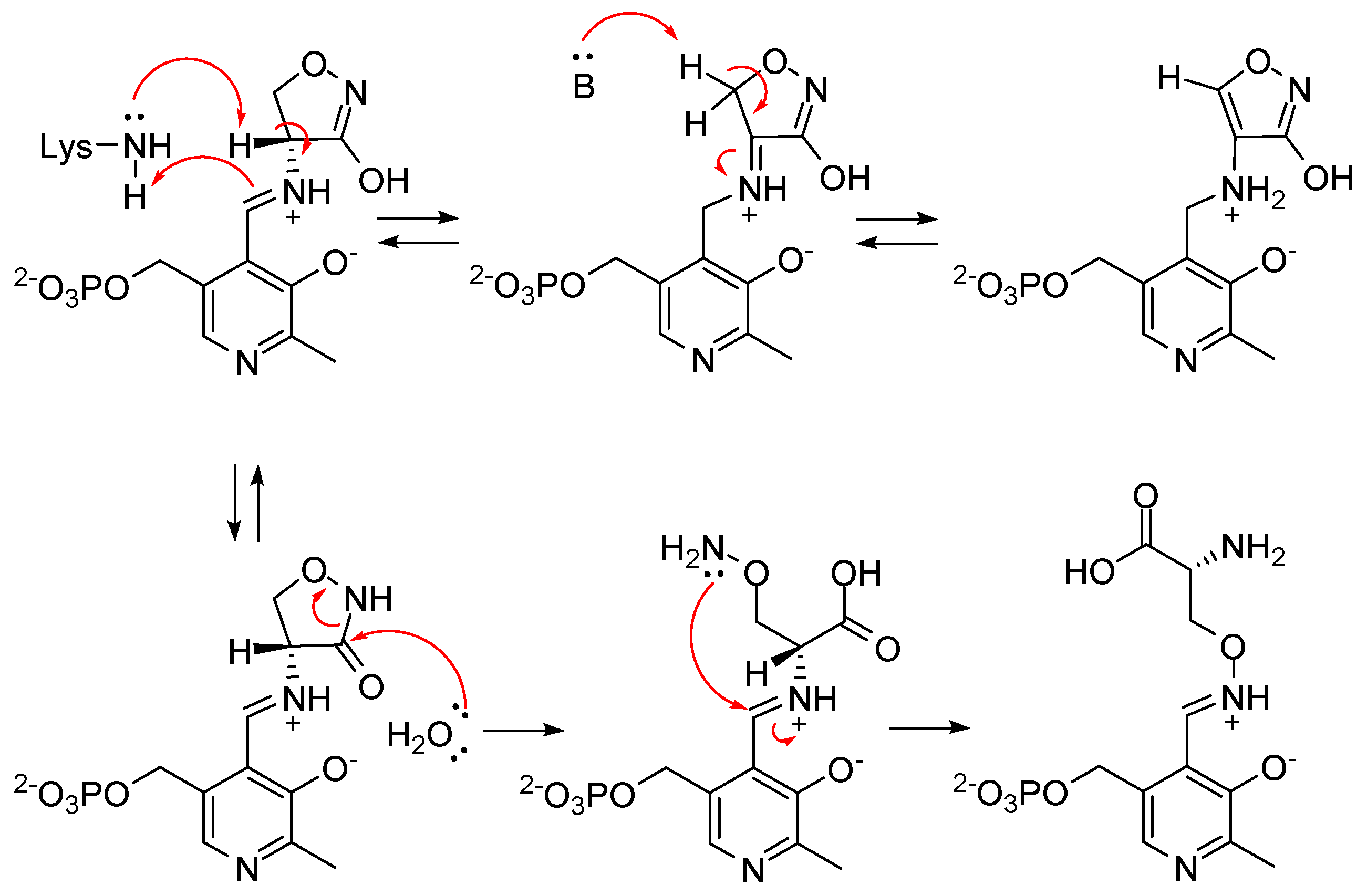
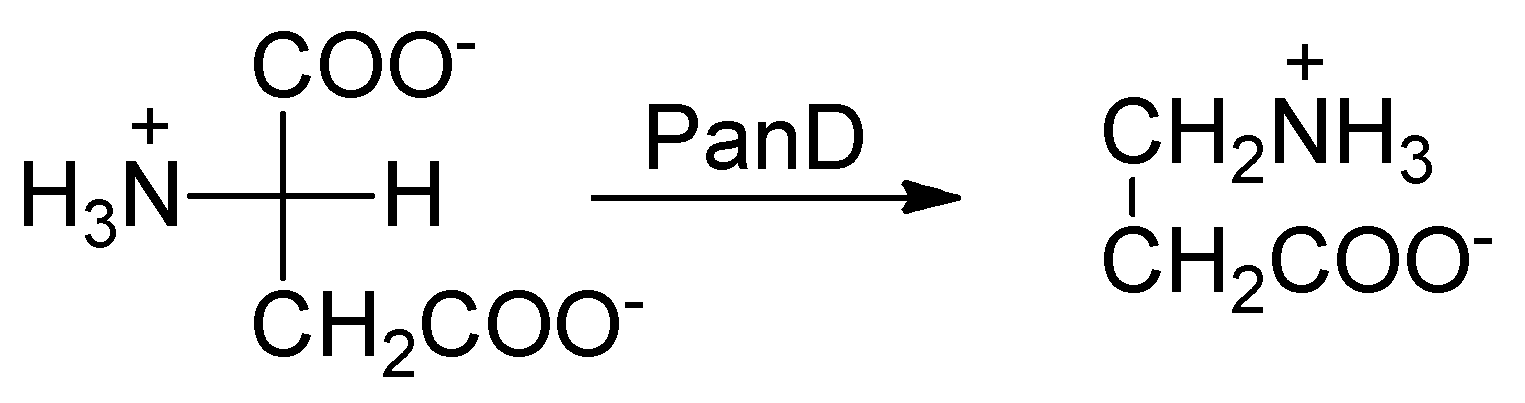
2.7. l-Aspartate-α-Decarboxylase (PanD)
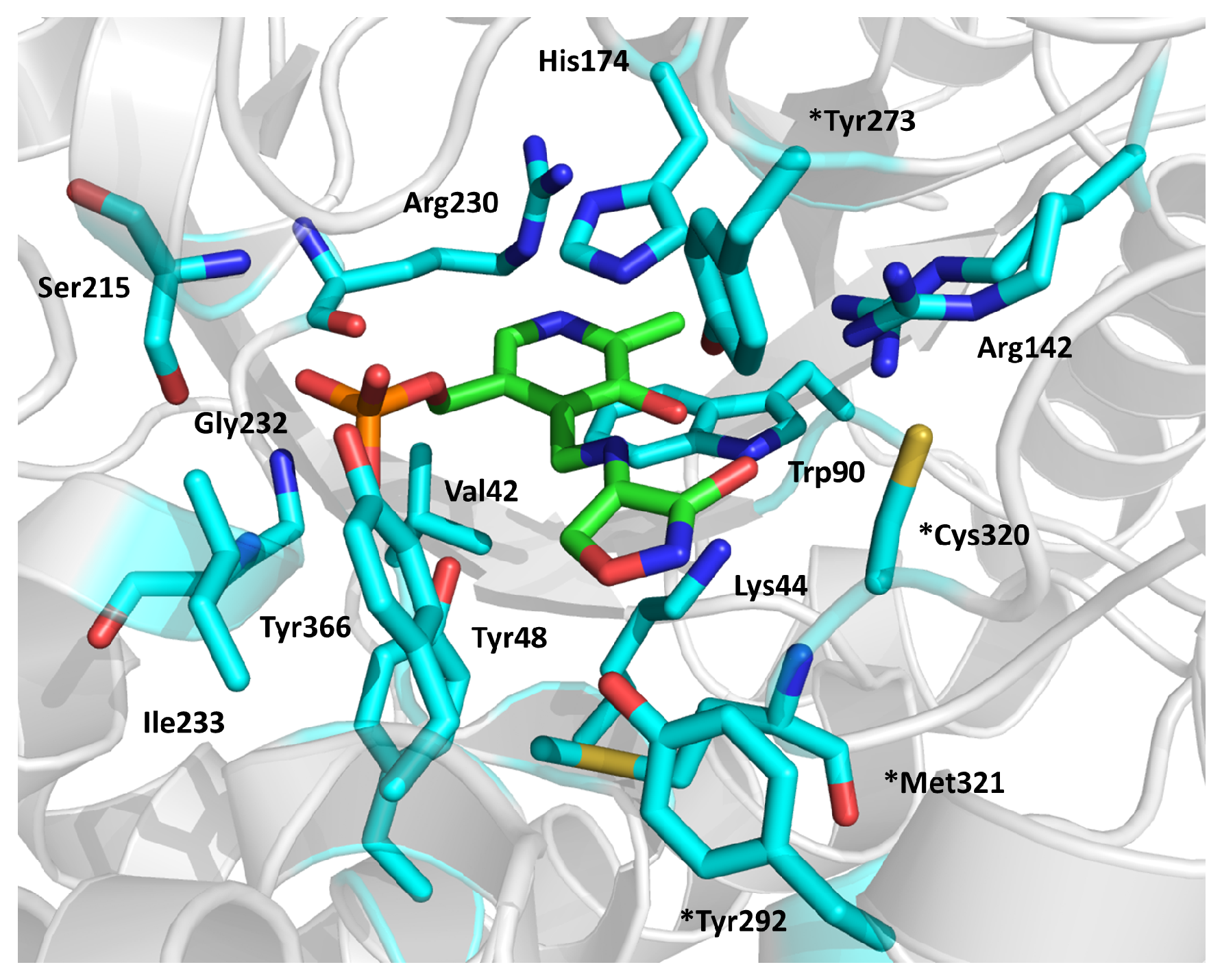
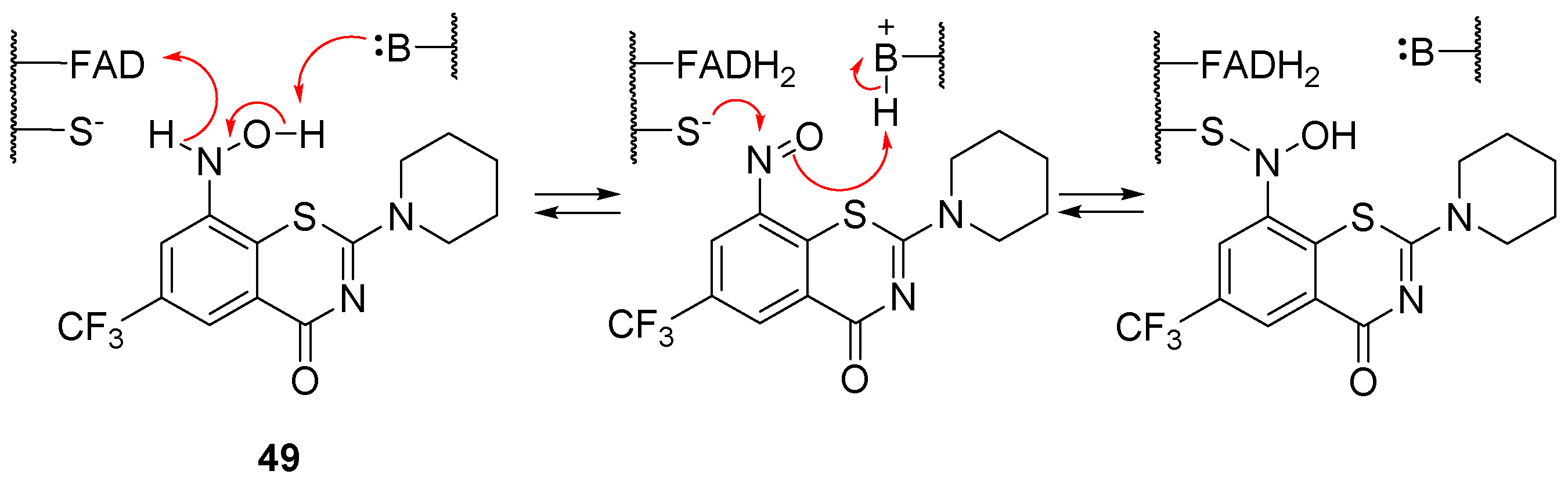
2.8. Decaprenylphosphoryl-β-d-ribose-2′-oxidase (DprE1)
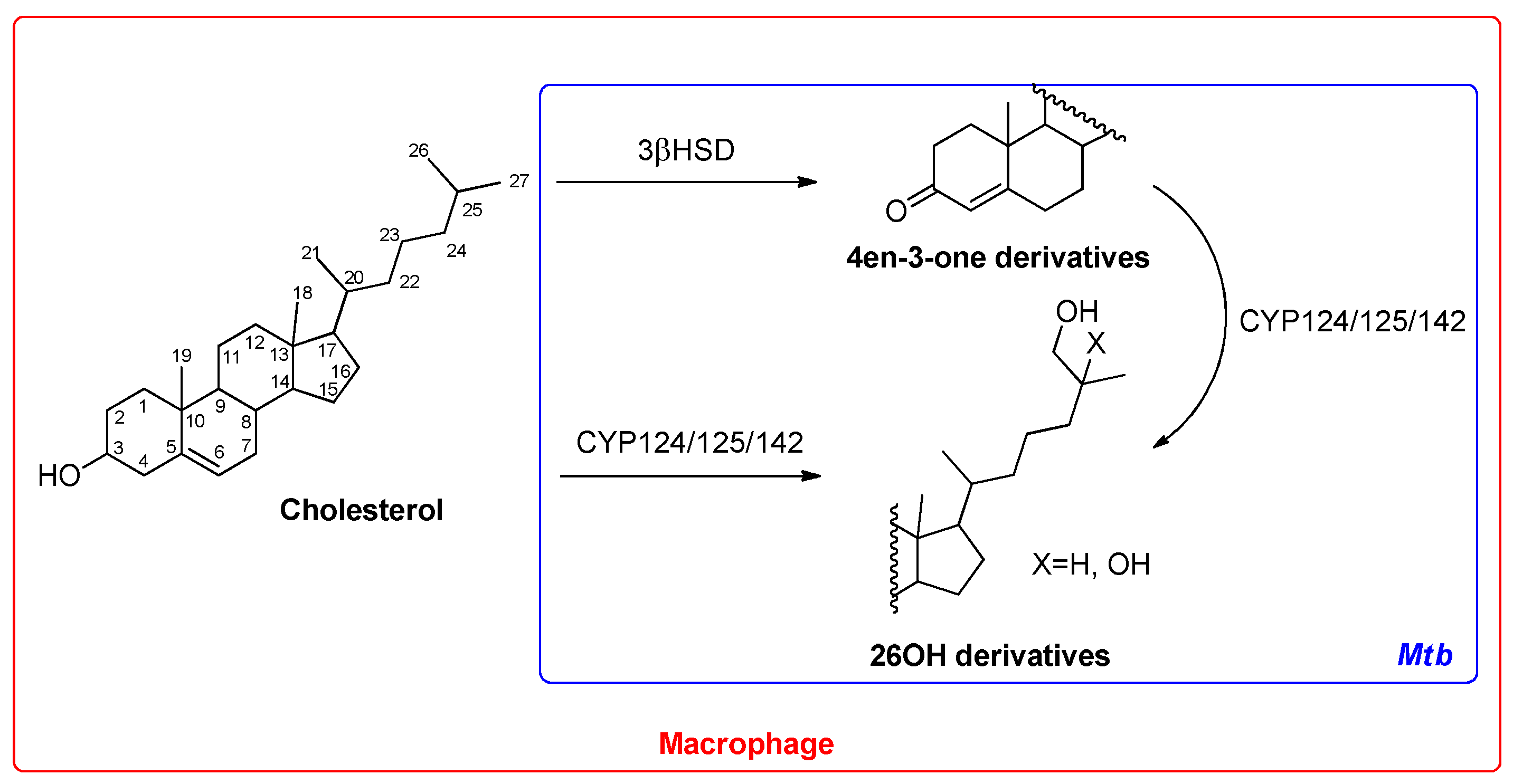
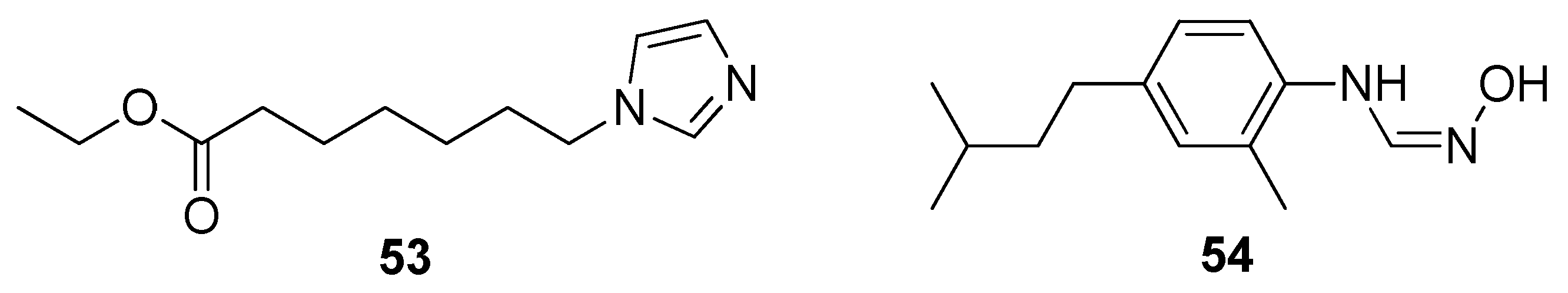
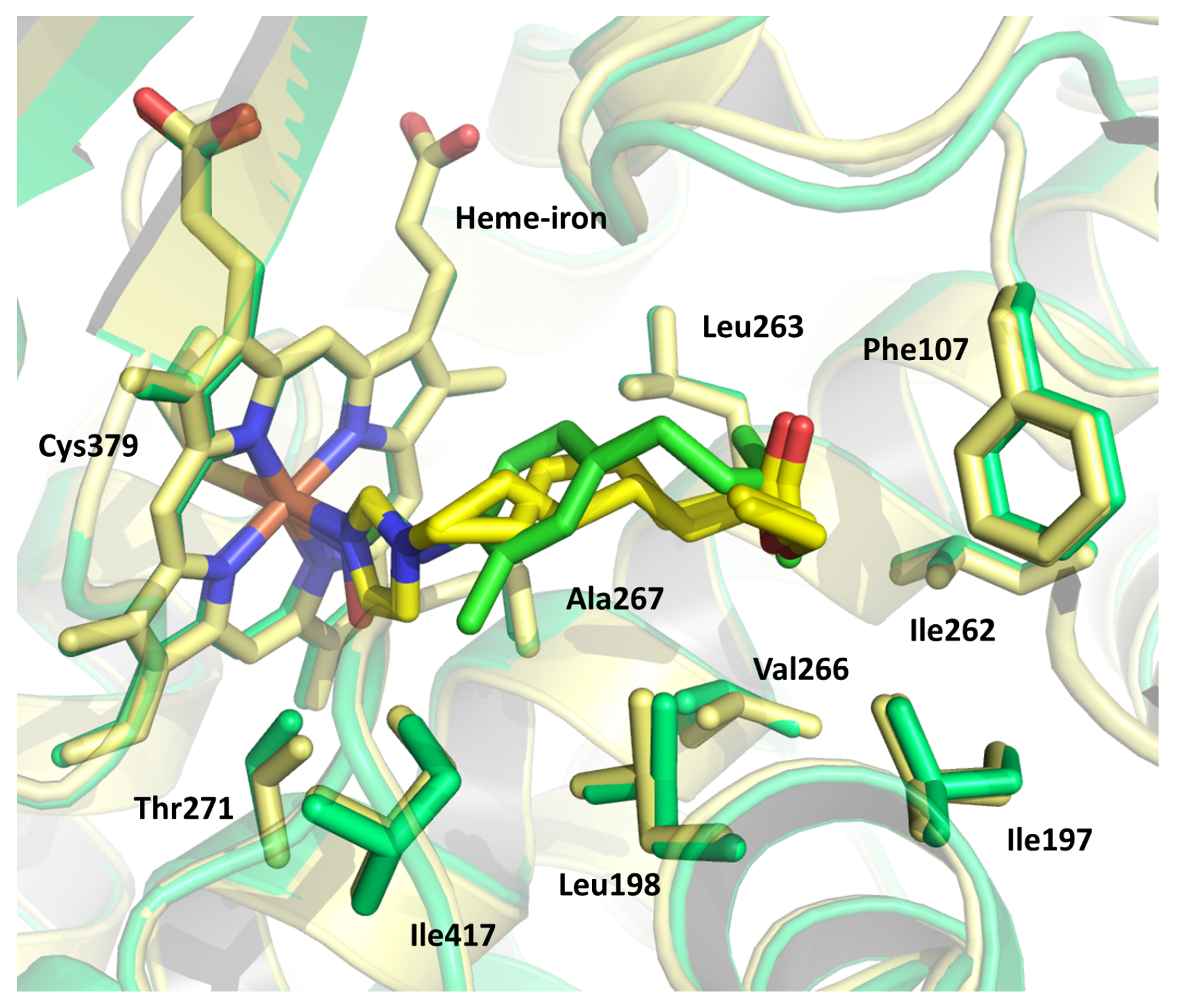
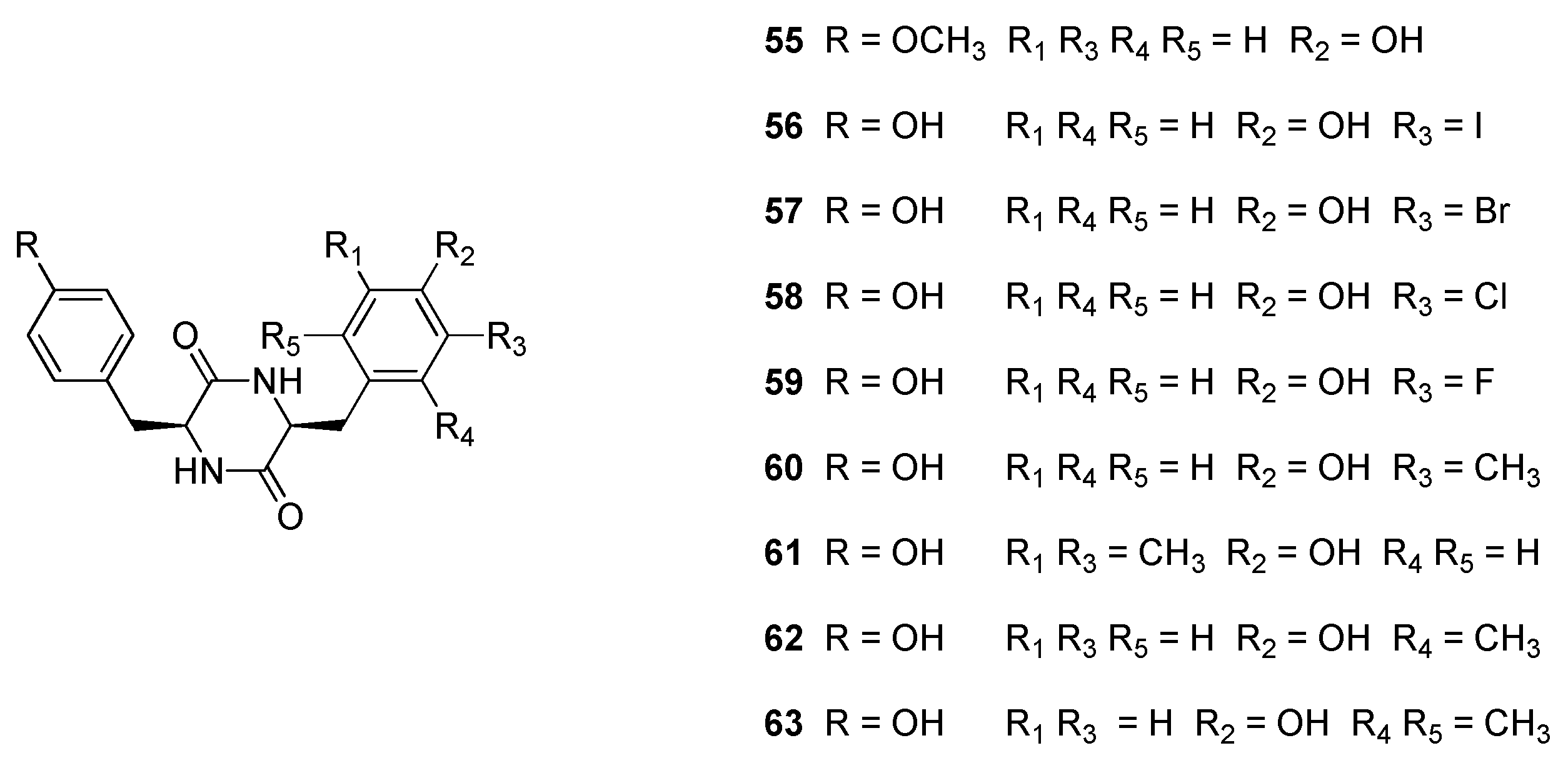
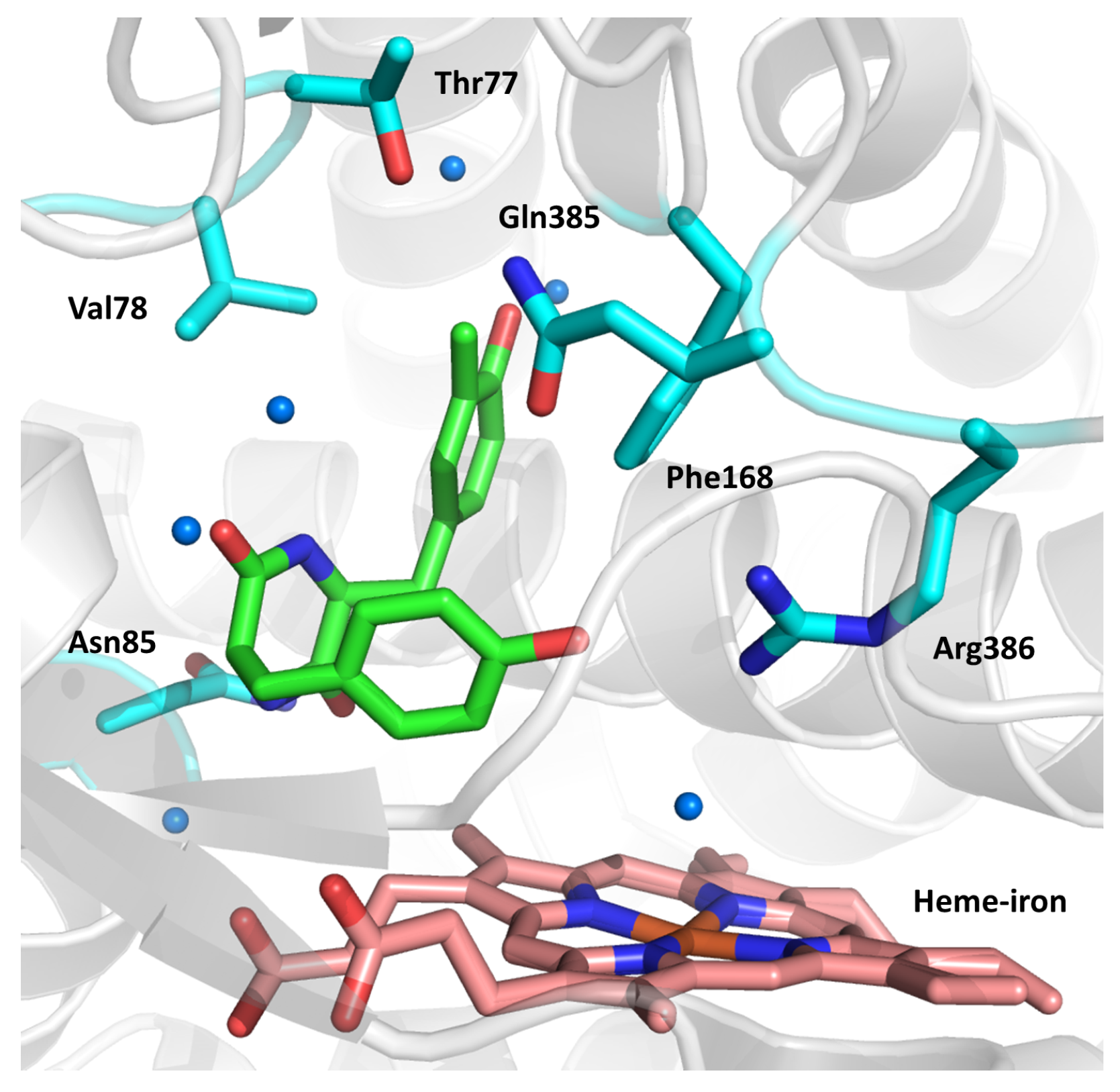
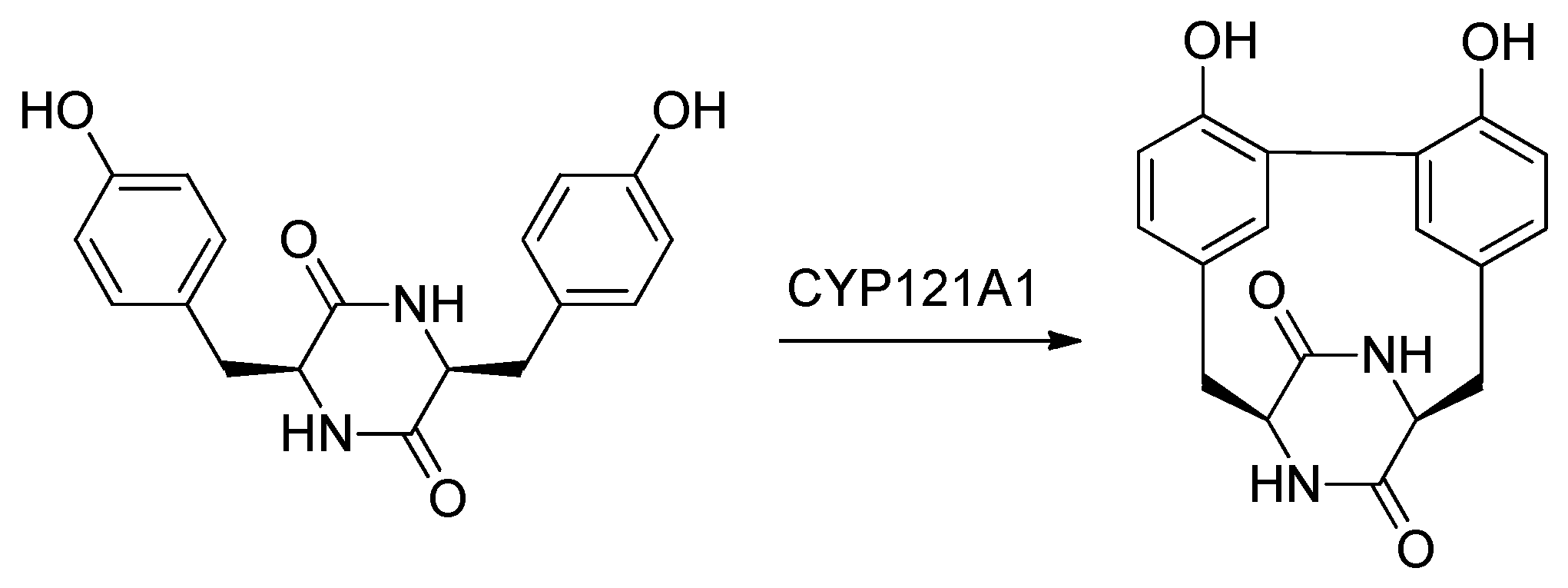
2.9. Cytochromes CYP124 and CYP121
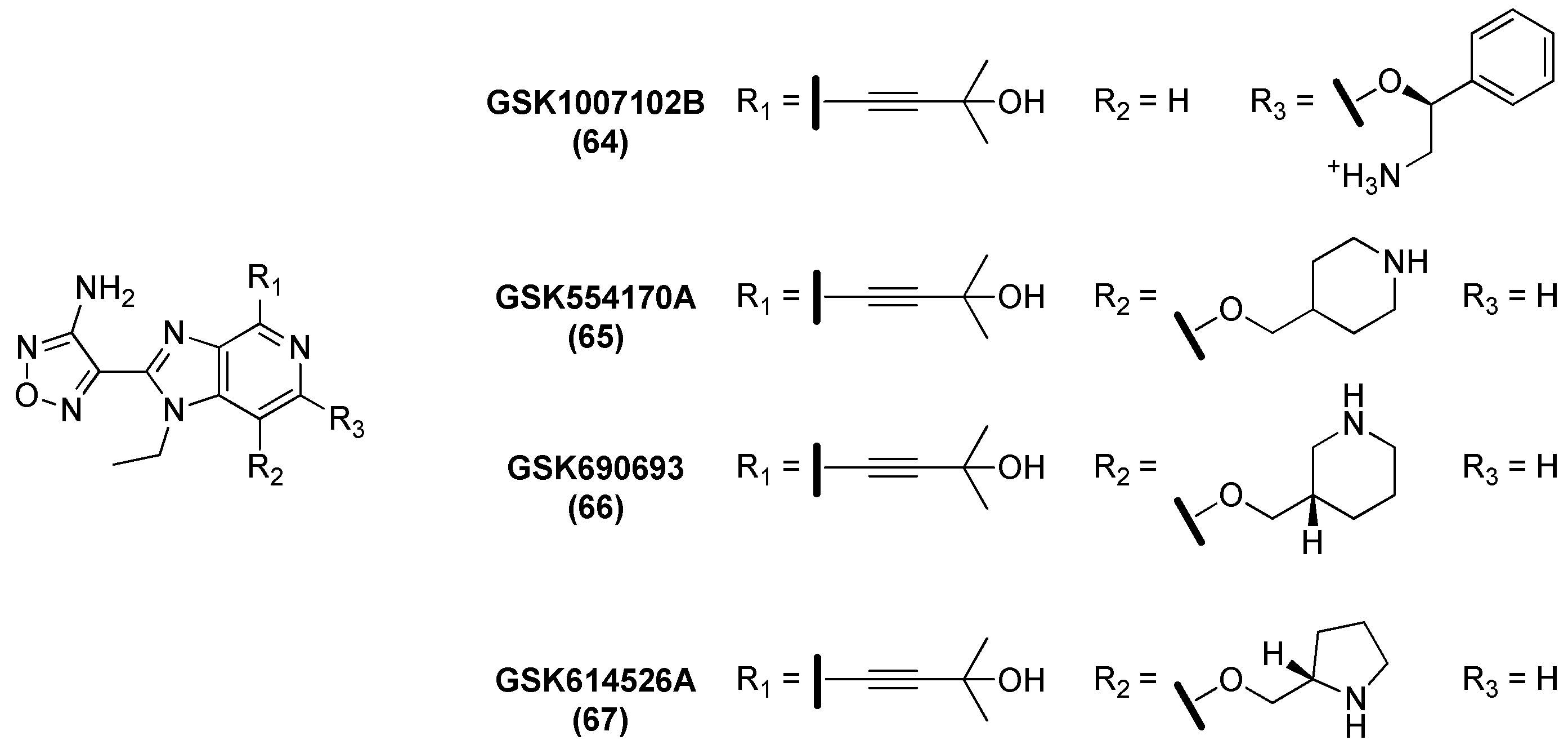
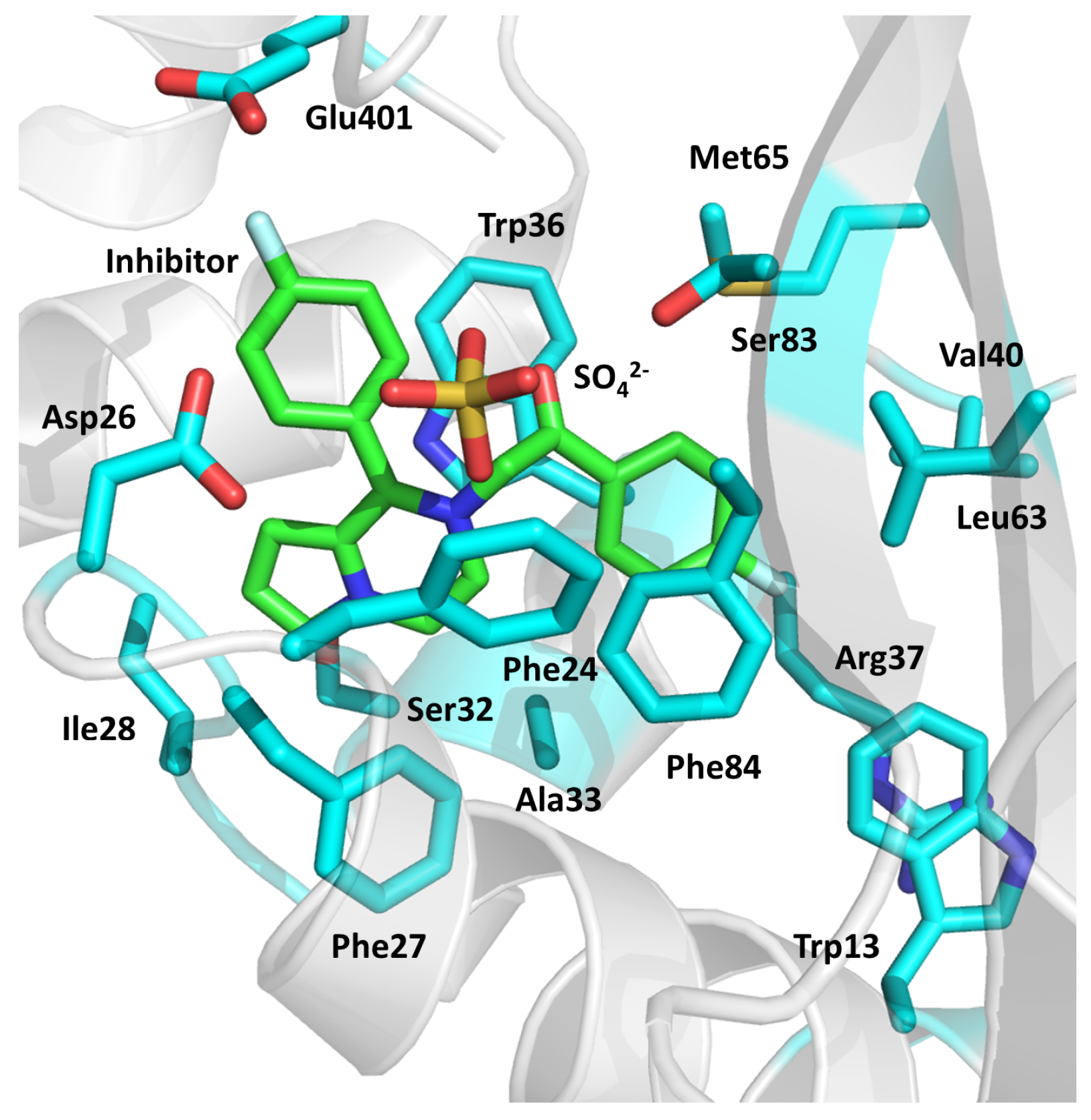
2.10. Ser/Thr Protein Kinase B (PknB)
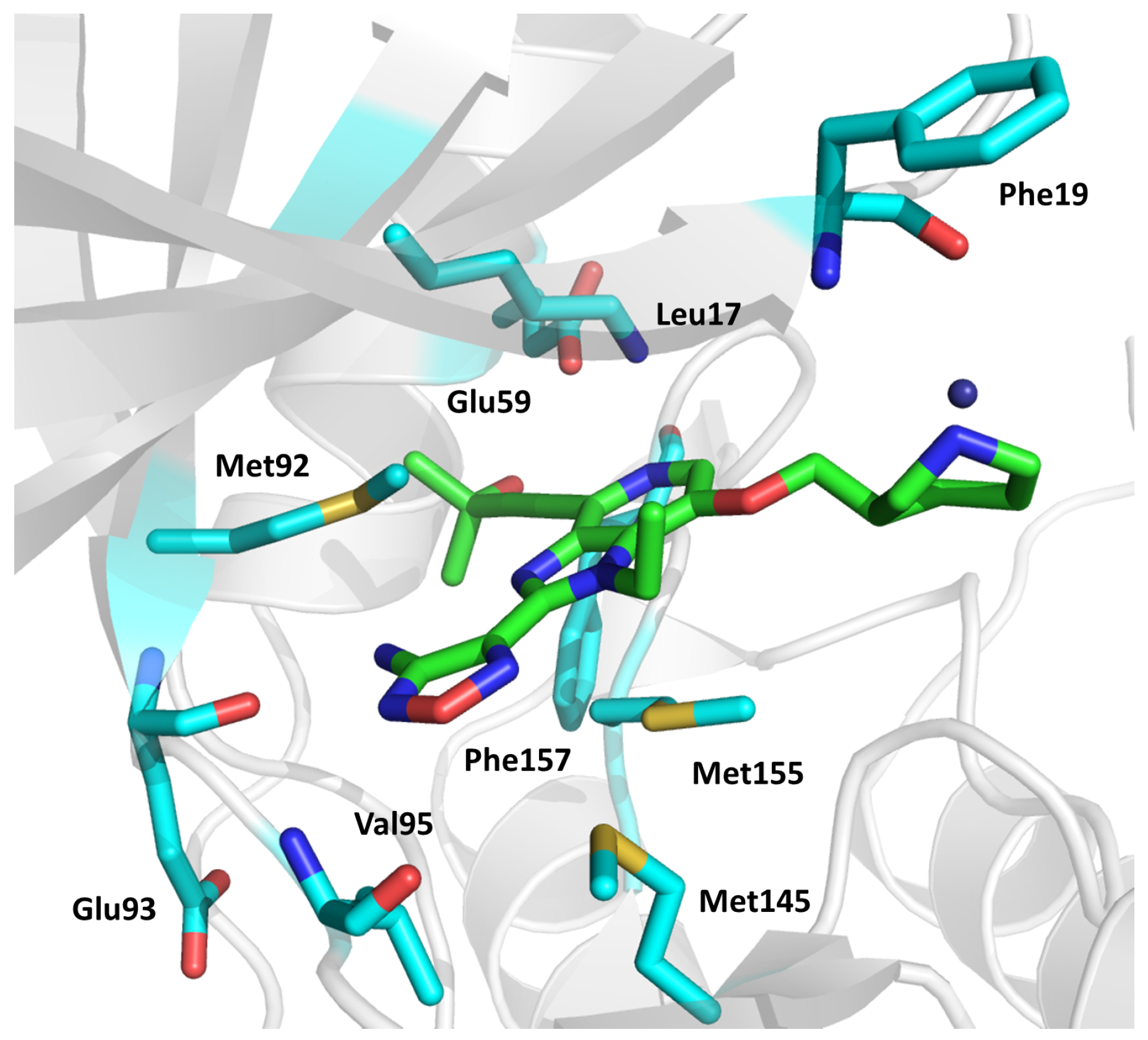
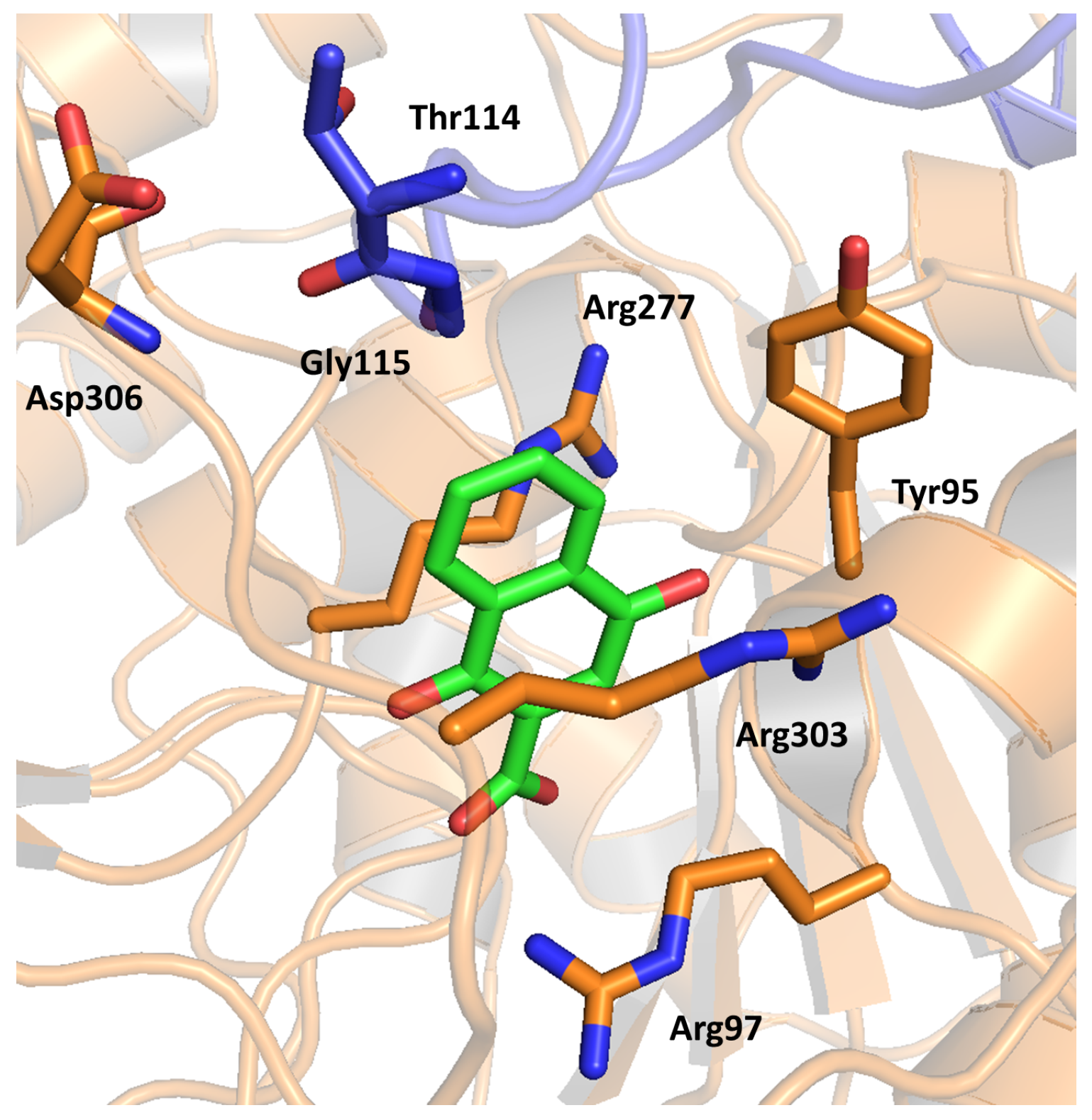
2.11. Adenosine Kinase (AdoK)
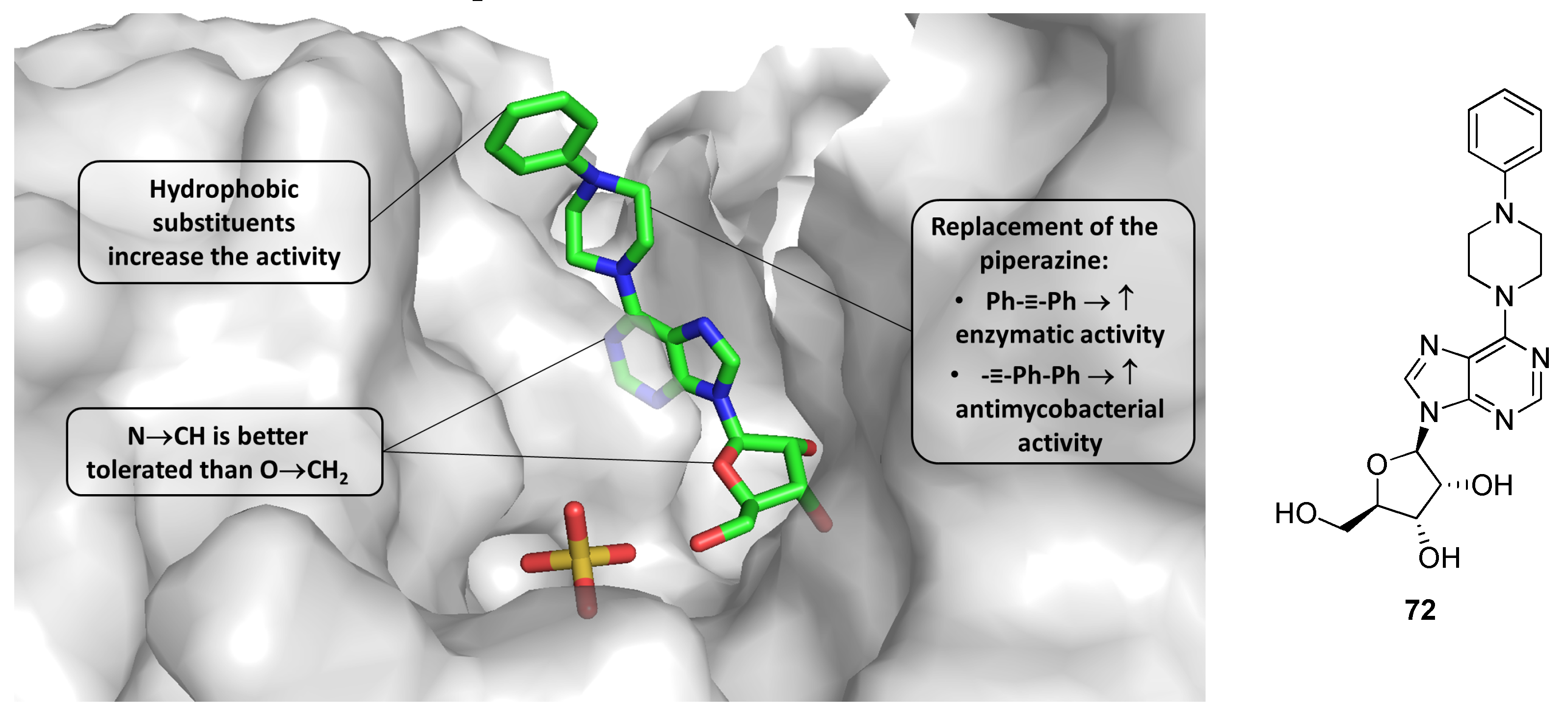
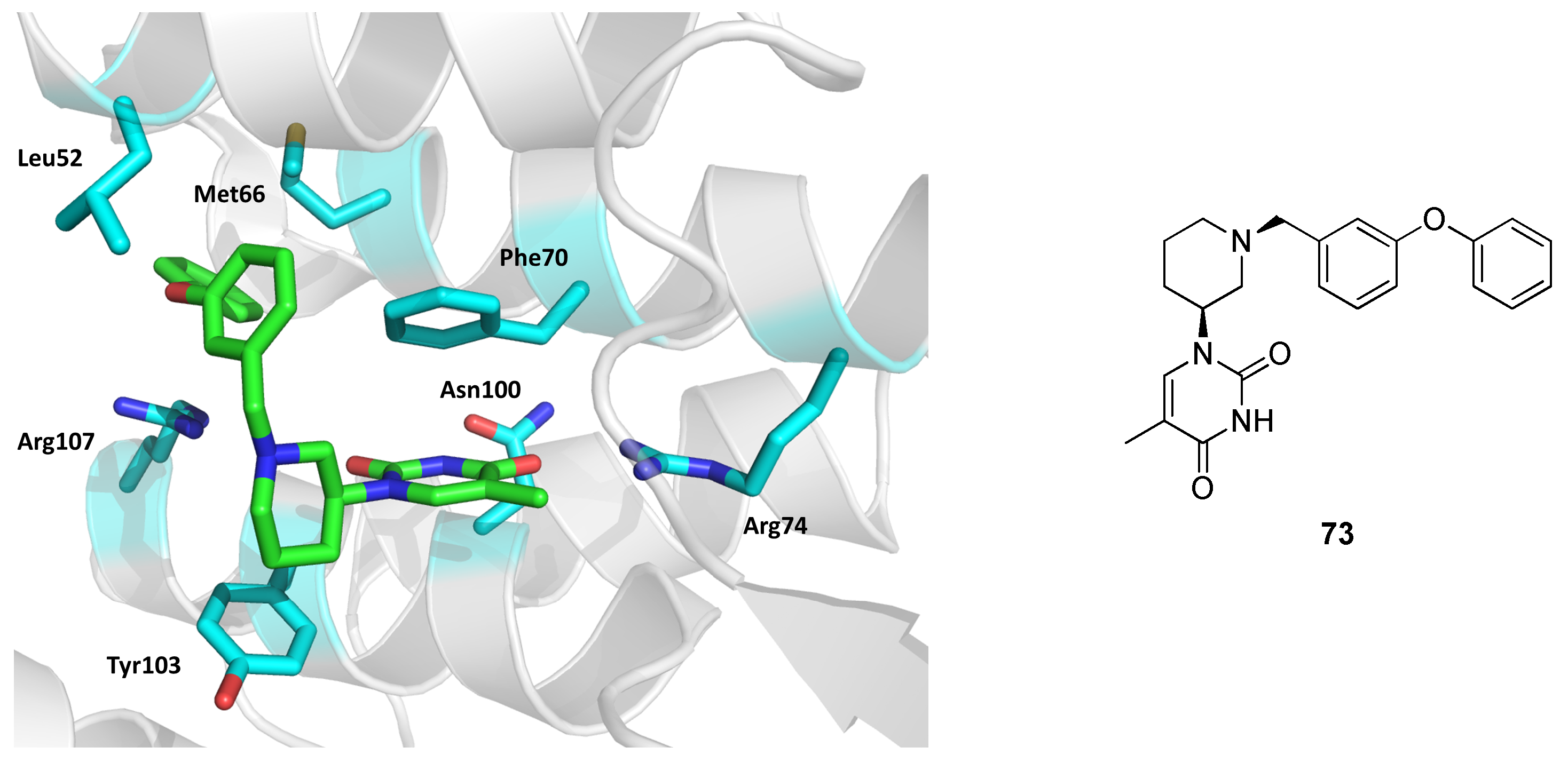
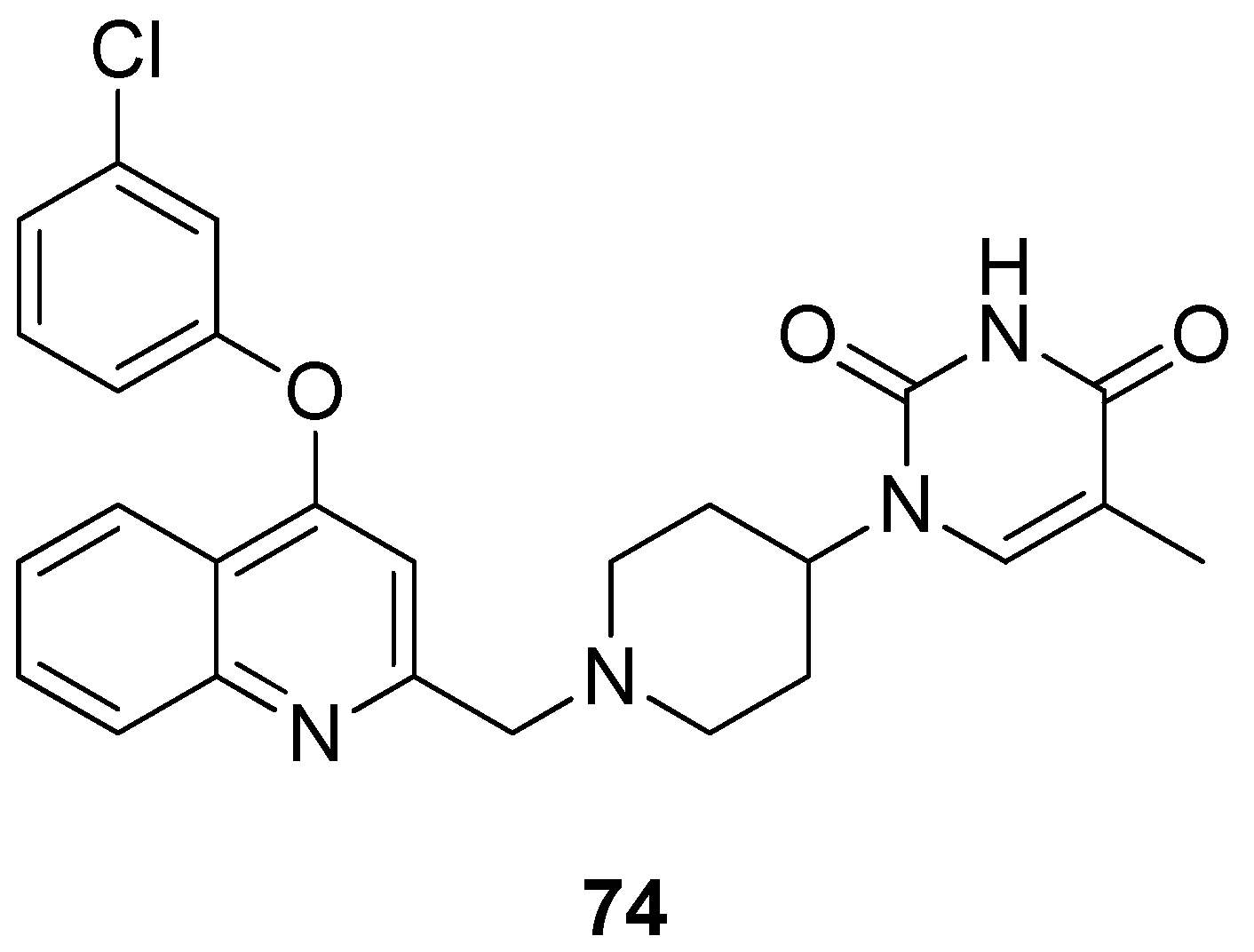
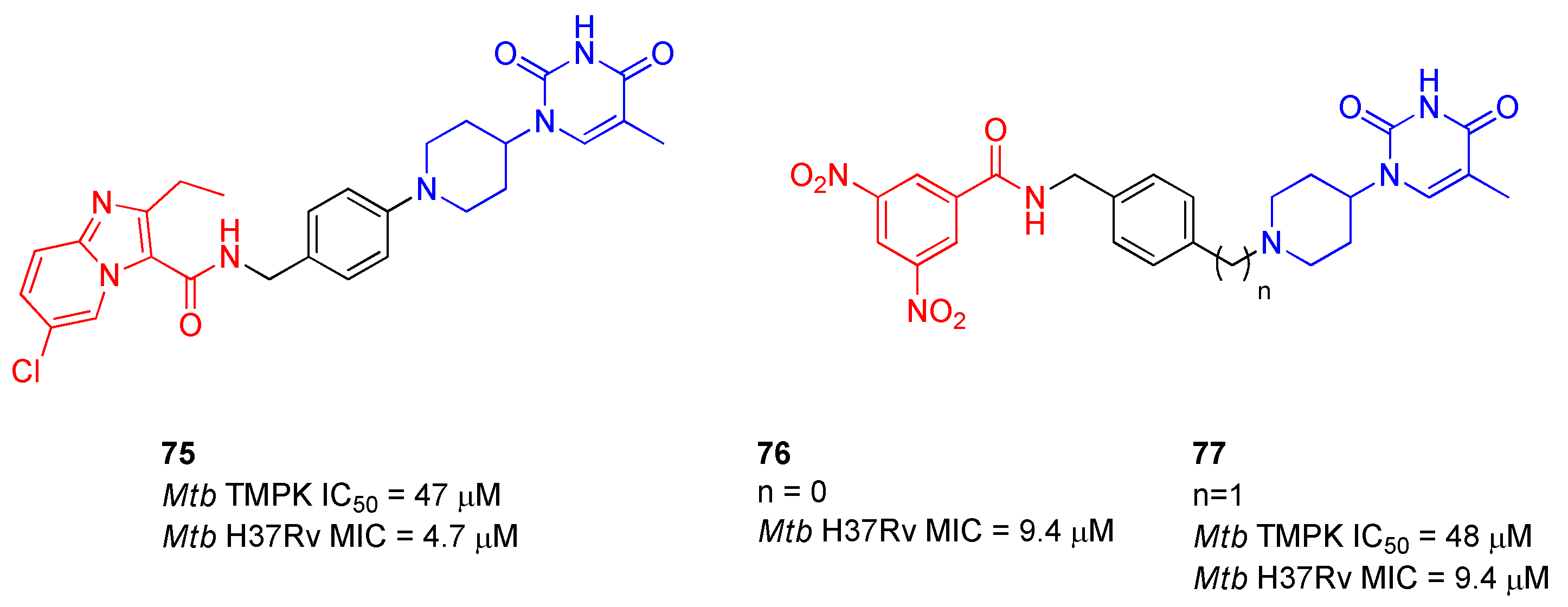
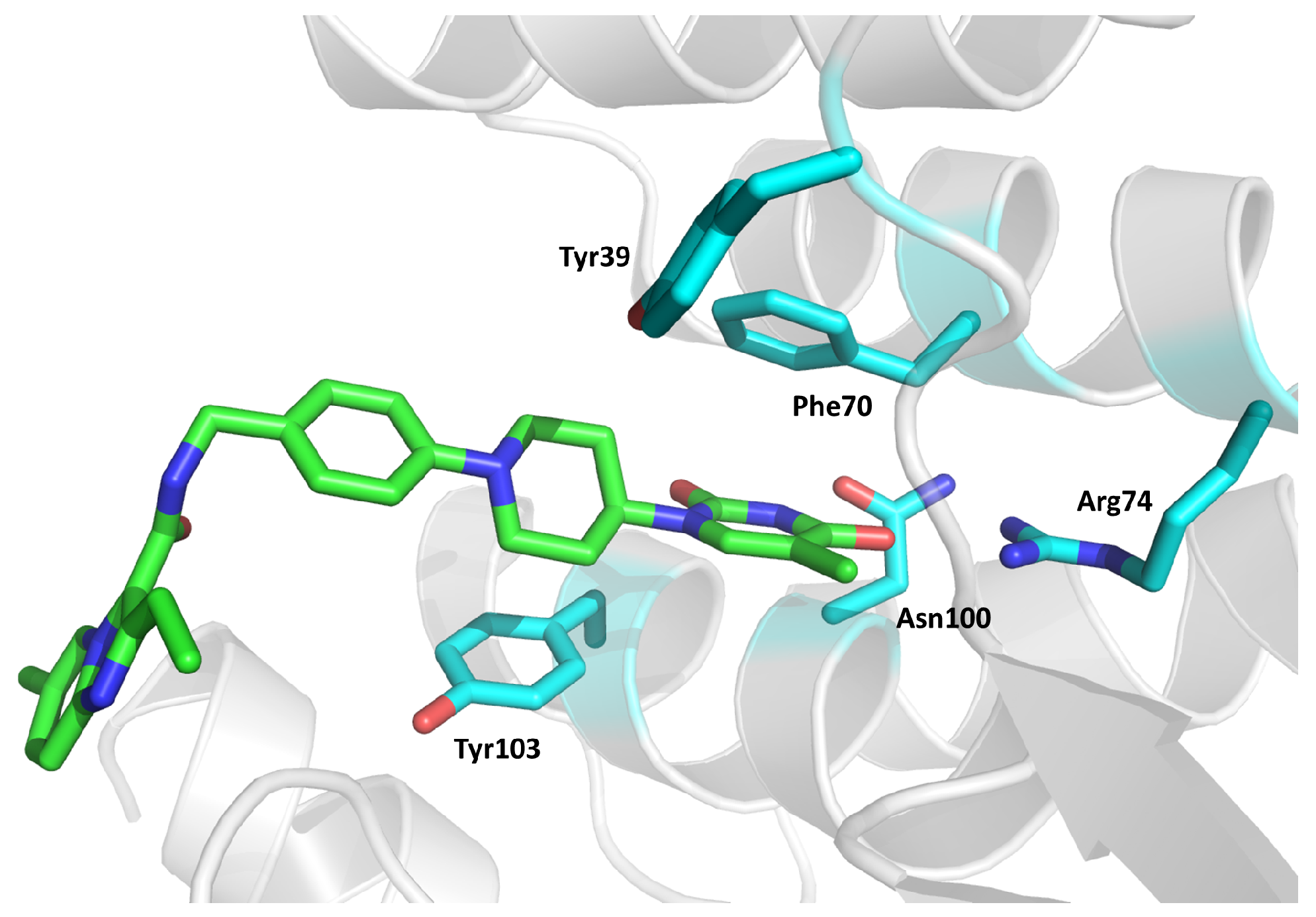
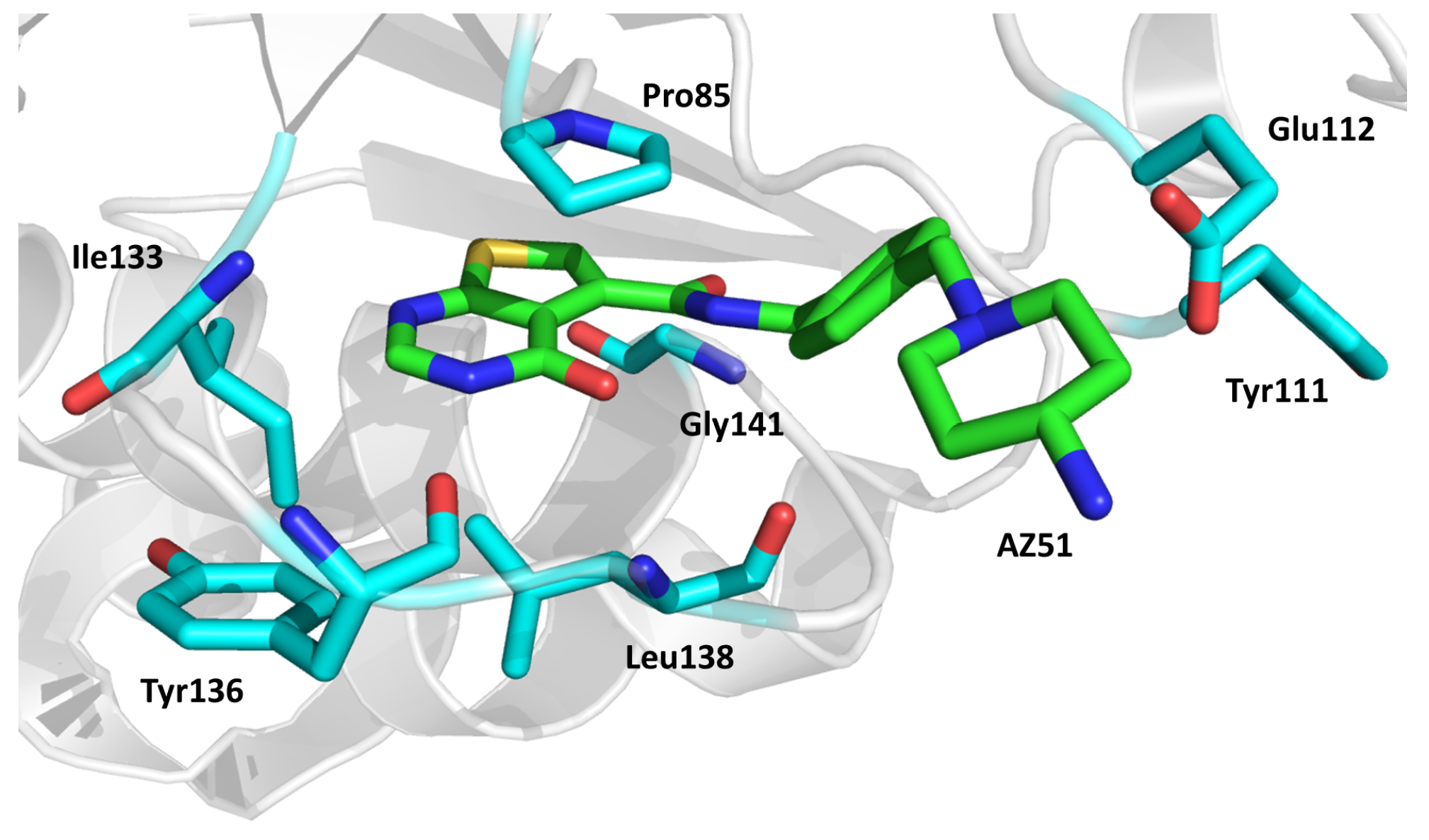
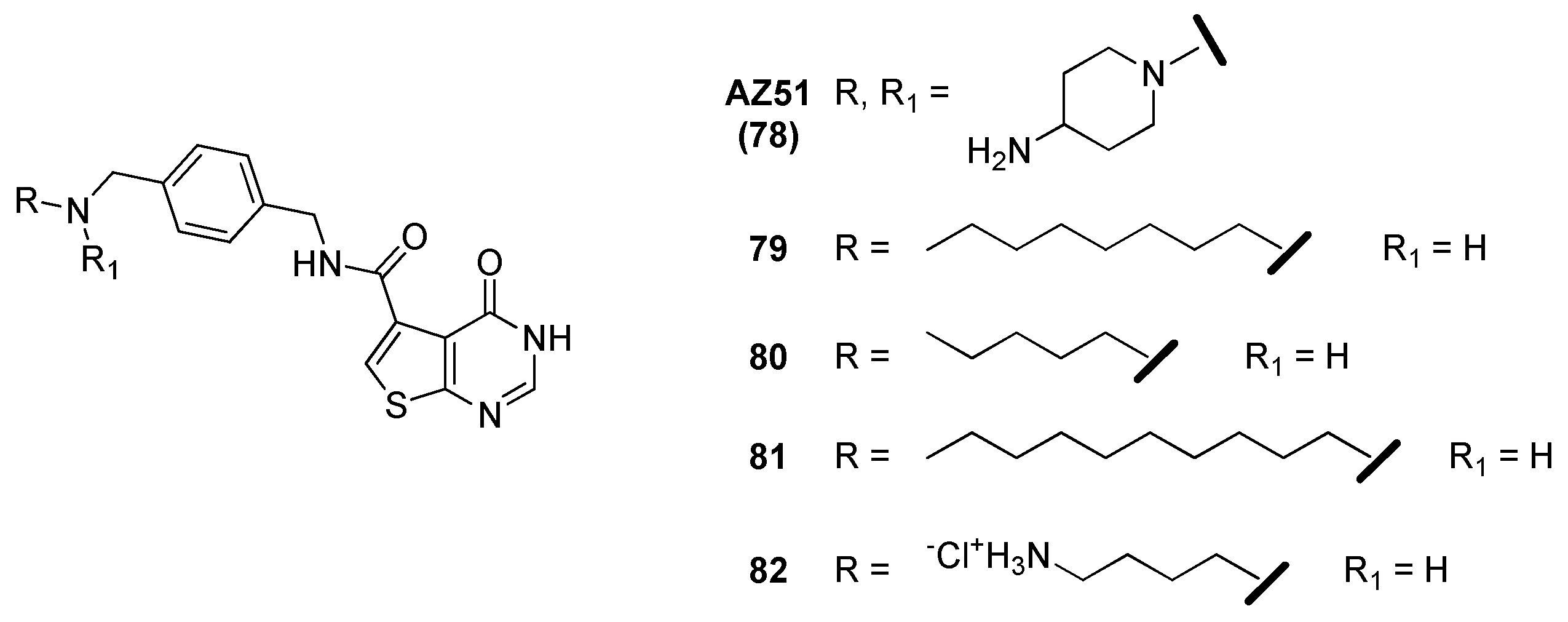
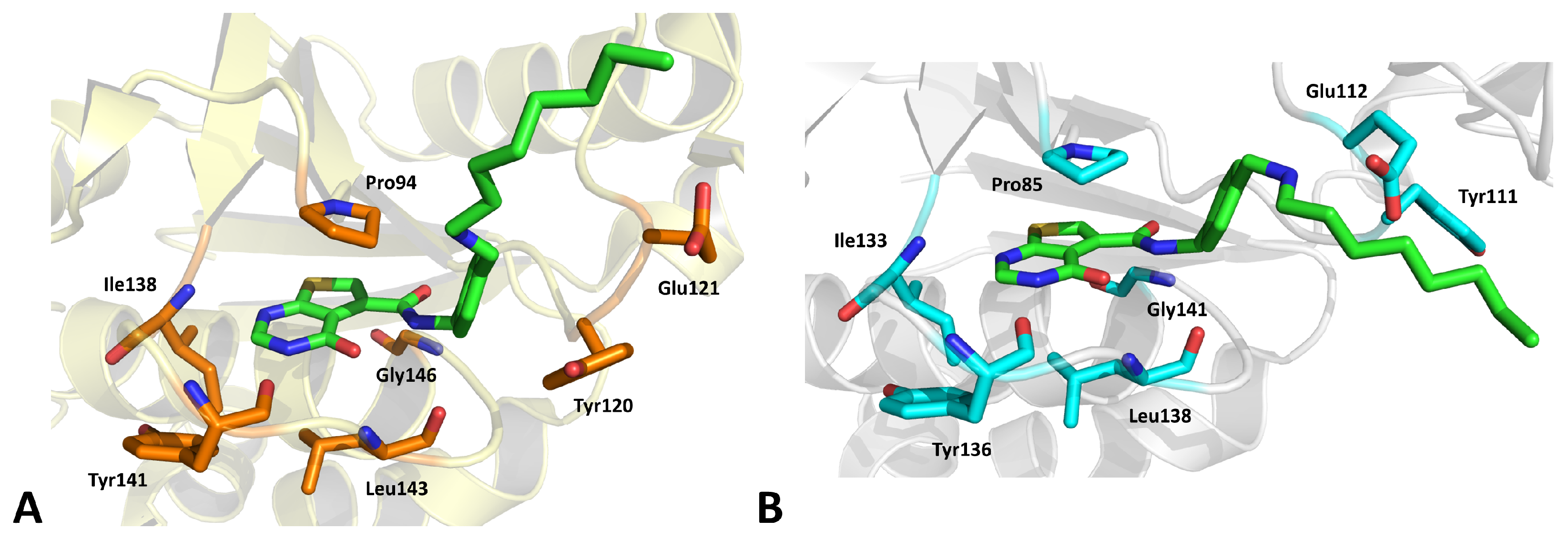
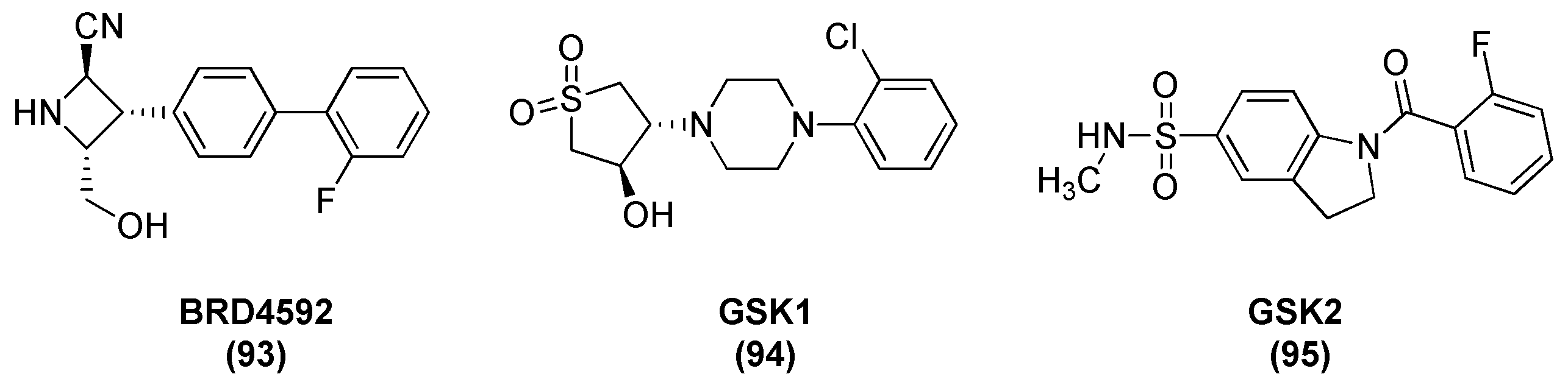
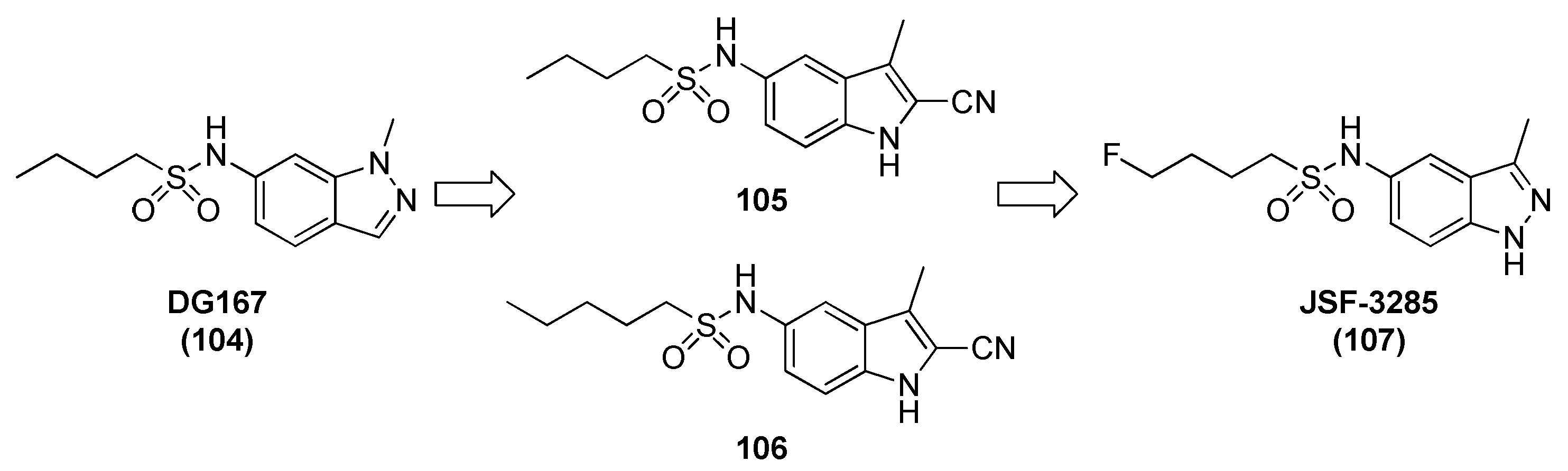
2.12. Thymidylate Kinase (TMPK)
2.13. tRNA (guanine37-N1)-methyltransferase (TrmD)
2.14. Nicotinic Acid Mononucleotide (NaMN) Adenylyltransferase (NadD)
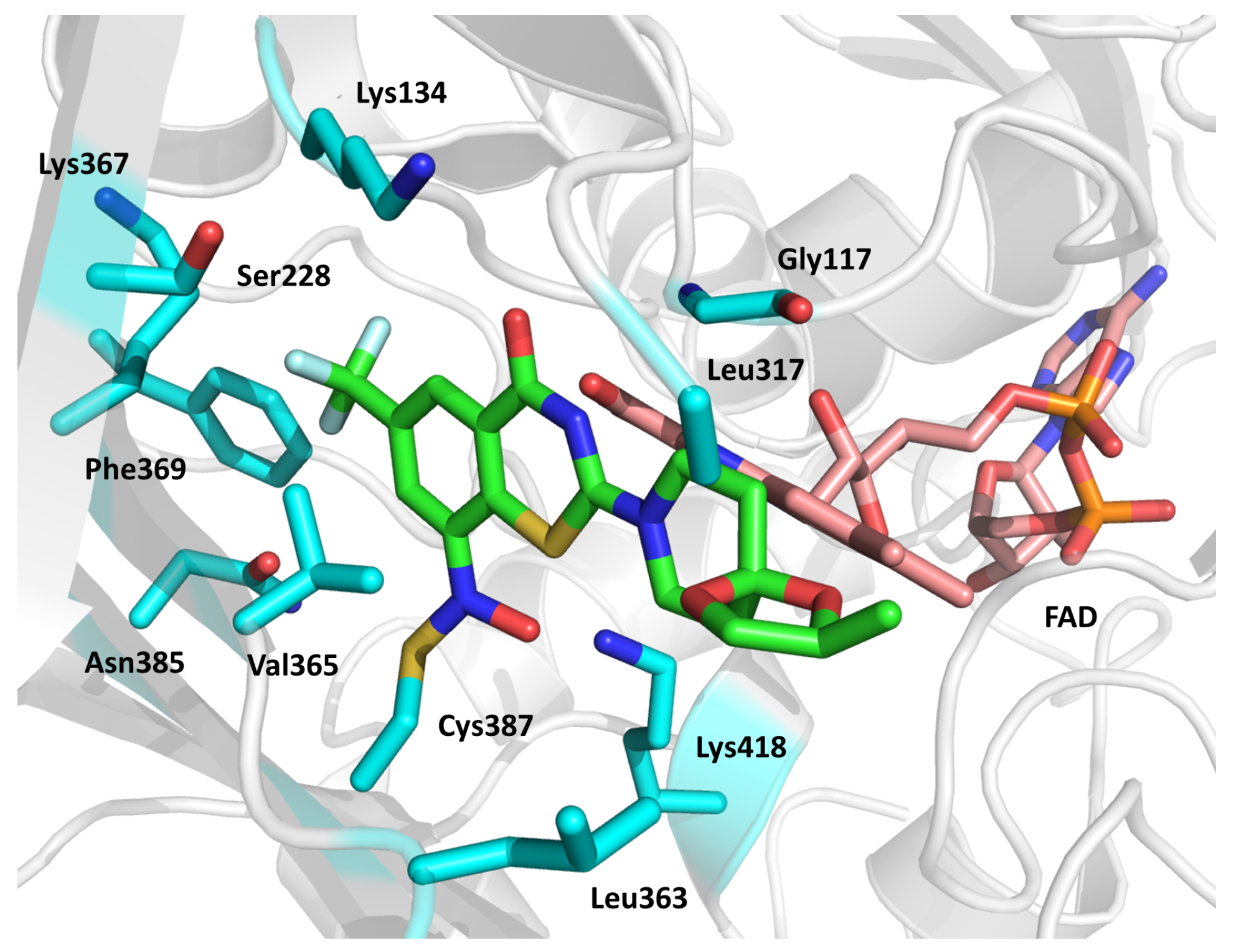
2.15. Enhanced Intracellular Survival (Eis) Transferase
2.16. 2-Succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexadiene-1-carboxylate Synthase (MenD)
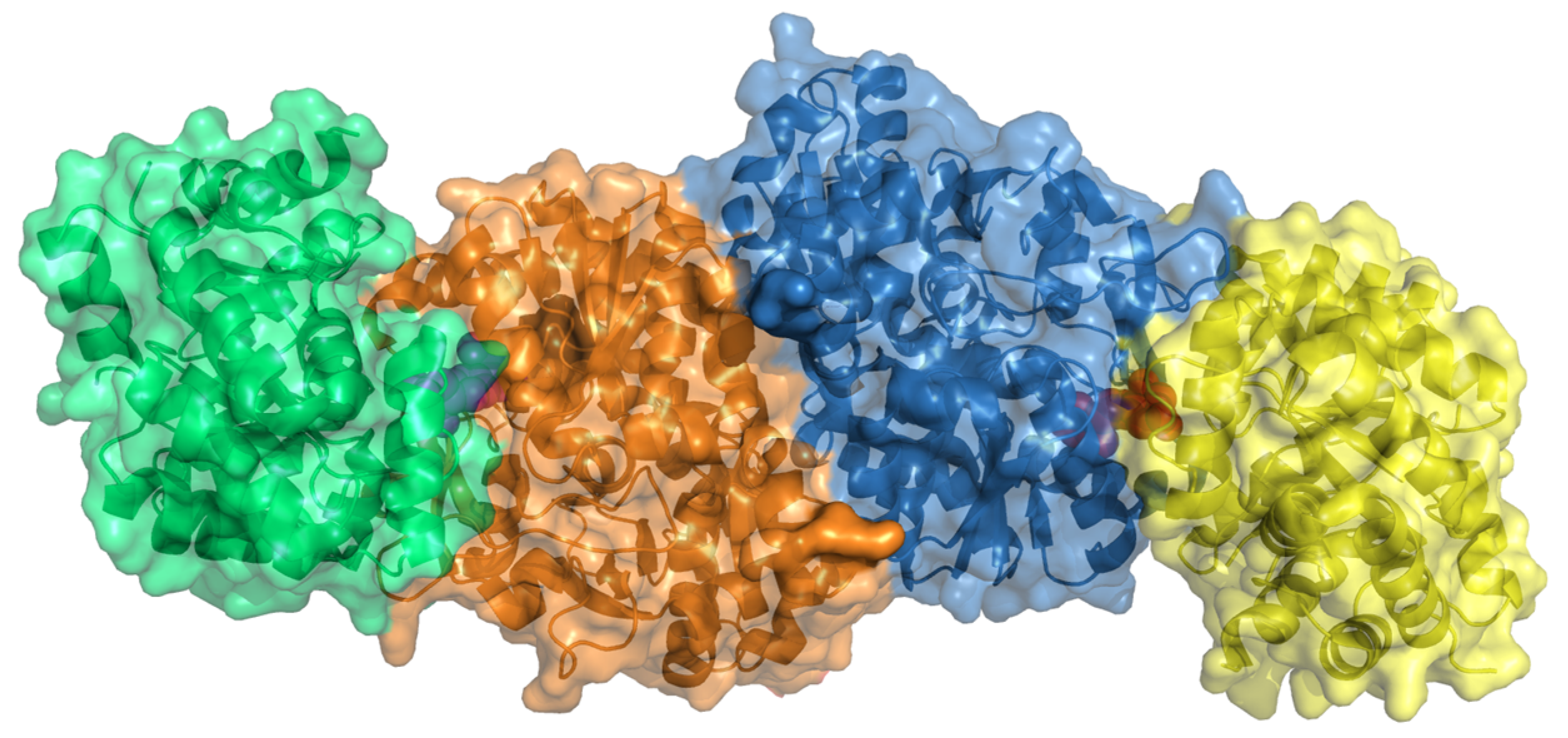
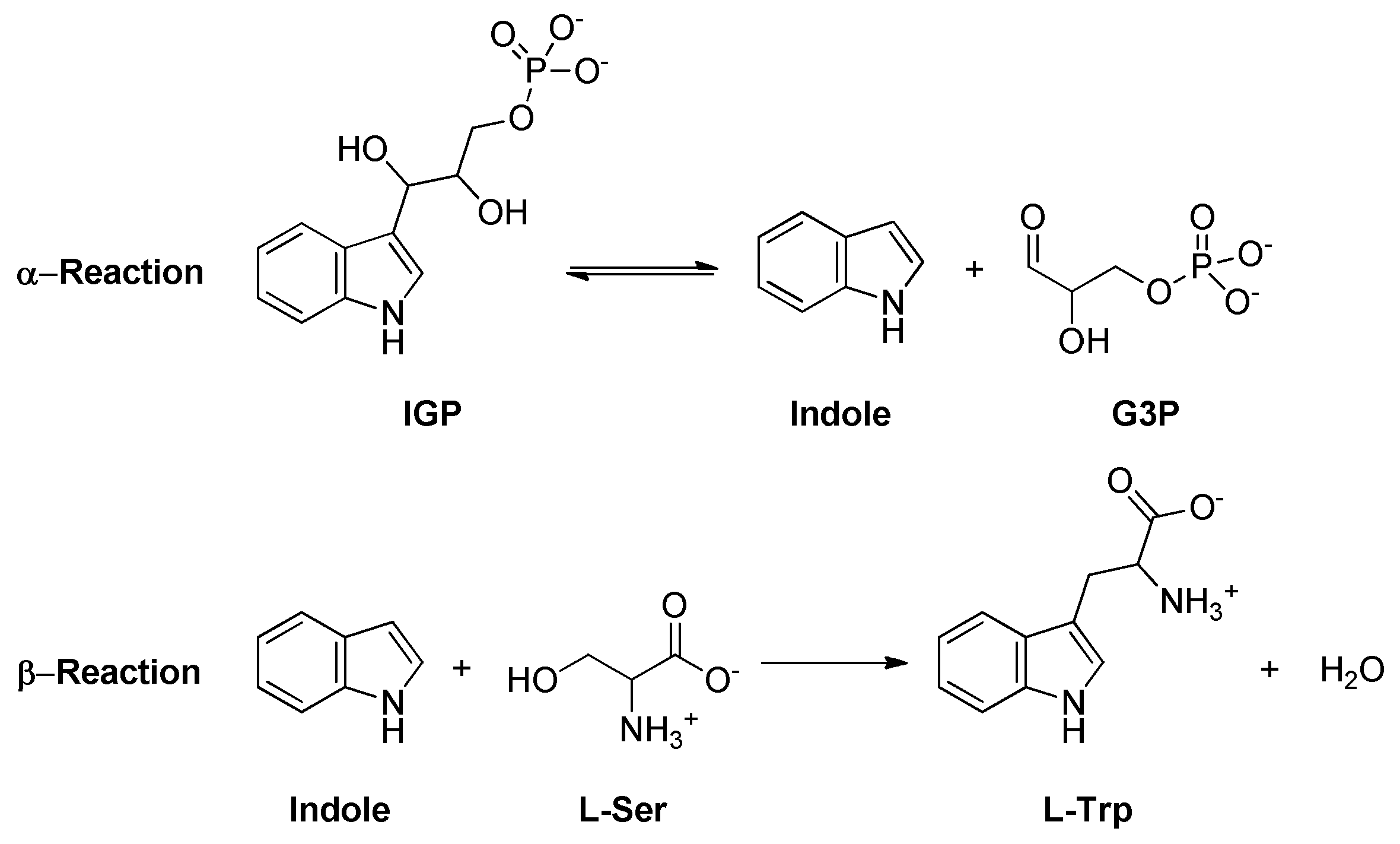
2.17. Tryptophan Synthase (TrpAB)
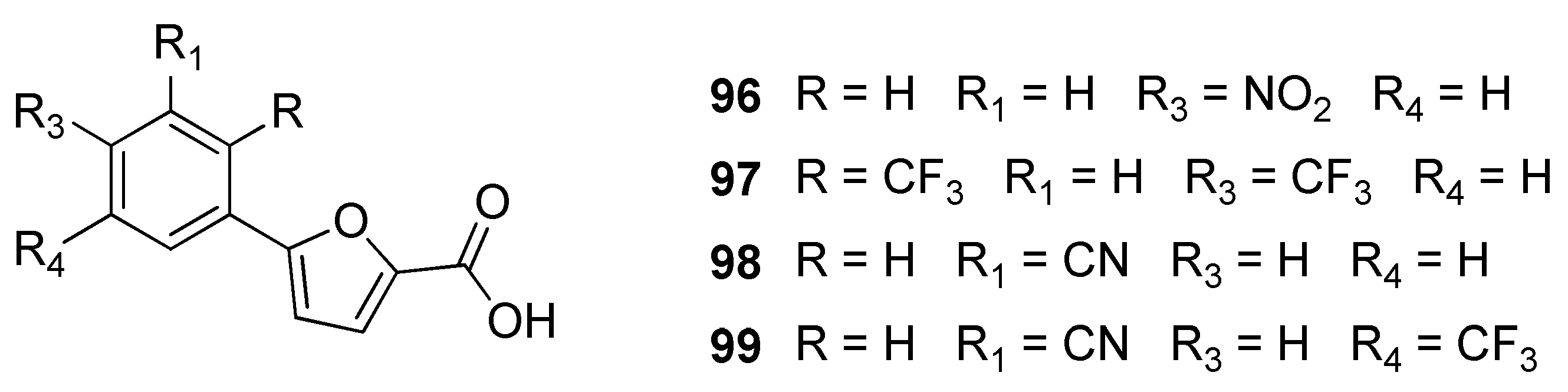
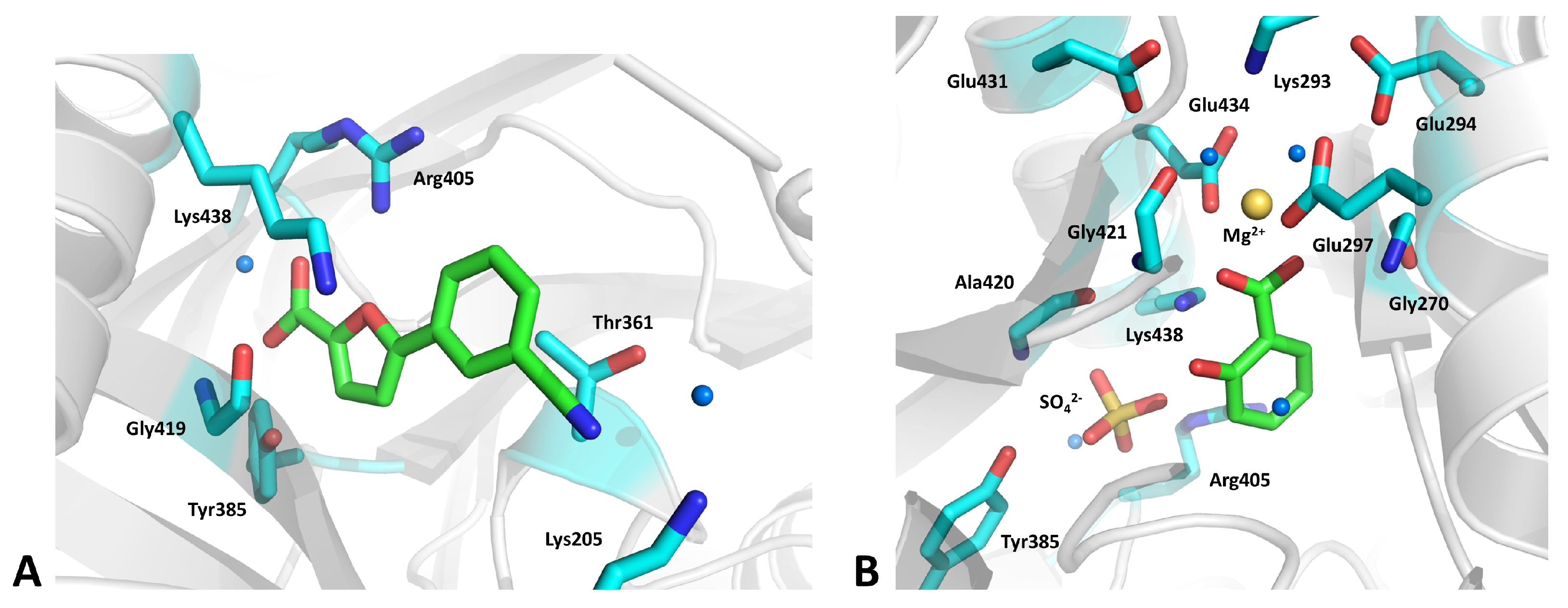
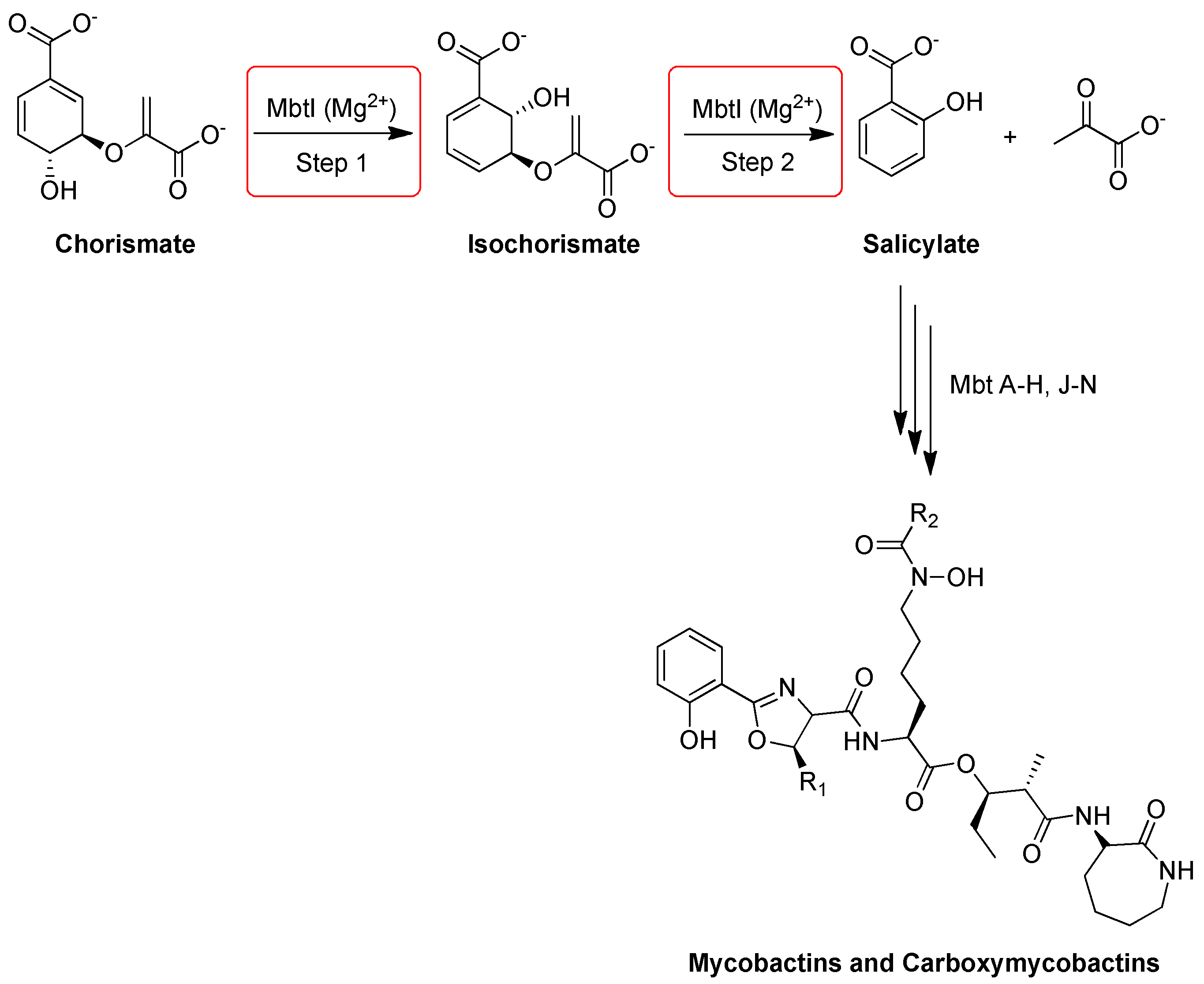
2.18. Salicylate Synthase (MbtI)
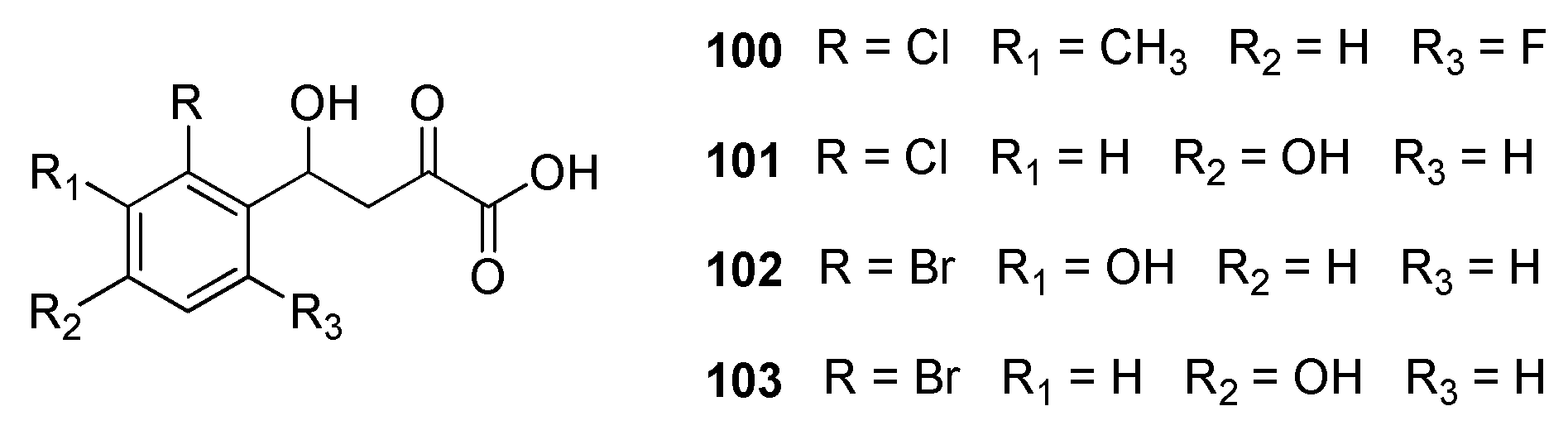
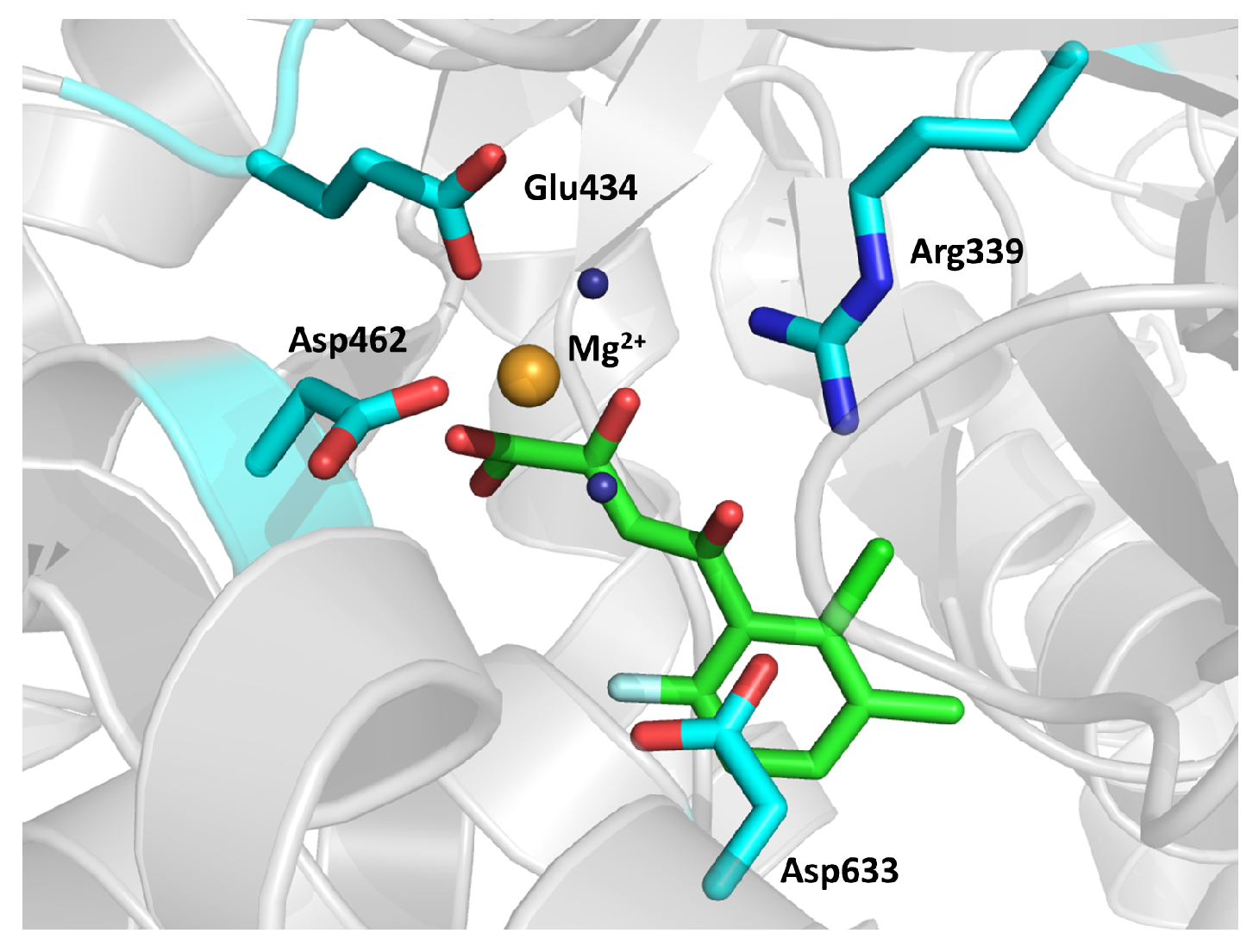
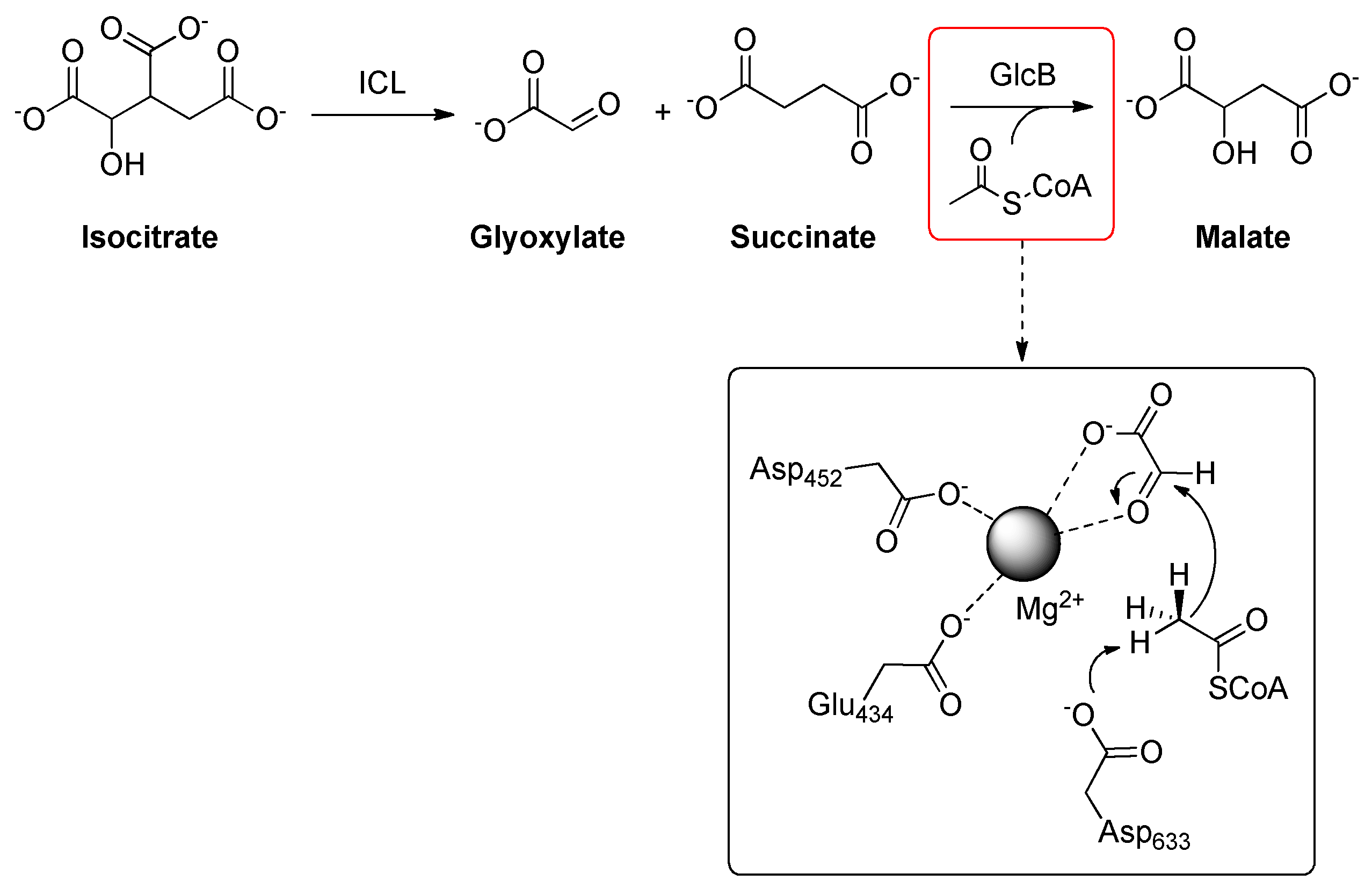
2.19. Malate Synthase G (GlcB)
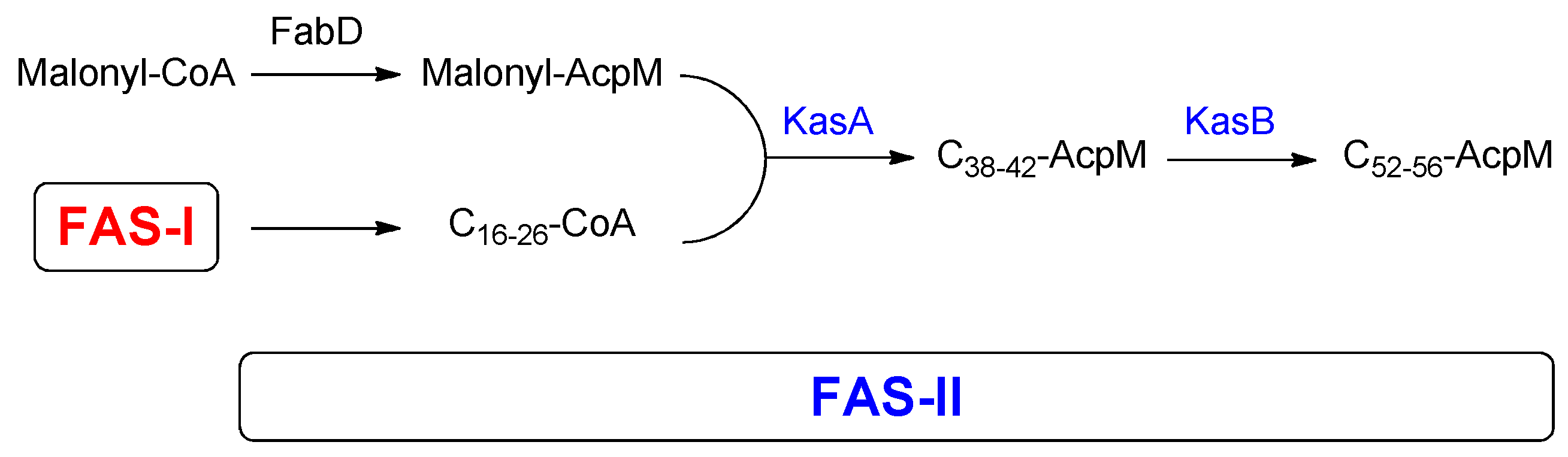
2.20. β-ketoacyl-AcpM Synthase (KasA)
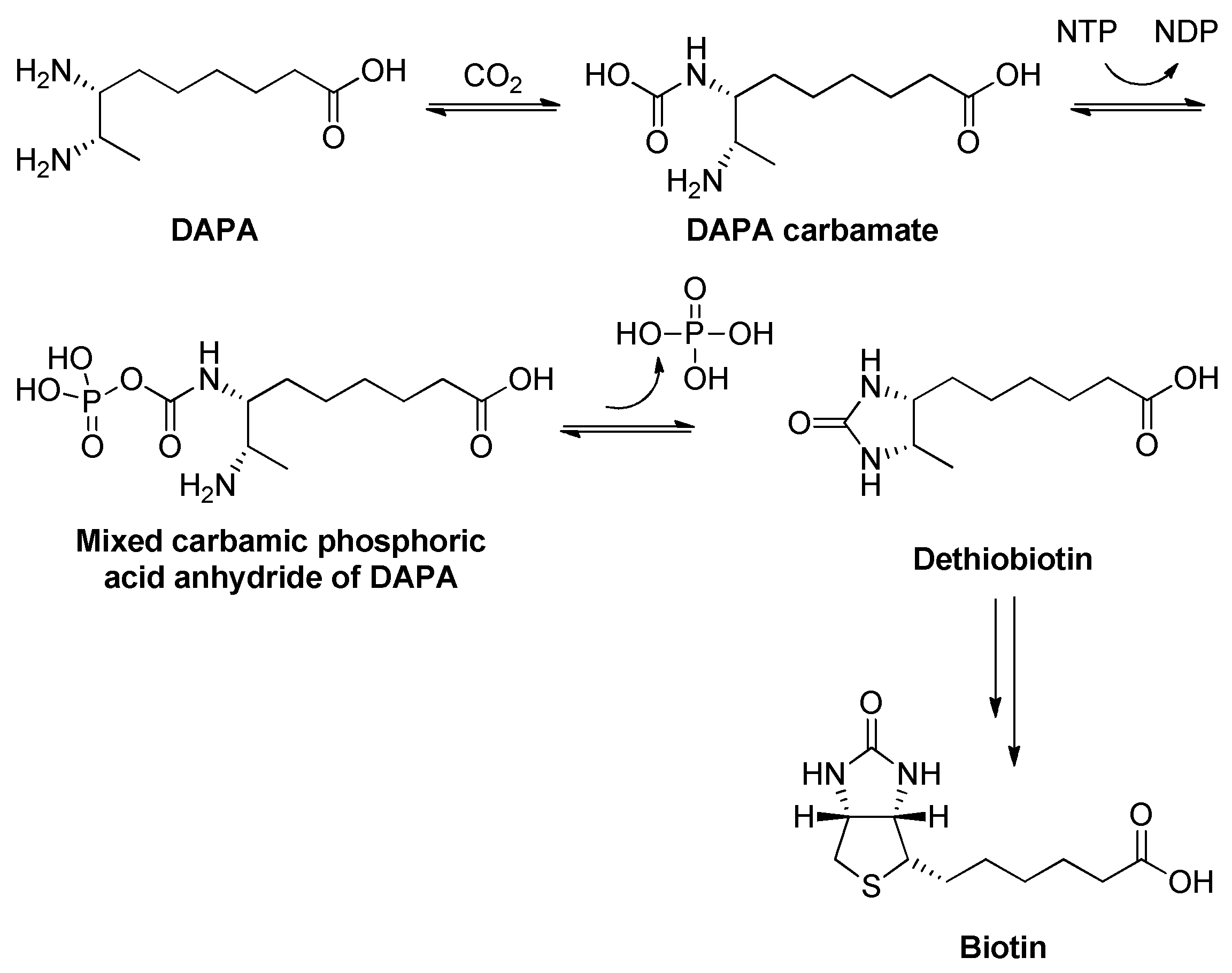
2.21. Dethiobiotin Synthase (DTBS)
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Programme, G.T. Global Tuberculosis Report 2021; World Health Organization [Online]: Geneva, Switzerland, 14 October 2021; Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 14 November 2021).
- Yuan, T.; Sampson, N.S. Hit generation in TB drug discovery: From genome to granuloma. Chem. Rev. 2018, 118, 1887–1916. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Zhang, X. Protein targets for structure-based anti-Mycobacterium tuberculosis drug discovery. Protein Cell 2010, 1, 435–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney, J.D.; Höner zu Bentrup, K.; Muñoz-Elías, E.J.; Miczak, A.; Chen, B.; Chan, W.T.; Swenson, D.; Sacchettini, J.C.; Jacobs, W.R.; Russell, D.G. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406, 735–738. [Google Scholar] [CrossRef]
- Sacchettini, J.C.; McKinney, J.D.; Russell, D.G.; Jacobs, W.R., Jr.; Sharma, V.; Sharma, S.; Hener zu Bentrup, K. Isocitrate Lyase Enzyme from Mycobacterium tuberculosis and Inhibitory Agents to Combat Persistent Infection. US Patent WO2002033118, 30 May 2003. [Google Scholar]
- Kwon, S.; Chun, H.L.; Ha, H.J.; Lee, S.Y.; Park, H.H. Heterogeneous multimeric structure of isocitrate lyase in complex with succinate and itaconate provides novel insights into its inhibitory mechanism. PLoS ONE 2021, 16, e0251067. [Google Scholar] [CrossRef] [PubMed]
- Kwai, B.X.C.; Collins, A.J.; Middleditch, M.J.; Sperry, J.; Bashiri, G.; Leung, I.K.H. Itaconate is a covalent inhibitor of the Mycobacterium tuberculosis isocitrate lyase. RSC Med. Chem. 2021, 12, 57–61. [Google Scholar] [CrossRef]
- Ruetz, M.; Campanello, G.C.; Purchal, M.; Shen, H.; McDevitt, L.; Gouda, H.; Wakabayashi, S.; Zhu, J.; Rubin, E.J.; Warncke, K.; et al. Itaconyl-CoA forms a stable biradical in methylmalonyl-CoA mutase and derails its activity and repair. Science 2019, 366, 589–593. [Google Scholar] [CrossRef]
- Barron, M. Pernicious anemia and tuberculosis: Is there an antagonism? JAMA 1933, 100, 1590. [Google Scholar] [CrossRef]
- Kasbekar, M.; Fischer, G.; Mott, B.T.; Yasgar, A.; Hyvönen, M.; Boshoff, H.I.M.; Abell, C.; Barry, C.E.; Thomas, C.J. Selective small molecule inhibitor of the Mycobacterium tuberculosis fumarate hydratase reveals an allosteric regulatory site. Proc. Natl. Acad. Sci. USA 2016, 113, 7503–7508. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, A.J.; Libardo, M.D.J.; Kasbekar, M.; Brear, P.D.; Fischer, G.; Thomas, C.J.; Barry, C.E.; Boshoff, H.I.M.; Coyne, A.G.; Abell, C. Targeting of fumarate hydratase from Mycobacterium tuberculosis using allosteric inhibitors with a dimeric-binding mode. J. Med. Chem. 2019, 62, 10586–10604. [Google Scholar] [CrossRef]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.T.; Li, H.J.; Yu, W.; Shah, S.; Bommineni, G.R.; Perrone, V.; Garcia-Diaz, M.; Tonge, P.J.; Simmerling, C. Rational modulation of the induced-fit conformational change for slow-onset inhibition in Mycobacterium tuberculosis InhA. Biochemistry 2015, 54, 4683–4691. [Google Scholar] [CrossRef] [Green Version]
- Prati, F.; Zuccotto, F.; Fletcher, D.; Convery, M.A.; Fernandez-Menendez, R.; Bates, R.; Encinas, L.; Zeng, J.; Chung, C.-W.; De Dios Anton, P.; et al. Screening of a novel fragment library with functional complexity against Mycobacterium tuberculosis InhA. ChemMedChem 2018, 13, 672–677. [Google Scholar] [CrossRef] [Green Version]
- Kamsri, P.; Hanwarinroj, C.; Phusi, N.; Pornprom, T.; Chayajarus, K.; Punkvang, A.; Suttipanta, N.; Srimanote, P.; Suttisintong, K.; Songsiriritthigul, C.; et al. Discovery of new and potent InhA inhibitors as antituberculosis agents: Structure-based virtual screening validated by biological assays and X-ray crystallography. J. Chem. Inf. Model. 2020, 60, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, M.; Mendes, V.; Vistal, R.G.; Dias, D.M.G.; Záhorszká, M.; Mikušová, K.; Korduláková, J.; Coyne, A.G.; Blundell, T.L.; Abell, C. Fragment-based design of Mycobacterium tuberculosis InhA inhibitors. J. Med. Chem. 2020, 63, 4749–4761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajian, B.; Scocchera, E.; Shoen, C.; Krucinska, J.; Viswanathan, K.; G-Dayanandan, N.; Erlandsen, H.; Estrada, A.; Mikušová, K.; Korduláková, J.; et al. Drugging the folate pathway in Mycobacterium tuberculosis: The role of multi-targeting agents. Cell Chem. Biol. 2019, 26, 781–791.e6. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.A.; Hammer, A.; Libreros-Zúñiga, G.A.; Chavez-Pacheco, S.M.; Tyrakis, P.; de Oliveira, G.S.; Kirkman, T.; El Bakali, J.; Rocco, S.A.; Sforça, M.L.; et al. Using a fragment-based approach to identify alternative chemical scaffolds targeting dihydrofolate reductase from Mycobacterium tuberculosis. ACS Infect. Dis. 2020, 6, 2192–2201. [Google Scholar] [CrossRef]
- de Chiara, C.; Homšak, M.; Prosser, G.A.; Douglas, H.L.; Garza-Garcia, A.; Kelly, G.; Purkiss, A.G.; Tate, E.W.; de Carvalho, L.P.S. D-Cycloserine destruction by alanine racemase and the limit of irreversible inhibition. Nat. Chem. Biol. 2020, 16, 686–694. [Google Scholar] [CrossRef]
- Sun, Q.; Li, X.; Perez, L.M.; Shi, W.; Zhang, Y.; Sacchettini, J.C. The molecular basis of pyrazinamide activity on Mycobacterium tuberculosis PanD. Nat. Commun. 2020, 11, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.; Rudolph, I.; Möllmann, U.; Voigt, K.; Chung, C.-W.; Singh, O.M.P.; Rees, M.; Mendoza-Losana, A.; Bates, R.; Ballell, L.; et al. Novel insight into the reaction of nitro, nitroso and hydroxylamino benzothiazinones and of benzoxacinones with Mycobacterium tuberculosis DprE1. Sci. Rep. 2018, 8, 13473. [Google Scholar] [CrossRef] [Green Version]
- Varaksa, T.; Bukhdruker, S.; Grabovec, I.; Marin, E.; Kavaleuski, A.; Gusach, A.; Kovalev, K.; Maslov, I.; Luginina, A.; Zabelskii, D.; et al. Metabolic fate of human immunoactive sterols in Mycobacterium tuberculosis. J. Mol. Biol. 2021, 433, 166763. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; McLean, K.J.; Poddar, H.; Selvam, I.R.; Nagalingam, G.; Triccas, J.A.; Levy, C.W.; Munro, A.W.; Hutton, C.A. Structure-activity relationships of cyclo(l-Tyrosyl-l-tyrosine) derivatives binding to Mycobacterium tuberculosis CYP121: Iodinated analogues promote shift to high-spin adduct. J. Med. Chem. 2019, 62, 9792–9805. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.; Polycarpou, E.; Lack, N.A.; Evangelopoulos, D.; Sieg, C.; Halman, A.; Bhakta, S.; Eleftheriadou, O.; McHugh, T.D.; Keany, S.; et al. Investigation of the mycobacterial enzyme HsaD as a potential novel target for anti-tubercular agents using a fragment-based drug design approach. Br. J. Pharmacol. 2017, 174, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Sammartino, J.C.; Costantino, L.; Gelain, A.; Meneghetti, F.; Villa, S.; Chiarelli, L.R. An overview on the potential antimycobacterial agents targeting serine/threonine protein kinases from Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2019, 19, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Young, T.A.; Delagoutte, B.; Endrizzi, J.A.; Falick, A.M.; Alber, T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 2003, 10, 168–174. [Google Scholar] [CrossRef]
- Wlodarchak, N.; Teachout, N.; Beczkiewicz, J.; Procknow, R.; Schaenzer, A.J.; Satyshur, K.; Pavelka, M.; Zuercher, W.; Drewry, D.; Sauer, J.-D.; et al. In silico screen and structural analysis identifies bacterial kinase inhibitors which act with β-lactams to inhibit mycobacterial growth. Mol. Pharm. 2018, 15, 5410–5426. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.A.; Dang, Q.; Zhou, N.E.; Guthrie, L.M.; Snavely, T.C.; Dong, W.; Loesch, K.A.; Suzuki, T.; You, L.; Wang, W.; et al. Structure-guided drug design of 6-substituted adenosine analogues as potent inhibitors of Mycobacterium tuberculosis adenosine kinase. J. Med. Chem. 2019, 62, 4483–4499. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Merceron, R.; Gracia, B.; Quintana, A.L.; Risseeuw, M.D.P.; Hulpia, F.; Cos, P.; Aínsa, J.A.; Munier-Lehmann, H.; Savvides, S.N.; et al. Structure guided lead generation toward nonchiral M. tuberculosis thymidylate kinase inhibitors. J. Med. Chem. 2018, 61, 2753–2775. [Google Scholar] [CrossRef]
- Jian, Y.; Merceron, R.; De Munck, S.; Forbes, H.E.; Hulpia, F.; Risseeuw, M.D.P.; van Hecke, K.; Savvides, S.N.; Munier-Lehmann, H.; Boshoff, H.I.M.; et al. Endeavors towards transformation of M. tuberculosis thymidylate kinase (MtbTMPK) inhibitors into potential antimycobacterial agents. Eur. J. Med. Chem. 2020, 206, 112659. [Google Scholar] [CrossRef]
- Kang, S.; Kim, R.Y.; Seo, M.J.; Lee, S.; Kim, Y.M.; Seo, M.; Seo, J.J.; Ko, Y.; Choi, I.; Jang, J.; et al. Lead optimization of a novel series of imidazo1,2-apyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 2014, 57, 5293–5305. [Google Scholar] [CrossRef]
- Zhong, W.; Pasunooti, K.K.; Balamkundu, S.; Wong, Y.H.; Nah, Q.; Gadi, V.; Gnanakalai, S.; Chionh, Y.H.; McBee, M.E.; Gopal, P.; et al. Thienopyrimidinone derivatives that inhibit bacterial trna (guanine37-n1)-methyltransferase (TrmD) by restructuring the active site with a tyrosine-flipping mechanism. J. Med. Chem. 2019, 62, 7788–7805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, H.J.; Kim, H.-W.; Yoon, H.-J.; Lee, B.I.; Suh, S.W.; Yang, J.K. Crystal structure of tRNA(m1G37)methyltransferase: Insights into tRNA recognition. EMBO J. 2003, 22, 2593–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterman, A.L.; Rodionova, I.; Li, X.; Sergienko, E.; Ma, C.-T.; Catanzaro, A.; Pettigrove, M.E.; Reed, R.W.; Gupta, R.; Rohde, K.H.; et al. Novel antimycobacterial compounds suppress NAD biogenesis by targeting a unique pocket of namn adenylyltransferase. ACS Chem. Biol. 2019, 14, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Punetha, A.; Ngo, H.X.; Holbrook, S.Y.L.; Green, K.D.; Willby, M.J.; Bonnett, S.A.; Krieger, K.; Dennis, E.K.; Posey, J.E.; Parish, T.; et al. Structure-guided optimization of inhibitors of acetyltransferase Eis from Mycobacterium tuberculosis. ACS Chem. Biol. 2020, 15, 1581–1594. [Google Scholar] [CrossRef]
- Ngo, H.X.; Green, K.D.; Gajadeera, C.S.; Willby, M.J.; Holbrook, S.Y.L.; Hou, C.; Garzan, A.; Mayhoub, A.S.; Posey, J.E.; Tsodikov, O.V.; et al. Potent 1,2,4-triazino5,6 bindole-3-thioether inhibitors of the kanamycin resistance enzyme Eis from Mycobacterium tuberculosis. ACS Infect. Dis. 2018, 4, 1030–1040. [Google Scholar] [CrossRef]
- Green, K.D.; Punetha, A.; Hou, C.; Garneau-Tsodikova, S.; Tsodikov, O.V. Probing the robustness of inhibitors of tuberculosis aminoglycoside resistance enzyme Eis by mutagenesis. ACS Infect. Dis. 2019, 5, 1772–1778. [Google Scholar] [CrossRef]
- Bashiri, G.; Nigon, L.V.; Jirgis, E.N.M.; Ho, N.A.T.; Stanborough, T.; Dawes, S.S.; Baker, E.N.; Bulloch, E.M.M.; Johnston, J.M. Allosteric regulation of menaquinone (vitamin K2) biosynthesis in the human pathogen Mycobacterium tuberculosis. J. Biol. Chem. 2020, 295, 3759–3770. [Google Scholar] [CrossRef] [Green Version]
- Michalska, K.; Chang, C.; Maltseva, N.I.; Jedrzejczak, R.; Robertson, G.T.; Gusovsky, F.; McCarren, P.; Schreiber, S.L.; Nag, P.P.; Joachimiak, A. Allosteric inhibitors of Mycobacterium tuberculosis tryptophan synthase. Protein Sci. 2020, 29, 779–788. [Google Scholar] [CrossRef]
- Dunn, M.F. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch. Biochem. Biophys. 2012, 519, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Wellington, S.; Nag, P.P.; Michalska, K.; Johnston, S.E.; Jedrzejczak, R.P.; Kaushik, V.K.; Clatworthy, A.E.; Siddiqi, N.; McCarren, P.; Bajrami, B.; et al. A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 2017, 13, 943–950. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Cox, J.A.G.; Fütterer, K.; Rullas, J.; Ortega-Muro, F.; Loman, N.J.; Moynihan, P.J.; Pérez-Herrán, E.; Jiménez, E.; Esquivias, J.; et al. Inhibiting mycobacterial tryptophan synthase by targeting the inter-subunit interface. Sci. Rep. 2017, 7, 9430. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Mori, M.; Beretta, G.; Gelain, A.; Pini, E.; Sammartino, J.C.; Stelitano, G.; Barlocco, D.; Costantino, L.; Lapillo, M.; et al. New insight into structure-activity of furan-based salicylate synthase (MbtI) inhibitors as potential antitubercular agents. J. Enzyme Inhib. Med. Chem. 2019, 34, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Stelitano, G.; Gelain, A.; Pini, E.; Chiarelli, L.R.; Sammartino, J.C.; Poli, G.; Tuccinardi, T.; Beretta, G.; Porta, A.; et al. Shedding X-ray light on the role of magnesium in the activity of Mycobacterium tuberculosis salicylate synthase (MbtI) for drug design. J. Med. Chem. 2020, 63, 7066–7080. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Barlocco, D.; Pini, E.; Meneghetti, F.; Villa, S. Synthesis, characterization, and biological evaluation of new derivatives targeting MbtI as antitubercular agents. Pharmaceuticals 2021, 14, 155. [Google Scholar] [CrossRef]
- Ellenbarger, J.F.; Krieger, I.V.; Huang, H.-L.; Gómez-Coca, S.; Ioerger, T.R.; Sacchettini, J.C.; Wheeler, S.E.; Dunbar, K.R. Anion-π interactions in computer-aided drug design: Modeling the inhibition of malate synthase by phenyl-diketo acids. J. Chem. Inf. Model. 2018, 58, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Schmoldt, A.; Benthe, H.F.; Haberland, G. Digitoxin metabolism by rat liver microsomes. Biochem. Pharmacol. 1975, 24, 1639–1641. [Google Scholar] [CrossRef] [Green Version]
- Krieger, I.V.; Freundlich, J.S.; Gawandi, V.B.; Roberts, J.P.; Gawandi, V.B.; Sun, Q.; Owen, J.L.; Fraile, M.T.; Huss, S.I.; Lavandera, J.-L.; et al. Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem. Biol. 2012, 19, 1556–1567. [Google Scholar] [CrossRef] [Green Version]
- Inoyama, D.; Awasthi, D.; Capodagli, G.C.; Tsotetsi, K.; Sukheja, P.; Zimmerman, M.; Li, S.-G.; Jadhav, R.; Russo, R.; Wang, X.; et al. A preclinical candidate targeting Mycobacterium tuberculosis KasA. Cell Chem. Biol. 2020, 27, 560–570.e10. [Google Scholar] [CrossRef]
- Slayden, R.A.; Barry, C.E. The role of KasA and KasB in the biosynthesis of meromycolic acids and isoniazid resistance in Mycobacterium tuberculosis. Tuberculosis 2002, 82, 149–160. [Google Scholar] [CrossRef]
- Kumar, P.; Capodagli, G.C.; Awasthi, D.; Shrestha, R.; Maharaja, K.; Sukheja, P.; Li, S.-G.; Inoyama, D.; Zimmerman, M.; Ho Liang, H.P.; et al. Synergistic lethality of a binary inhibitor of Mycobacterium tuberculosis KasA. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahams, K.A.; Chung, C.-W.; Ghidelli-Disse, S.; Rullas, J.; Rebollo-López, M.J.; Gurcha, S.S.; Cox, J.A.G.; Mendoza, A.; Jiménez-Navarro, E.; Martínez-Martínez, M.S.; et al. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 2016, 7, 12581. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.; Esquivias, J.; Fernández-Menéndez, R.; Pérez, A.; Guardia, A.; Escribano, J.; Rivero, C.; Vimal, M.; Cacho, M.; de Dios-Antón, P.; et al. Exploring the SAR of the β-ketoacyl-acp synthase inhibitor GSK3011724A and optimization around a genotoxic metabolite. ACS Infect. Dis. 2020, 6, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Salaemae, W.; Yap, M.Y.; Wegener, K.L.; Booker, G.W.; Wilce, M.C.J.; Polyak, S.W. Nucleotide triphosphate promiscuity in Mycobacterium tuberculosis dethiobiotin synthetase. Tuberculosis 2015, 95, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Schumann, N.C.; Lee, K.J.; Thompson, A.P.; Salaemae, W.; Pederick, J.L.; Avery, T.; Gaiser, B.I.; Hodgkinson-Bean, J.; Booker, G.W.; Polyak, S.W.; et al. Inhibition of Mycobacterium tuberculosis dethiobiotin synthase (MtDTBS): Toward next-generation antituberculosis agents. ACS Chem. Biol. 2021. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray crystallography and drug discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, J.-P.; Chari, A.; Ciferri, C.; Liu, W.-T.; Rémigy, H.-W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef]
- Rout, M.P.; Sali, A. Principles for integrative structural biology studies. Cell 2019, 177, 1384–1403. [Google Scholar] [CrossRef]






































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, M.; Villa, S.; Ciceri, S.; Colombo, D.; Ferraboschi, P.; Meneghetti, F. An Outline of the Latest Crystallographic Studies on Inhibitor-Enzyme Complexes for the Design and Development of New Therapeutics against Tuberculosis. Molecules 2021, 26, 7082. https://doi.org/10.3390/molecules26237082
Mori M, Villa S, Ciceri S, Colombo D, Ferraboschi P, Meneghetti F. An Outline of the Latest Crystallographic Studies on Inhibitor-Enzyme Complexes for the Design and Development of New Therapeutics against Tuberculosis. Molecules. 2021; 26(23):7082. https://doi.org/10.3390/molecules26237082
Chicago/Turabian StyleMori, Matteo, Stefania Villa, Samuele Ciceri, Diego Colombo, Patrizia Ferraboschi, and Fiorella Meneghetti. 2021. "An Outline of the Latest Crystallographic Studies on Inhibitor-Enzyme Complexes for the Design and Development of New Therapeutics against Tuberculosis" Molecules 26, no. 23: 7082. https://doi.org/10.3390/molecules26237082