
Deficiency in Androgen Receptor Aggravates Traumatic Brain Injury-Induced Pathophysiology and Motor Deficits in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
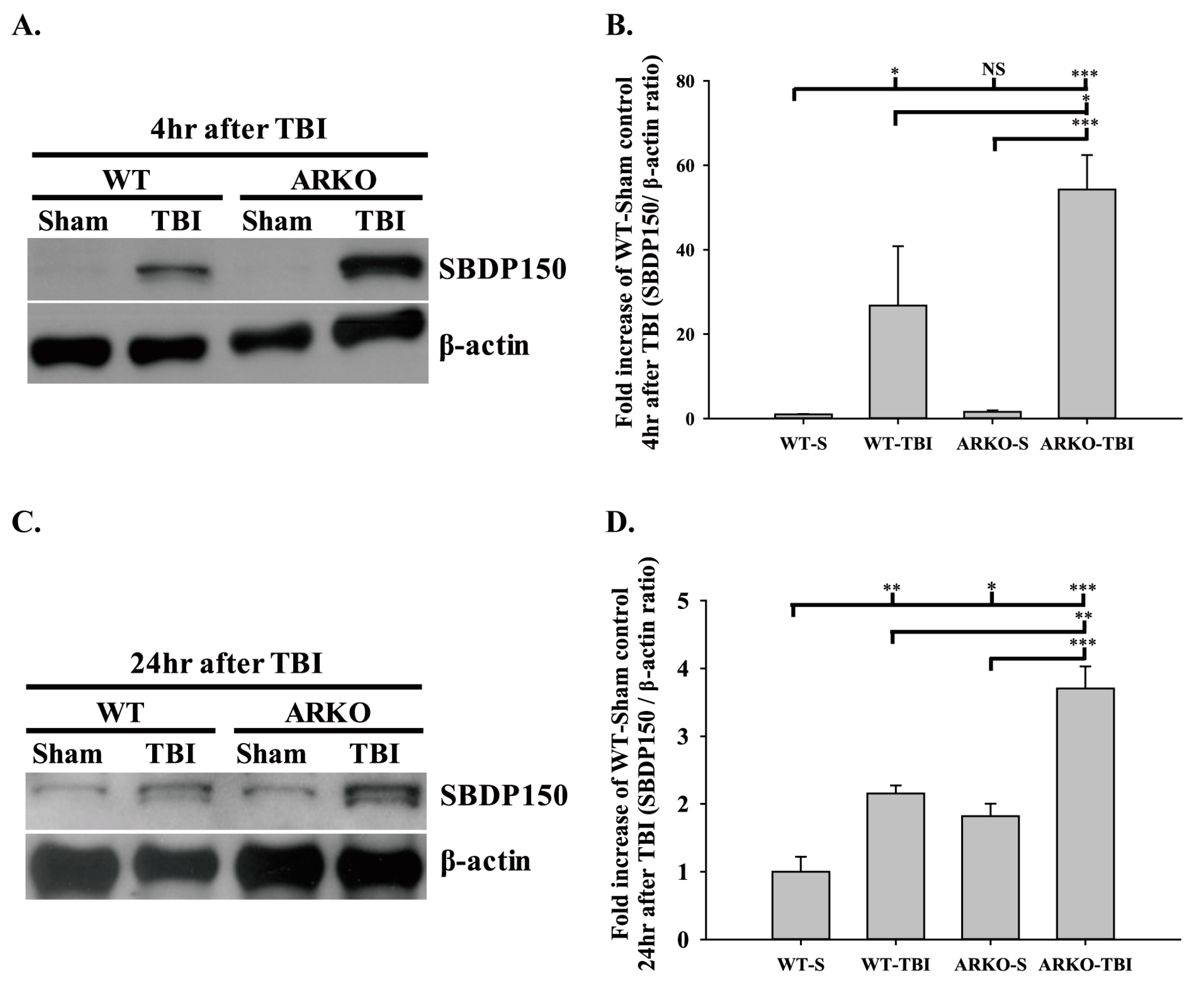
2.1. Effects of Androgen Receptor Knockout on SBDP150 Expression in Mice following TBI
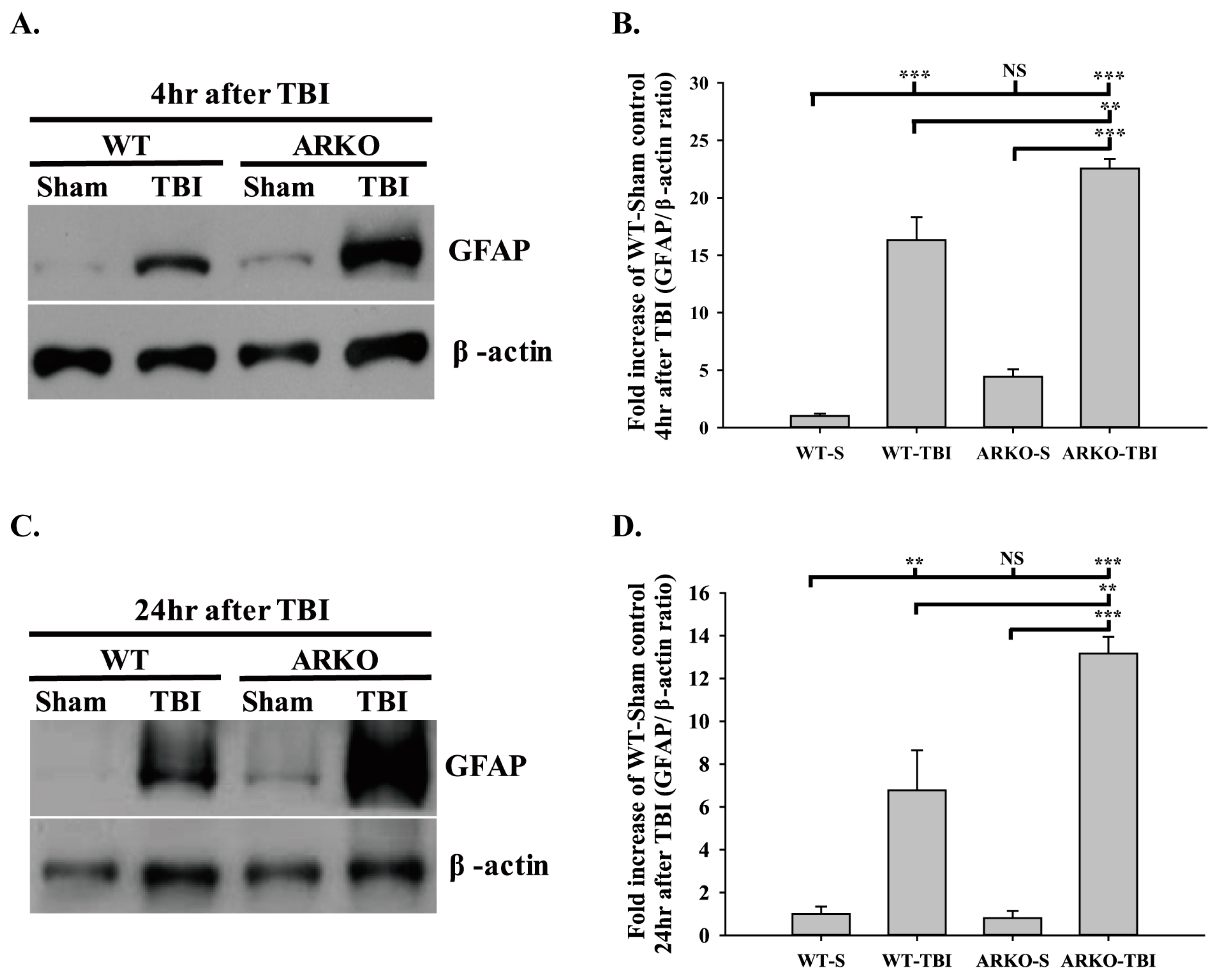
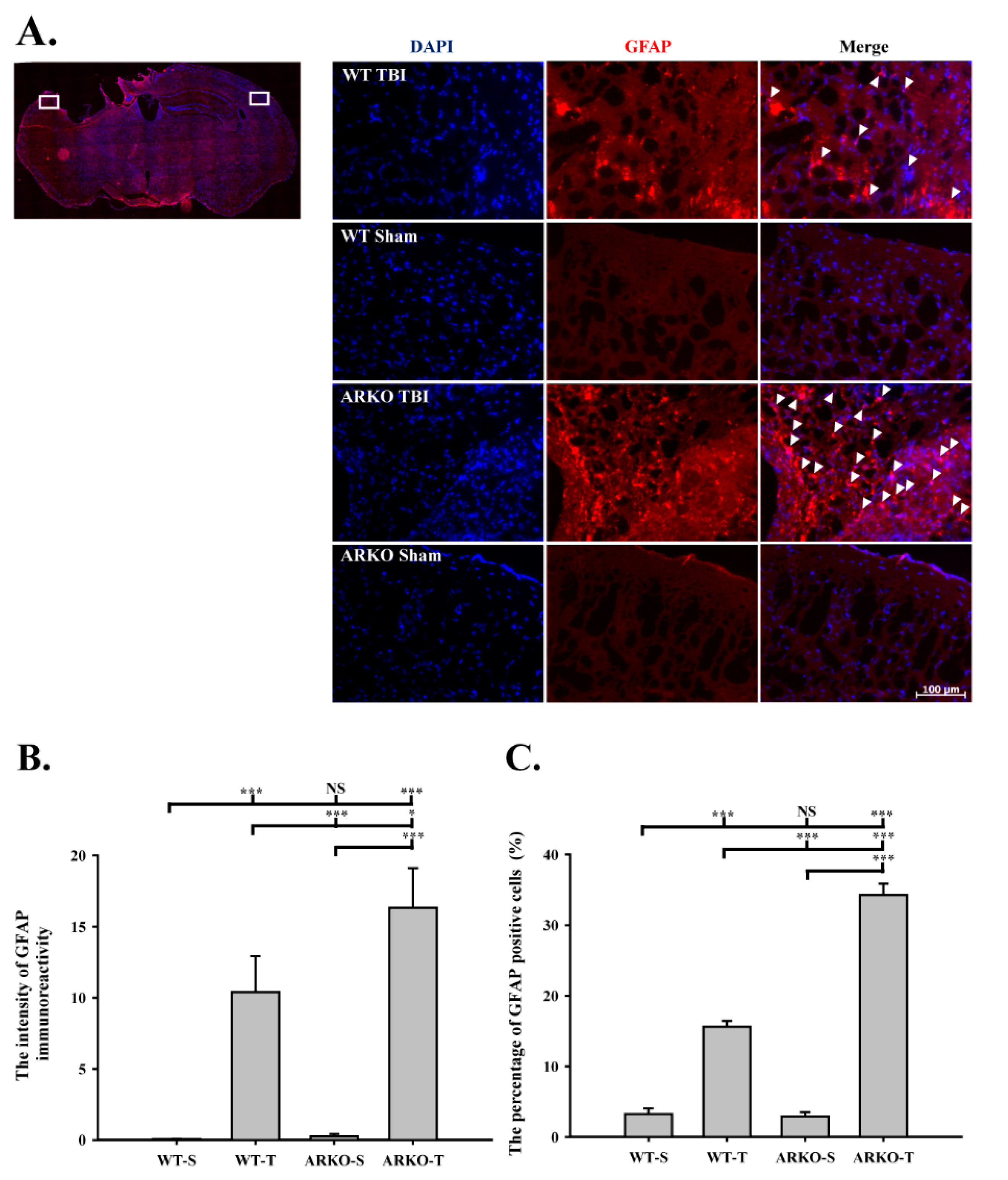
2.2. Effects of Androgen Receptor Knockout on GFAP Expression in Mice following TBI
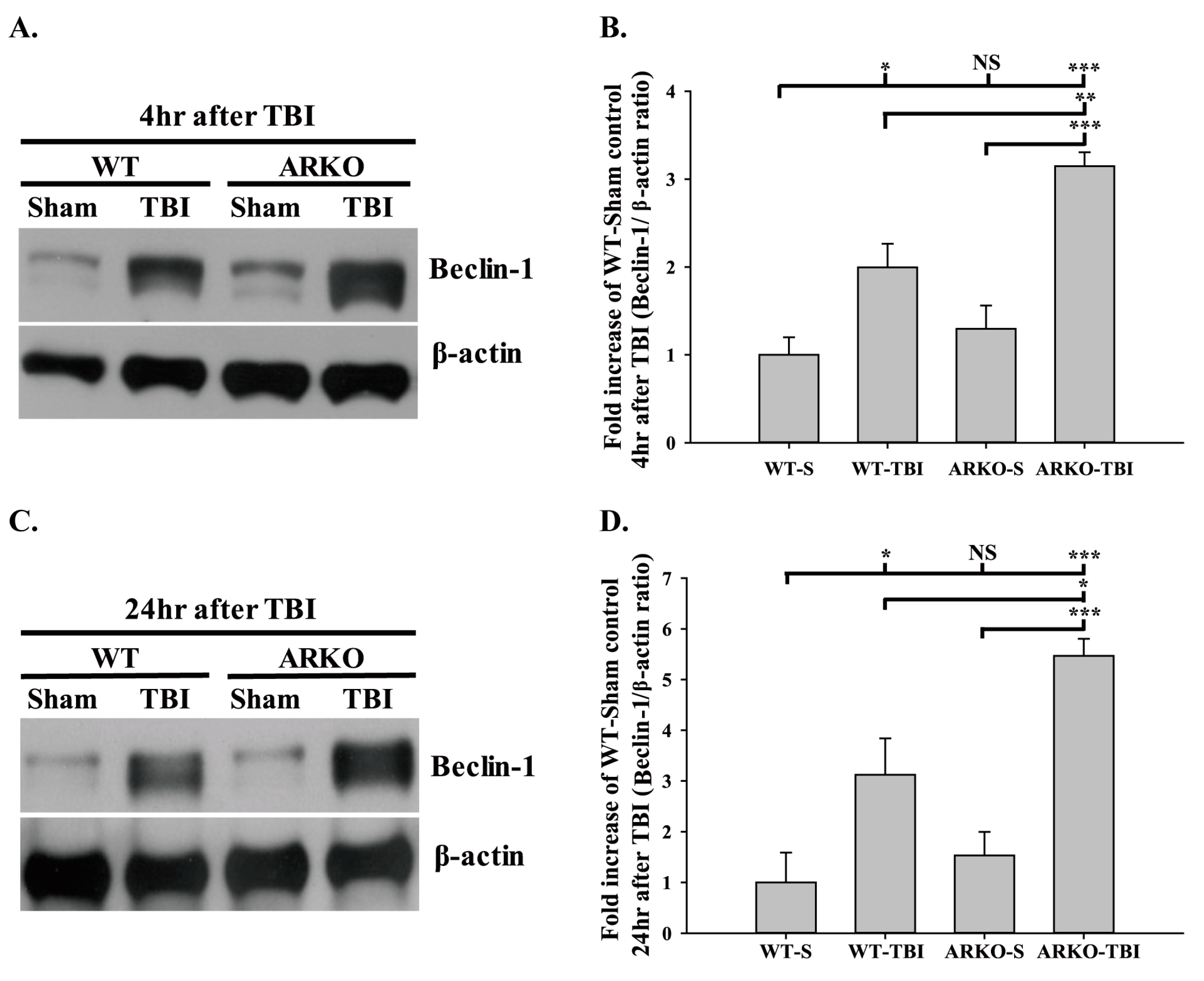
2.3. Effects of Androgen Receptor Knockout on Beclin-1 Expression in Mice following TBI
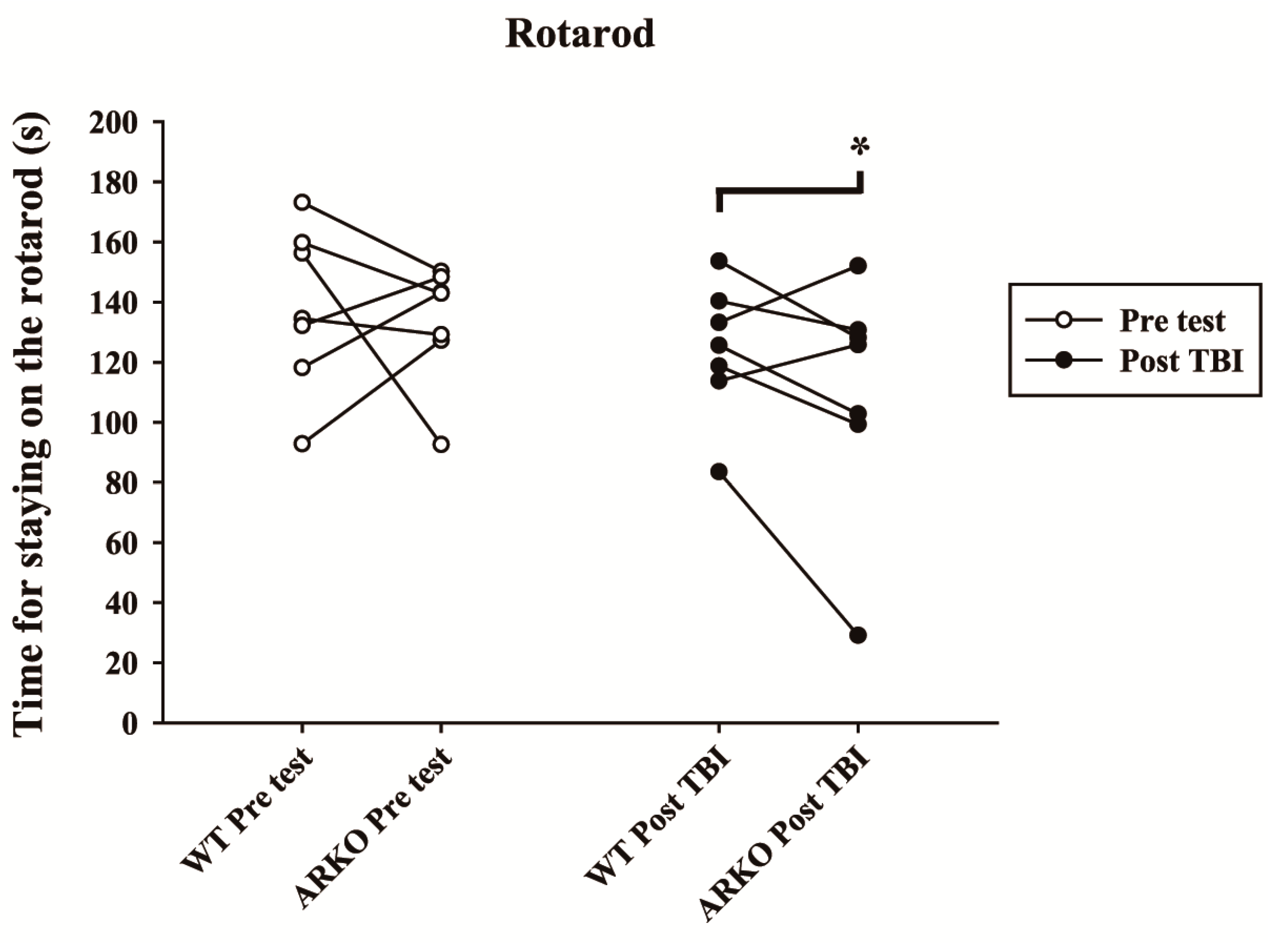
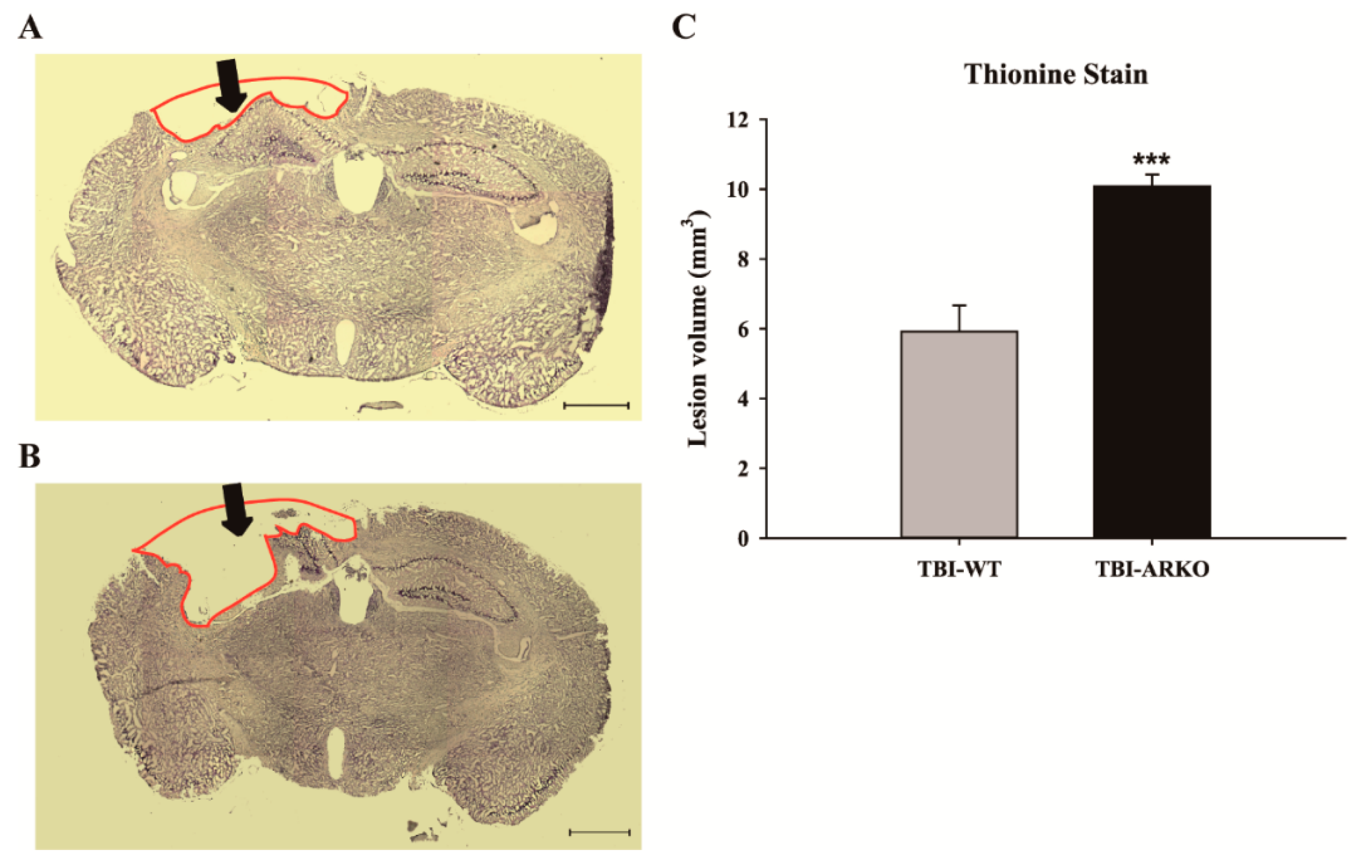
2.4. Androgen Receptor Knockout Affects the Motor Behavioral Outcomes and Lesion Volumes in Mice following TBI
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Experimental Design and Procedures
4.3. Genotyping
4.4. Polymerase Chain Reaction
4.5. Controlled Cortical Impact
4.6. Western Blot
4.7. Rotarod Test
4.8. Immunohistochemistry
4.9. Thionine Staining
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Langlois, J.A.; Rutland-Brown, W.; Wald, M.M. The epidemiology and impact of traumatic brain injury: A brief overview. J. Head Trauma Rehabil. 2006, 21, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Ganau, M.; Syrmos, N.; Paris, M.; Ganau, L.; Ligarotti, G.K.I.; Moghaddamjou, A.; Chibbaro, S.; Soddu, A.; Ambu, R.; Prisco, L. Current and Future Applications of Biomedical Engineering for Proteomic Profiling: Predictive Biomarkers in Neuro-Traumatology. Medicines 2018, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Horneman, G.; Emanuelson, I. Cognitive outcome in children and young adults who sustained severe and moderate traumatic brain injury 10 years earlier. Brain Inj. 2009, 23, 907–914. [Google Scholar] [CrossRef]
- Gavett, B.E.; Stern, R.A.; Cantu, R.C.; Nowinski, C.J.; McKee, A.C. Mild traumatic brain injury: A risk factor for neurodegeneration. Alzheimers Res. 2010, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Delgado, R.E.; Peacock, K.; Elizondo, B.; Wells, M.; Grafman, J.H.; Pugh, M.J. A Family’s Affair: Caring for Veterans with Penetrating Traumatic Brain Injury. Mil. Med. 2018, 183, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Bell, J.D.; Baker, A.J. Traumatic brain injury: Can the consequences be stopped? CMAJ 2008, 178, 1163–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitnay, G.A.; Zitnay, K.M.; Povlishock, J.T.; Hall, E.D.; Marion, D.W.; Trudel, T.; Zafonte, R.D.; Zasler, N.; Nidiffer, F.D.; DaVanzo, J.; et al. Traumatic brain injury research priorities: The Conemaugh International Brain Injury Symposium. J. Neurotrauma 2008, 25, 1135–1152. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging. Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.E. Mechanisms of traumatic brain injury: Biomechanical, structural and cellular considerations. Crit. Care Nurs. Q 2000, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Masel, B.E.; DeWitt, D.S. Traumatic Brain Injury: A Disease Process, Not an Event. J. Neurotraum. 2010, 27, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Sharma, S. Recent Advances In Pathophysiology Of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 2020, 9, 2698. [Google Scholar] [CrossRef] [PubMed]
- Bsat, S.; Chanbour, H.; Bsat, A.; Alomari, S.; Moussalem, C.; Houshiemy, M.N.E.; Omeis, I. Clinical utility of degradomics as predictors of complications and clinical outcome in aneurysmal subarachnoid hemorrhage. J. Integr. Neurosci. 2021, 20, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Riederer, B.M.; Zagon, I.S.; Goodman, S.R. Brain spectrin(240/235) and brain spectrin(240/235E): Two distinct spectrin subtypes with different locations within mammalian neural cells. J. Cell Biol. 1986, 102, 2088–2097. [Google Scholar] [CrossRef] [Green Version]
- Pike, B.R.; Flint, J.; Dutta, S.; Johnson, E.; Wang, K.K.; Hayes, R.L. Accumulation of non-erythroid alpha II-spectrin and calpain-cleaved alpha II-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J. Neurochem. 2001, 78, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Mondello, S.; Robicsek, S.A.; Gabrielli, A.; Brophy, G.M.; Papa, L.; Tepas, J.; Robertson, C.; Buki, A.; Scharf, D.; Jixiang, M.; et al. alphaII-spectrin breakdown products (SBDPs): Diagnosis and outcome in severe traumatic brain injury patients. J. Neurotrauma 2010, 27, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.K.W. Calpain and caspase: Can you tell the difference? Trends Neurosci. 2000, 23, 20–26. [Google Scholar] [CrossRef]
- DeDominicis, K.E.; Hwang, H.; Cartagena, C.M.; Shear, D.A.; Boutté, A.M. Cerebrospinal Fluid Biomarkers Are Associated With Glial Fibrillary Acidic Protein and αII-spectrin Breakdown Products in Brain Tissues Following Penetrating Ballistic-Like Brain Injury in Rats. Front. Neurol. 2018, 9, 490. [Google Scholar] [CrossRef]
- Mehta, T.; Fayyaz, M.; Giler, G.E.; Kaur, H.; Raikwar, S.P.; Kempuraj, D.; Selvakumar, G.P.; Ahmed, M.E.; Thangavel, R.; Zaheer, S.; et al. Current Trends in Biomarkers for Traumatic Brain Injury. Open Access J. Neurol. Neurosurg. 2020, 12, 86–94. [Google Scholar]
- Matsumoto, T.; Sakari, M.; Okada, M.; Yokoyama, A.; Takahashi, S.; Kouzmenko, A.; Kato, S. The androgen receptor in health and disease. Annu. Rev. Physiol. 2013, 75, 201–224. [Google Scholar] [CrossRef] [PubMed]
- Young, W.J.; Chang, C. Ontogeny and autoregulation of androgen receptor mRNA expression in the nervous system. Endocrine 1998, 9, 79–88. [Google Scholar] [CrossRef]
- Swift-Gallant, A.; Duarte-Guterman, P.; Hamson, D.K.; Ibrahim, M.; Monks, D.A.; Galea, L.A.M. Neural androgen receptors affect the number of surviving new neurons in the adult dentate gyrus of male mice. J. Neuroendocr. 2018. [Google Scholar] [CrossRef]
- Mhaouty-Kodja, S. Role of the androgen receptor in the central nervous system. Mol. Cell Endocrinol. 2018, 465, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, D.A.; Saunders, P.T.K.; McEwan, I.J. Androgens and androgen receptor: Above and beyond. Mol. Cell Endocrinol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Frye, C.A.; Edinger, K.; Sumida, K. Androgen administration to aged male mice increases anti-anxiety behavior and enhances cognitive performance. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 1049–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, J.; Le, Q.; Goodyer, C.; Gelfand, M.; Trifiro, M.; LeBlanc, A. Testosterone-mediated neuroprotection through the androgen receptor in human primary neurons. J. Neurochem. 2001, 77, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Champagne, N.; Beitel, L.K.; Goodyer, C.G.; Trifiro, M.; LeBlanc, A. Estrogen and androgen protection of human neurons against intracellular amyloid beta1-42 toxicity through heat shock protein 70. J. Neurosci. 2004, 24, 5315–5321. [Google Scholar] [CrossRef] [PubMed]
- Huo, D.S.; Sun, J.F.; Zhang, B.; Yan, X.S.; Wang, H.; Jia, J.X.; Yang, Z.J. Protective effects of testosterone on cognitive dysfunction in Alzheimer’s disease model rats induced by oligomeric beta amyloid peptide 1-42. J. Toxicol. Env. Health A 2016, 79, 856–863. [Google Scholar] [CrossRef]
- Huppenbauer, C.B.; Tanzer, L.; DonCarlos, L.L.; Jones, K.J. Gonadal steroid attenuation of developing hamster facial motoneuron loss by axotomy: Equal efficacy of testosterone, dihydrotestosterone, and 17-beta estradiol. J. Neurosci. 2005, 25, 4004–4013. [Google Scholar] [CrossRef] [PubMed]
- Young, T.P.; Hoaglin, H.M.; Burke, D.T. The role of serum testosterone and TBI in the in-patient rehabilitation setting. Brain Inj. 2007, 21, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Späni, C.B.; Braun, D.J.; Van Eldik, L.J. Sex-related responses after traumatic brain injury: Considerations for preclinical modeling. Front. Neuroendocr. 2018, 50, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.S.B.; Bayir, H.; Chu, C.T.; Alber, S.M.; Kochanek, P.M.; Watkins, S.C. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 2008, 4, 88–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.J.; Ding, Y.; Kohtz, D.S.; Mizushima, N.; Cristea, I.M.; Rout, M.P.; Chait, B.T.; Zhong, Y.; Heintz, N.; Yue, Z. Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 2006, 26, 8057–8068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2020, 14, 612757. [Google Scholar] [CrossRef]
- Bekker, M.; Abrahams, S.; Loos, B.; Bardien, S. Can the interplay between autophagy and apoptosis be targeted as a novel therapy for Parkinson’s disease? Neurobiol. Aging 2021, 100, 91–105. [Google Scholar] [CrossRef]
- Brattas, P.L.; Hersbach, B.A.; Madsen, S.; Petri, R.; Jakobsson, J.; Pircs, K. Impact of differential and time-dependent autophagy activation on therapeutic efficacy in a model of Huntington disease. Autophagy 2021, 17, 1316–1329. [Google Scholar] [CrossRef]
- Sadasivan, S.; Dunn, W.A., Jr.; Hayes, R.L.; Wang, K.K. Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem. Biophys. Res. Commun. 2008, 373, 478–481. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, R.; Yang, S.; Zhang, X.; Dai, J. Role of Microglia Autophagy in Microglia Activation After Traumatic Brain Injury. World Neurosurg. 2017, 100, 351–360. [Google Scholar] [CrossRef]
- Au, A.K.; Aneja, R.K.; Bayir, H.; Bell, M.J.; Janesko-Feldman, K.; Kochanek, P.M.; Clark, R.S.B. Autophagy Biomarkers Beclin 1 and p62 are Increased in Cerebrospinal Fluid after Traumatic Brain Injury. Neurocrit. Care 2017, 26, 348–355. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Balduini, W.; Carloni, S.; Buonocore, G. Autophagy in hypoxia-ischemia induced brain injury: Evidence and speculations. Autophagy 2009, 5, 221–223. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.L.; Li, B.X.; Li, Q.Q.; Chen, X.P.; Sun, Y.X.; Bao, H.J.; Dai, D.K.; Shen, Y.W.; Xu, H.F.; Ni, H.; et al. Autophagy Is Involved in Traumatic Brain Injury-Induced Cell Death and Contributes to Functional Outcome Deficits in Mice. Neuroscience 2011, 184, 54–63. [Google Scholar] [CrossRef]
- Sun, L.; Zhao, M.; Liu, M.; Su, P.; Zhang, J.; Li, Y.; Yang, X.; Wu, Z. Suppression of FoxO3a attenuates neurobehavioral deficits after traumatic brain injury through inhibiting neuronal autophagy. Behav. Brain Res. 2018, 337, 271–279. [Google Scholar] [CrossRef]
- Yeh, S.; Tsai, M.Y.; Xu, Q.; Mu, X.M.; Lardy, H.; Huang, K.E.; Lin, H.; Yeh, S.D.; Altuwaijri, S.; Zhou, X.; et al. Generation and characterization of androgen receptor knockout (ARKO) mice: An in vivo model for the study of androgen functions in selective tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 13498–13503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, K.; Davey, R.A.; Zajac, J.D. Human androgen deficiency: Insights gained from androgen receptor knockout mouse models. Asian J. 2014, 16, 169–177. [Google Scholar]
- Zhang, Z.; Larner, S.F.; Liu, M.C.; Zheng, W.; Hayes, R.L.; Wang, K.K. Multiple alphaII-spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathways. Apoptosis 2009, 14, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Cardali, S.; Maugeri, R. Detection of alphaII-spectrin and breakdown products in humans after severe traumatic brain injury. J. Neurosurg. Sci. 2006, 50, 25–31. [Google Scholar] [PubMed]
- Brophy, G.M.; Pineda, J.A.; Papa, L.; Lewis, S.B.; Valadka, A.B.; Hannay, H.J.; Heaton, S.C.; Demery, J.A.; Liu, M.C.; Tepas, J.J.; et al. alphaII-Spectrin breakdown product cerebrospinal fluid exposure metrics suggest differences in cellular injury mechanisms after severe traumatic brain injury. J. Neurotrauma 2009, 26, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Zoltewicz, J.S.; Mondello, S.; Yang, B.; Newsom, K.J.; Kobeissy, F.; Yao, C.; Lu, X.C.; Dave, J.R.; Shear, D.A.; Schmid, K.; et al. Biomarkers track damage after graded injury severity in a rat model of penetrating brain injury. J. Neurotrauma 2013, 30, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.C.; Gardiner, R.A.; Hooper, J.D.; Johnson, D.W.; Gobe, G.C. Molecular cell biology of androgen receptor signalling. Int. J. Biochem. Cell Biol. 2010, 42, 813–827. [Google Scholar] [CrossRef]
- Rothman, M.S.; Arciniegas, D.B.; Filley, C.M.; Wierman, M.E. The neuroendocrine effects of traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 2007, 19, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Johann, S.; Beyer, C. Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J. Steroid Biochem. Mol. Biol. 2013, 137, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Izzo, G.; Tirelli, A.; Angrisani, E.; Cannaviello, G.; Cannaviello, L.; Puzziello, A.; Vatrella, A.; Vitale, M. Pituitary dysfunction and its association with quality of life in traumatic brain injury. Int. J. Surg. 2016, 28, S103–S108. [Google Scholar] [CrossRef]
- Bianchi, V.E.; Rizzi, L.; Bresciani, E.; Omeljaniuk, R.J.; Torsello, A. Androgen Therapy in Neurodegenerative Diseases. J. Endocr. Soc. 2020, 4, bvaa120. [Google Scholar] [CrossRef]
- Xiao, L.; Jordan, C.L. Sex differences, laterality, and hormonal regulation of androgen receptor immunoreactivity in rat hippocampus. Horm. Behav. 2002, 42, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Willems, A.; Batlouni, S.R.; Esnal, A.; Swinnen, J.V.; Saunders, P.T.; Sharpe, R.M.; França, L.R.; De Gendt, K.; Verhoeven, G. Selective ablation of the androgen receptor in mouse sertoli cells affects sertoli cell maturation, barrier formation and cytoskeletal development. PLoS ONE 2010, 5, e14168. [Google Scholar] [CrossRef] [Green Version]
- Hussain, R.; Ghoumari, A.M.; Bielecki, B.; Steibel, J.; Boehm, N.; Liere, P.; Macklin, W.B.; Kumar, N.; Habert, R.; Mhaouty-Kodja, S.; et al. The neural androgen receptor: A therapeutic target for myelin repair in chronic demyelination. Brain A J. Neurol. 2013, 136, 132–146. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef]
- Osier, N.D.; Carlson, S.W.; DeSana, A.; Dixon, C.E. Chronic Histopathological and Behavioral Outcomes of Experimental Traumatic Brain Injury in Adult Male Animals. J. Neurotrauma 2015, 32, 1861–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, E.M.; Su, D.; Lopez-Velazquez, L.; Haus, D.L.; Perez, H.; Lacuesta, G.A.; Anderson, A.J.; Cummings, B.J. Functional assessment of long-term deficits in rodent models of traumatic brain injury. Regen. Med. 2013, 8, 483–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, R.E.; Thorndyke, E.C., 3rd. Patterns of Behavioral Deficits in Rodents Following Brain Injury Across Species, Gender, and Experimental Model. Acta Neurochir. Suppl. 2016, 121, 71–75. [Google Scholar] [PubMed]
- Hamm, R.J.; Pike, B.R.; O’Dell, D.M.; Lyeth, B.G.; Jenkins, L.W. The rotarod test: An evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J. Neurotrauma 1994, 11, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Abou-El-Hassan, H.; Sukhon, F.; Assaf, E.J.; Bahmad, H.; Abou-Abbass, H.; Jourdi, H.; Kobeissy, F.H. Degradomics in Neurotrauma: Profiling Traumatic Brain Injury. Methods Mol. Biol. 2017, 1598, 65–99. [Google Scholar]
- Yan, X.X.; Jeromin, A.; Jeromin, A. Spectrin Breakdown Products (SBDPs) as Potential Biomarkers for Neurodegenerative Diseases. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 2012, 1, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Yokobori, S.; Hosein, K.; Burks, S.; Sharma, I.; Gajavelli, S.; Bullock, R. Biomarkers for the clinical differential diagnosis in traumatic brain injury-a systematic review. Cns. Neurosci. 2013, 19, 556–565. [Google Scholar] [CrossRef]
- Ringger, N.C.; O’Steen, B.E.; Brabham, J.G.; Silver, X.; Pineda, J.; Wang, K.K.; Hayes, R.L.; Papa, L. A novel marker for traumatic brain injury: CSF alphaII-spectrin breakdown product levels. J. Neurotrauma 2004, 21, 1443–1456. [Google Scholar] [CrossRef]
- Pineda, J.A.; Lewis, S.B.; Valadka, A.B.; Papa, L.; Hannay, H.J.; Heaton, S.C.; Demery, J.A.; Liu, M.C.; Aikman, J.M.; Akle, V.; et al. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J. Neurotrauma 2007, 24, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.D.; Sullivan, P.G.; Gibson, T.R.; Pavel, K.M.; Thompson, B.M.; Scheff, S.W. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: More than a focal brain injury. J. Neurotraum 2005, 22, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Kobeissy, F.H.; Liu, M.C.; Yang, Z.H.; Zhang, Z.Q.; Zheng, W.R.; Glushakova, O.; Mondello, S.; Anagli, J.; Hayes, R.L.; Wang, K.K.W. Degradation of beta II-Spectrin Protein by Calpain-2 and Caspase-3 Under Neurotoxic and Traumatic Brain Injury Conditions. Mol. Neurobiol. 2015, 52, 696–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, M.E.; Requena, D.F.; Davis, L.J.; Metzger, R.R.; Bennett, K.S.; Morita, D.; Niedzwecki, C.; Yang, Z.; Wang, K.K. Alpha II Spectrin breakdown products in immature Sprague Dawley rat hippocampus and cortex after traumatic brain injury. Brain Res. 2014, 1574, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Korley, F.K.; Nikolian, V.C.; Williams, A.M.; Dennahy, I.S.; Weykamp, M.; Alam, H.B. Valproic Acid Treatment Decreases Serum Glial Fibrillary Acidic Protein and Neurofilament Light Chain Levels in Swine Subjected to Traumatic Brain Injury. J. Neurotrauma 2018. [Google Scholar] [CrossRef]
- Raghu, V.K.; Horvat, C.M.; Kochanek, P.M.; Fink, E.L.; Clark, R.S.B.; Benos, P.V.; Au, A.K. Neurological Complications Acquired During Pediatric Critical Illness: Exploratory “Mixed Graphical Modeling” Analysis Using Serum Biomarker Levels. Pediatric Crit. Care Med. 2021, 22, 906–914. [Google Scholar] [CrossRef]
- Toman, E.; Harrisson, S.; Belli, T. Biomarkers in traumatic brain injury: A review. J. R. Army Med. Corps 2016, 162, 103–108. [Google Scholar] [CrossRef]
- Cullen, D.K.; Simon, C.M.; LaPlaca, M.C. Strain rate-dependent induction of reactive astrogliosis and cell death in three-dimensional neuronal-astrocytic co-cultures. Brain Res. 2007, 1158, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, W.; Edelmann, W.; Bieri, P.L.; Chiu, F.C.; Cowan, N.J.; Kucherlapati, R.; Raine, C.S. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron 1996, 17, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Bogoslovsky, T.; Wilson, D.; Chen, Y.; Hanlon, D.; Gill, J.; Jeromin, A.; Song, L.; Moore, C.; Gong, Y.; Kenney, K.; et al. Increases of Plasma Levels of Glial Fibrillary Acidic Protein, Tau, and Amyloid β up to 90 Days after Traumatic Brain Injury. J. Neurotrauma 2017, 34, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Korley, F.; Pauls, Q.; Yeatts, S.D.; Jones, C.M.C.; Corbett-Valade, E.; Silbergleit, R.; Frankel, M.; Barsan, W.; Cahill, N.D.; Bazarian, J.J.; et al. Progesterone Treatment Does Not Decrease Serum Levels of Biomarkers of Glial and Neuronal Cell Injury in Moderate and Severe Traumatic Brain Injury Subjects: A Secondary Analysis of the Progesterone for Traumatic Brain Injury, Experimental Clinical Treatment (ProTECT) III Trial. J. Neurotrauma 2021, 38, 1953–1960. [Google Scholar]
- Mondello, S.; Shear, D.A.; Bramlett, H.M.; Dixon, C.E.; Schmid, K.E.; Dietrich, W.D.; Wang, K.K.W.; Hayes, R.L.; Glushakova, O.; Catania, M.; et al. Insight into Pre-Clinical Models of Traumatic Brain Injury Using Circulating Brain Damage Biomarkers: Operation Brain Trauma Therapy. J. Neurotraum. 2016, 33, 595–605. [Google Scholar] [CrossRef]
- Jones, K.J.; Coers, S.; Storer, P.D.; Tanzer, L.; Kinderman, N.B. Androgenic regulation of the central glia response following nerve damage. J. Neurobiol. 1999, 40, 560–573. [Google Scholar] [CrossRef]
- Barreto, G.; Veiga, S.; Azcoitia, I.; Garcia-Segura, L.M.; Garcia-Ovejero, D. Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: Role of its metabolites, oestradiol and dihydrotestosterone. Eur. J. Neurosci. 2007, 25, 3039–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ovejero, D.; Veiga, S.; Garcia-Segura, L.M.; Doncarlos, L.L. Glial expression of estrogen and androgen receptors after rat brain injury. J. Comp. Neurol. 2002, 450, 256–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccariglia, S.; Cargnoni, A.; Silini, A.R.; Parolini, O. Autophagy: A potential key contributor to the therapeutic action of mesenchymal stem cells. Autophagy 2020, 16, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Diskin, T.; Tal-Or, P.; Erlich, S.; Mizrachy, L.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J. Neurotrauma 2005, 22, 750–762. [Google Scholar] [CrossRef]
- Shen, M.; Wang, S.; Wen, X.; Han, X.R.; Wang, Y.J.; Zhou, X.M.; Zhang, M.H.; Wu, D.M.; Lu, J.; Zheng, Y.L. Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed. Pharm. 2017, 95, 885–893. [Google Scholar] [CrossRef]
- Wang, C.Q.; Ye, Y.; Chen, F.; Han, W.C.; Sun, J.M.; Lu, X.; Guo, R.; Cao, K.; Zheng, M.J.; Liao, L.C. Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience 2017, 343, 30–38. [Google Scholar] [CrossRef]
- Feng, Y.; Cui, C.; Liu, X.; Wu, Q.; Hu, F.; Zhang, H.; Ma, Z.; Wang, L. Protective Role of Apocynin via Suppression of Neuronal Autophagy and TLR4/NF-κB Signaling Pathway in a Rat Model of Traumatic Brain Injury. Neurochem. Res. 2017, 42, 3296–3309. [Google Scholar] [CrossRef]
- Feng, Y.; Cui, Y.; Gao, J.L.; Li, M.H.; Li, R.; Jiang, X.H.; Tian, Y.X.; Wang, K.J.; Cui, C.M.; Cui, J.Z. Resveratrol attenuates neuronal autophagy and inflammatory injury by inhibiting the TLR4/NF-κB signaling pathway in experimental traumatic brain injury. Int. J. Mol. Med. 2016, 37, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef]
- Ortiz-Rodriguez, A.; Arevalo, M.A. The Contribution of Astrocyte Autophagy to Systemic Metabolism. Int. J. Mol. Sci. 2020, 21, 2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, X.; Sun, Y.; Sun, X.; Zhou, Y.; Bian, Y.; Shu, Z.; Ding, J.; Lu, M.; Hu, G. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Tian, F.; Wang, S.; Wang, F.; Xiong, L. Astrocyte Autophagy Flux Protects Neurons Against Oxygen-Glucose Deprivation and Ischemic/Reperfusion Injury. Rejuvenation Res. 2018, 21, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular cloning: A Laboratory Manual; Cold Spring Harbor: New York, NY, USA, 1989. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Chen, Y.-C.; Hwang, L.-L.; Yang, L.-Y.; Lu, D.-Y. Deficiency in Androgen Receptor Aggravates Traumatic Brain Injury-Induced Pathophysiology and Motor Deficits in Mice. Molecules 2021, 26, 6250. https://doi.org/10.3390/molecules26206250
Chen Y-H, Chen Y-C, Hwang L-L, Yang L-Y, Lu D-Y. Deficiency in Androgen Receptor Aggravates Traumatic Brain Injury-Induced Pathophysiology and Motor Deficits in Mice. Molecules. 2021; 26(20):6250. https://doi.org/10.3390/molecules26206250
Chicago/Turabian StyleChen, Yu-Hsin, Yen-Chou Chen, Ling-Ling Hwang, Liang-Yo Yang, and Dah-Yuu Lu. 2021. "Deficiency in Androgen Receptor Aggravates Traumatic Brain Injury-Induced Pathophysiology and Motor Deficits in Mice" Molecules 26, no. 20: 6250. https://doi.org/10.3390/molecules26206250