
Intermolecular Forces Driving Hexamethylenetetramine Co-Crystal Formation, a DFT and XRD Analysis


Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, Viale F. Stagno d’Alcontres 31, 98166 Messina, Italy
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(19), 5746; https://doi.org/10.3390/molecules26195746
Submission received: 18 July 2021
/
Revised: 7 September 2021
/
Accepted: 15 September 2021
/
Published: 22 September 2021
(This article belongs to the Special Issue Hybrid Compounds Tailoring at the Nanoscale for Biomedical Applications)
Abstract
:Interest in co-crystals formation has been constantly growing since their discovery, almost a century ago. Such success is due to the ability to tune the physical-chemical properties of the components in solid state by avoiding a change in their molecular structure. The properties influenced by the co-crystals formation range from an improvement of mechanical features and chemical stability to different solubility. In the scientific research area, the pharmacological field is undoubtedly one of those in which an expansion of the co-crystal knowledge can offer wide benefits. In this work, we described the crystalline structure of hexamethylenetetramine co-crystallized with the isophthalic acid, and we compared it with another co-crystal, showing the same components but different stoichiometry. To give a wider overview on the nature of the interactions behind the observed crystal packing and to rationalize the reasons of its formation, a computational analysis on such structures was carried out.
1. Introduction
One of the main limits in the development of highly efficient drug molecules regards their low aqueous solubility with the consequent limitation of the bioavailability. There are several possible approaches to solve such a problem and this improves the water solubility and the dissolution rate. One of the oldest strategies regards the size reduction of the particles of the drug, which influence the kinetic of the dissolution rate. However, valid and modern approaches to enhance the solubility of the drug can be pursued in different ways, by combining the size reduction with different formulations of the drug. One of the most effective strategies for enhancing the solubility of acid/basic drugs consists of the formation of salts, using an appropriate partner for the drug [1]. More recently explored possibilities consist of co-crystallize the active drug with another suitable component [2], drug delivery by a suitable carrier [3,4] or resort to the formation of polymorphs. It is nowadays well known that the arrangement adopted by species in their solid form brings a change of their physical-chemical properties and can have profound effects on the dissolution rate [5,6], stability, and bioavailability of that species. Such properties and the possibility to predict them led to an increased economic interest by pharmaceutical companies on the study of the basic principles that can influence co-crystals, polymorphs and pseudo-polymorphs formation. The kinetic factors in a crystallization process are linked to the operating conditions. Some of them can be easily controlled, like temperature of crystallization, some others such as the presence of impurities can be hardly predicted. Among all the possible crystals, only the ones with sufficient kinetic stability are commercially and academically important. Unfortunately, it is impossible to take into account all of the kinetic factors that can generate and influence new crystal forms. The possible forms that a species can assume in its crystalline state could be predicted by taking in consideration the supramolecular aspects of the crystallization process. A supramolecular approach allows one to build complex structures with programmed properties [7,8] and shapes [9] in solution, through the correct understanding of all the thermodynamic and kinetic processes which govern the system. Such approach can be used in a solid state to study all the crystallization paths which can give different outputs such polymorphs, co-crystals or salts. It is important to consider how the molecules arranging themselves to build an energetically favored crystal packing and which kind of non-covalent forces are involved in the process. By taking advantage of computational chemistry, it is possible to evaluate the thermodynamic stability of many possible structures, and among them, select the most thermodynamically plausible ones.
In this general framework, we examine and attempt to provide some rationalization to a new co-crystal structure of a hexamethylenetetramine (HMTA) and isophthalic acid (IPA) containing a water molecule linked through hydrogen bonds to the crystal lattice. The HMTA is widely used in pharmacology to treat bladder and kidney bacterial infections. When pH = 5.5 in the urine, the HMTA undergoes a degradation process, converting into formaldehyde [10].
2. Results and Discussions
2.1. Structure Description
The crystallographic data for compound 1 collected at room temperature are presented in Table 1. The asymmetric unit (AU) of the supramolecular complex is constituted by half a molecule of water, IPA and HMTA. These components lie on two adjacent crystallographic planes {040}. While the acid molecule lies entirely on the 040 crystallographic plane, the HTMA is cut in two by the plane passing through the atoms N3C10C12N1 (see Figure 1).
The evidence of the co-crystal nature of the compound 1 is found in the bond distances of carboxylic fragments C=O (2.208 Å) and C-OH (1.322 Å), which clearly indicate no acid deprotonation.
The whole molecular packing is mainly determined by an array of strong hydrogen bonds between all the components of compound 1, with a non-negligible contribution of p-p stacking interactions at 3.452 Å, involving two centrosymmetric IPA molecules (as shown in Figure 2).
The water molecule is involved in three hydrogen bonds (Ow…O3 (2.643(2) Å), Ow…N2(N2′) (2.822(1) Å)), assisted by two weaker electrostatic interactions with methylenic hydrogens, as depicted in Figure 3b. In the same picture (Figure 3a), the graph set (28) [11] motif generated by the HBs is illustrated. In the compound 1, each molecule of IPA is directly bonded via HB to a N3 atom (N3…H-O1, 2.727(2) Å) and to the water molecule.
In the literature, some other structures constituted by the same molecular components of compound 1 (IPA, HMTA and water) are reported. These structures can differ from each other via molecular packing (polymorphs), the presence of one or more water molecules (pseudo-polymorph) and different stoichiometry. In particular, the compound 2 (MIPVIQ [12]) shown in Figure 3c–d exhibits the same (28) interaction pattern of compound 1 (Figure 3a), showing a similar hydrogen network.
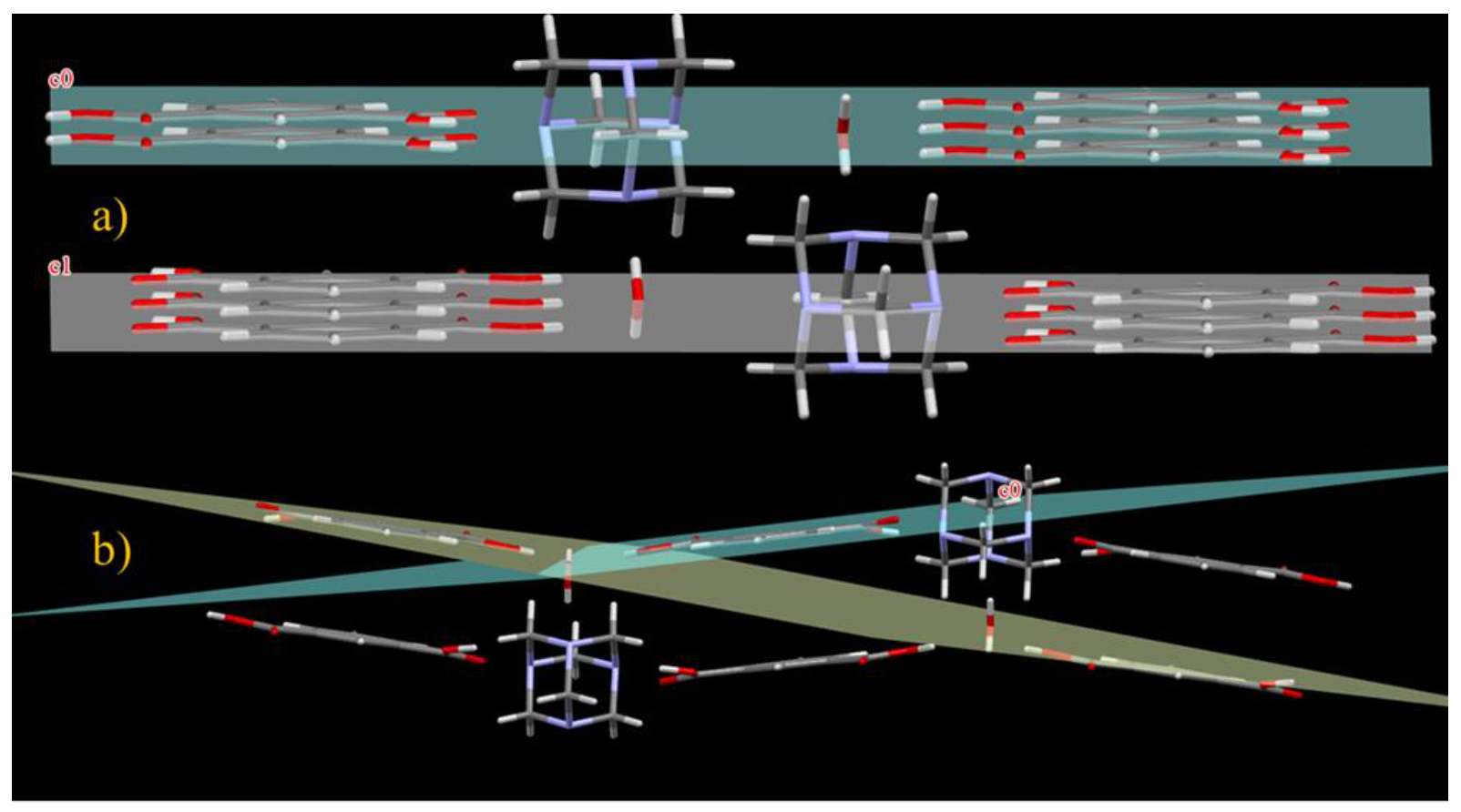
Although the two co-crystals exhibit essentially the same intermolecular interactions, their whole molecular packing is largely different, as can be seen in Figure 4a,b, where their spatial distribution is compared.
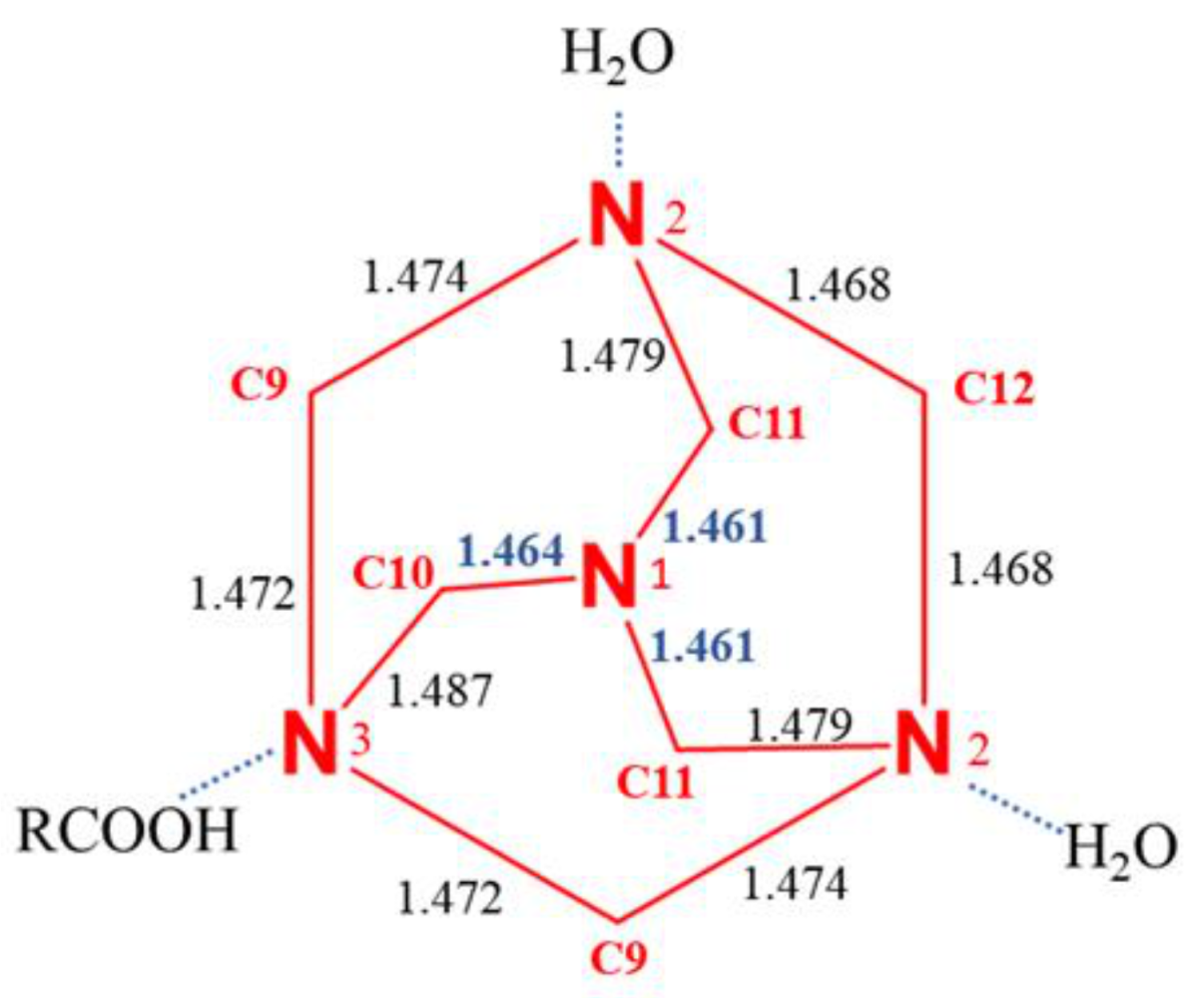
The absence of conformational flexibility makes the hexamethylenetetramine a molecule which is structurally very stiff, however we observed that the Td symmetry is altered by the different HBs that the four nitrogen atoms can form; thus, indicative differences in the N-C bond distances can be observed. Nitrogen atom N1 not involved in any interaction shows the shortest N-C bonds distances (1.462 Å), while the others, N2-C and N3-C, are 1.474 Å and 1.477 Å, respectively. Although small, the observed differences in N-C bond distances are systematic and are attributed to the different strength of the intermolecular interactions involving the different nitrogen atoms of the HMTA.
In compound 1, three nitrogen atoms of HTMA are involved in as many hydrogen bonds, the fourth nitrogen (N1) does not present any significant intermolecular interaction. While in the HTMA in compound 2, all the four nitrogen atoms are involved in HB interactions, two of them with the OH group of the carboxylate (2.745 Å of bond distance) and the other two with water molecules (2.777 Å and 2.803 Å).
It is well known that HMTA usually participates in only two strong hydrogen bonds [13,14,15,16,17]. Only in cases where HTMA interacts with rich hydrogen donors, can it form three or four hydrogen bonds [18]. Such behaviour could be substantially attributed to the lower electron donor attitude of the HMTA when involved in multiple hydrogen bonds. This behaviour showed by the HMTA fragment could be a key feature to explain the abundance of co-crystal structures with HMTA in literature, in comparison to salt crystals. When the HMTA becomes protonated, its ability to form strong hydrogen bonds with the other non-protonated nitrogen atoms decreases. In the CSD [19] there are only three compounds in which the HMTA has two protonated nitrogen atoms and no tris or tetra protonated compounds are present [20,21,22].
In the synthesis of compound 1, its co-crystal structure was predicted by using the ΔpKa rule, a general rule which help to predict crystal formation rather than salt. It is based on the relative values of pKa. In particular, when this difference (ΔpKa = pKa base–pKa acid) is greater than 2–3, the product obtained will be a salt [23,24]. The reliability of the prediction rules is important in several fields, with a particular focus in the pharmaceutical industry for the active pharmaceutical ingredients (APIs) with desirable properties. For complex 1, the ΔpKa value corresponds to 1.67 (5.13 for HMTA [25], 3.36 for IPA [26]).
2.2. Computational Details
The use of density functional theory (DFT) has become increasingly commonplace among chemists, providing them a useful tool for an accurate analysis of complex chemical species [27,28]. In this paper, single point energy was performed at CAM-B3LYP/6-311++G(d,p) level adding Grimme-dispersion terms (D3) [29] by means of Gaussian09 package [30]; a non-covalent interaction (NCI) was generated by Multiwfn code [31] with a high-quality grid and visualized using VMD [32]. Focusing on the crystal packing of compounds 1 and 2, an accurate matching could be essential in order to visualize the difference between the two co-crystalline aggregates. In these terms, the occurrence of electrostatically favorable intermolecular interactions represents a useful indicator of energy stability of the crystal [33]. Figure 5 and Figure 6 clarifies how HMTA assumes a different electrostatic behavior in the two-crystal packing. In compound 2, each nitrogen atom is involved in a strong hydrogen bond (blue pseudo circular surface) regarding two molecules of water and two IPA. As concerning compound 1, only three nitrogen atoms establish hydrogen bonds (two water molecules and one IPA). In the latter, it is worth noting that a hydrogen bond between carboxylic scaffold and tertiary amine nitrogen is cooperatively assisted by an electrostatically favorable long-range interaction between carbonyl sp2-oxygen and hydrogens on methylene group (HMTA).
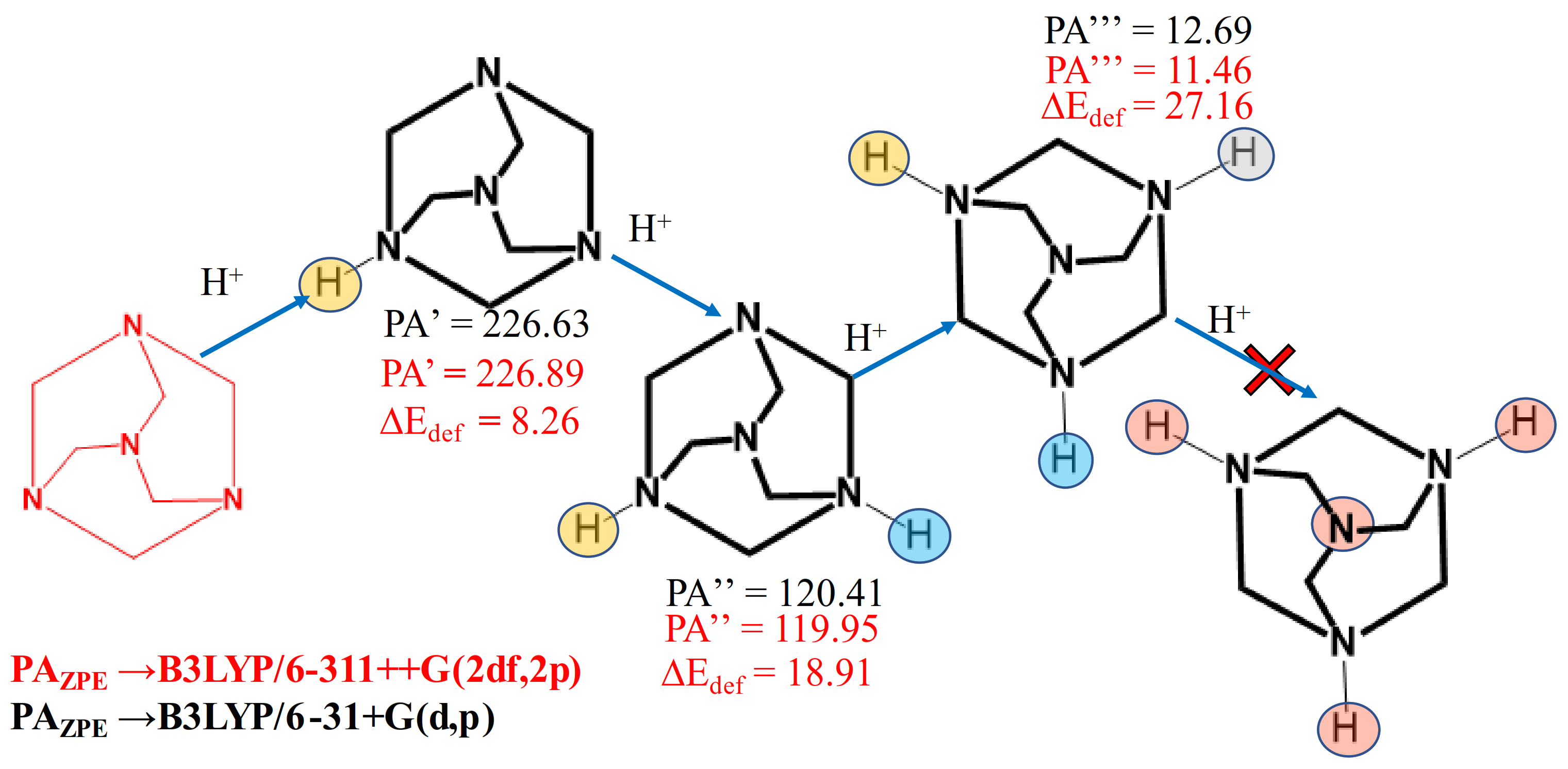
An important property that can be analysed by using computational methods is the proton affinity (PA). The proton affinity was considered as the negative of the protonation enthalpy (PA=-ΔH). The PA was computationally calculated at gas phase, at B3LYP/6-31+G(d,p) and B3LYP/ 6311++G(2df,2p) level. Scheme 1 shows the computed PA of mono, bis and tris-protonated forms. The values of the first three protonations, corrected for zero-point energy, together with the deformation energy, are reported in Scheme 1.
Deformation energy for each protonated species was computed as the difference between the ground state energy of previous neutral molecule or ions (HMTAnHn+) and the energy of the neutral molecule or ions with the same conformation of the optimized protonated species. As can be seen from the data shown in Scheme 1, the PA drastically decreases from the first to the third protonation. Although the calculated PA relative to the fourth protonation can be calculated, it is not reported, because its negative value has no chemical significance.
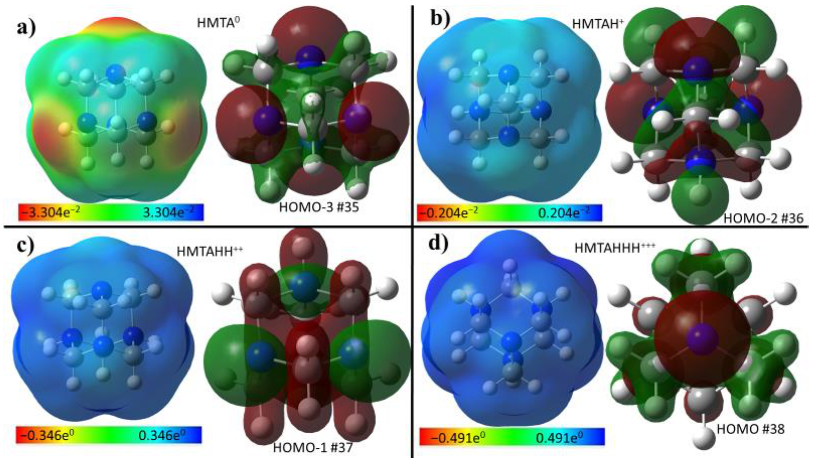
Figure 7 illustrates the molecular electrostatic potential surface (ESP) and the MOs mainly centred on the available lone-pair of nitrogen atoms for the neutral HMTA and the three protonated species. The ESP pictures clearly spotlight the low attitude of HMTA to multiple protonations.
3. Materials and Methods
Chemicals and solvents used were purchased from commercial suppliers (Sigma Aldrich, Merk Life Science S.r.l., Milan, Italy) and were used without further purification.
The crystal was prepared by adding an ethanol solution of IPA (around 1 mmol) to a solution of HMTA (1 mmol). The reaction was stirred for a few minutes and then the solvent was left to evaporate at room temperature, to obtain, in a few days, colorless crystals.
A suitable crystal was selected and mounted on a Bruker APEX-II CCD diffractometer. The crystal was kept at 296.15 K during data collection. Using Olex2 [34], the structure was solved with the SHELXT [35] structure solution program using intrinsic phasing and refined with the SHELXL [36] refinement package using least squares minimization. A SCXRD analysis was made using several crystals of the same sample, obtaining, for all the data acquired, the same crystallographic cell. Here, we presented the result with the best R factor.
All the crystallographic data of compound 1 are reported in the Supplementary Materials.
4. Conclusions
In summary, we synthesized and studied, through single crystal X ray diffraction, a new co-crystal formed by HMTA, IPA and a water molecule. The obtained co-crystal obeys to the semi-empyrical rule of ΔpKa, showing a value of 1.67, within the expected range of 0–3. Supported by a DFT analysis, the relative abundance of similar co-crystals in CSD was attributed to the energetic unfavored HBs when HMTA is mono or bis protonated. A comparison based on SCXRD data with a species sharing the same components was performed in order to highlight the reasons for their different 3D arrangements. The major contribution to such differences turned out to be the number of hydrogen bonds which involve the HMTA fragment (three in compound 1 and four in compound 2), whereas in compound 1, the nitrogen not directly involved in a hydrogen bond exhibits two long range hydrogen interactions.
Supplementary Materials
The following are available online. Table S1: Fractional Atomic Coordinates (×104) and Equivalent Isotropic Displacement Parameters (Å2 × 103) for compound 1. Ueq is defined as 1/3 of of the trace of the orthogonalised UIJ tensor, Table S2: Anisotropic Displacement Parameters (Å2 × 103) for compound 1. The Anisotropic displacement factor exponent takes the form: −2π2[h2a*2U11+2hka*b*U12+…], Table S3: Bond Lengths for compound 1, Table S4: Bond Angles for compound 1, Table S5: Hydrogen Bonds for compound 1, Table S6: Torsion Angles for compound 1, Table S7: Hydrogen Atom Coordinates (Å × 104) and Isotropic Displacement Parameters (Å2 × 103) for compound 1.
Author Contributions
Conceptualization, A.S., G.B. (Giuseppe Bruno); Computational methodology, G.B. (Giovanni Bella); software, Giovanni Bella; SCXRD analysis, F.N.; data curation, A.S.; writing—original draft preparation, A.S., G.B. (Giuseppe Bruno); writing—review and editing, A.S., G.B. (Giovanni Bella); visualization, G.B. (Giuseppe Bruno), F.N.; supervision, A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
References
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celentano, W.; Neri, G.; Distante, F.; Li, M.; Messa, P.; Chirizzi, C.; Chaabane, L.; De Campo, F.; Metrangolo, P.; Baldelli Bombelli, F.; et al. Design of fluorinated hyperbranched polyether copolymers for 19F MRI nanotheranostics. Polym. Chem. 2020, 11, 3951–3963. [Google Scholar] [CrossRef]
- Neri, G.; Corsaro, C.; Fazio, E. Plasmon-Enhanced Controlled Drug Release from Ag-PMA Capsules. Molecules 2020, 25, 2267. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Olsen, K.W. Molecular Dynamics of Drug Crystal Dissolution: Simulation of Acetaminophen Form I in Water. Mol. Pharm. 2013, 10, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Elts, E.; Greiner, M.; Briesen, H. In Silico Prediction of Growth and Dissolution Rates for Organic Molecular Crystals: A Multiscale Approach. Crystals 2017, 7, 288. [Google Scholar] [CrossRef] [Green Version]
- Nastasi, F.; Santoro, A.; Serroni, S.; Campagna, S.; Kaveevivitchai, N.; Thummel, R.P. Early photophysical events of a ruthenium(ii) molecular dyad capable of performing photochemical water oxidation and of its model compounds. Photochem. Photobiol. Sci. 2019, 18, 2164–2173. [Google Scholar] [CrossRef]
- Mancuso, A.; Barattucci, A.; Bonaccorsi, P.; Giannetto, A.; La Ganga, G.; Musarra-Pizzo, M.; Salerno, T.M.G.; Santoro, A.; Sciortino, M.T.; Puntoriero, F.; et al. Carbohydrates and Charges on Oligo(phenylenethynylenes): Towards the Design of Cancer Bullets. Chem. A Eur. J. 2018, 24, 16972–16976. [Google Scholar] [CrossRef]
- Santoro, A.; Holub, J.; Fik-Jaskółka, M.A.; Vantomme, G.; Lehn, J.-M. Dynamic Helicates Self-Assembly from Homo- and Heterotopic Dynamic Covalent Ligand Strands. Chem. A Eur. J. 2020, 26, 15664–15671. [Google Scholar] [CrossRef]
- Sauberan, J.B.; Bradley, J.S. 292—Antimicrobial Agents. In Principles and Practice of Pediatric Infectious Diseases, 5th ed.; Long, S.S., Prober, C.G., Fischer, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1499–1531. [Google Scholar]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Li, W.; Zhang, J.-P.; Tong, M.-L.; Chen, X.-M. Cocrystallization of Hexamethylenetetramine (hmt) with Dicarboxylic Acids. Crystal Structures of Molecular Adducts of hmt with Three Different Dicarboxylic Acids. Aust. J. Chem. 2001, 54, 213–217. [Google Scholar] [CrossRef]
- Tong, M.-L.; Zheng, S.-L.; Chen, X.-M. Self-Assembly of Two- and Three-Dimensional Coordination Networks with Hexamethylenetetramine and Different Silver(I) Salts. Chem. A Eur. J. 2000, 6, 3729–3738. [Google Scholar] [CrossRef]
- Zheng, S.-L.; Tong, M.-L.; Yu, X.-L.; Chen, X.-M. Syntheses and structures of six chain-, ladder- and grid-like co-ordination polymers constructed from μ-hexamethylenetetramine and silver salts. J. Chem. Soc. Dalton Trans. 2001, 5, 586–592. [Google Scholar] [CrossRef]
- Carlucci, L.; Ciani, G.; Proserpio, D.M.; Rizzato, S. Structural Properties and Topological Diversity of Polymeric Ag(I)-hexamethylenetetramine Complexes: Self-Assembly of Three Novel Two-Dimensional Coordination Networks and Their Supramolecular Interactions. J. Solid State Chem. 2000, 152, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Lough, A.J.; Wheatley, P.S.; Ferguson, G.; Glidewell, C. Hydrogen-bonding behaviour of benzene-1,2,4,5-tetracarboxylic acid: Supramolecular structures of different dimensionality in the 2:1 adducts formed with 4,4’-bipyridyl and hexamethylenetetramine. Acta Crystallogr. Sect. B 2000, 56, 261–272. [Google Scholar] [CrossRef]
- Zheng, S.-L.; Tong, M.-L.; Zhu, H.-L.; Chen, X.-M. Four two-dimensional highly undulating silver(I)–hexamethylenetetramine co-ordination networks containing micropores. New J. Chem. 2001, 25, 1425–1429. [Google Scholar] [CrossRef]
- Blažzević, N.; Kolbah, D.; Belin, B.; Šunjić, V.; Kajfež, F. Hexamethylenetetramine, A Versatile Reagent in Organic Synthesis. Synthesis 1979, 1979, 161–176. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2019, B72, 171–179. [Google Scholar]
- Morita, H.; Tsunashima, R.; Nishihara, S.; Inoue, K.; Omura, Y.; Suzuki, Y.; Kawamata, J.; Hoshino, N.; Akutagawa, T. Ferroelectric Behavior of a Hexamethylenetetramine-Based Molecular Perovskite Structure. Angew. Chem. Int. Ed. 2019, 58, 9184–9187. [Google Scholar] [CrossRef] [PubMed]
- Zaręba, J.K.; Białek, M.J.; Janczak, J.; Zoń, J.; Dobosz, A. Extending the Family of Tetrahedral Tectons: Phenyl Embraces in Supramolecular Polymers of Tetraphenylmethane-based Tetraphosphonic Acid Templated by Organic Bases. Cryst. Growth Des. 2014, 14, 6143–6153. [Google Scholar] [CrossRef]
- Wei, X.; Guang-Ming, B.; Ting-Ting, S.; Chun-Yan, H.; Hou-Qun, Y. Crystal structure of 1,3,5,7-tetraazaadamantane-1,3-diium 2,5-dicarboxyterephthalate, C16H18N4O8. New Cryst. Struct. 2018, 233, 535–536. [Google Scholar] [CrossRef]
- Stahl, P.H.; Wermuth, C.G. (Eds.) Handbook of Pharmaceutical Salts: Properties Selection and Use; Verlag Helvetica Chimica Ac-ta/Wiley-VCH: Zurich, Switzerland, 2002; Volume 7, pp. 222–223. ISBN 3-906-390-26-8. [Google Scholar]
- Cruz-Cabeza, A.J. Acid–base crystalline complexes and the pKa rule. CrystEngComm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Bjerrum, J.; Sillén, L.G.; Schwarzenbach, G.K.; Berecki-Biedermann, C. Stability constants of metal-ion complexes, with solubility products of inorganic substances. In Inorganic Ligands; Chemical Society: London, UK, 1958. [Google Scholar]
- Braude, E.A.; Nachod, F.C. (Eds.) Determination of Organic Structures by Physical Methods, 1st ed.; Academic Press: Amsterdam, The Netherlands, 1955; pp. 1–824. [Google Scholar]
- Fresch, E.; Peruffo, N.; Trapani, M.; Cordaro, M.; Bella, G.; Castriciano, M.A.; Collini, E. The effect of hydrogen bonds on the ultrafast relaxation dynamics of a BODIPY dimer. J. Chem. Phys. 2021, 154, 084201. [Google Scholar] [CrossRef] [PubMed]
- Bella, G.; Santoro, A.; Cordaro, M.; Nicolò, F.; Bruno, G. Isoxazolone Reactivity Explained by Computed Electronic Structure Analysis. Chin. J. Chem. 2020, 38, 163–168. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision A.2; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bella, G.; Santoro, A.; Nicolò, F.; Bruno, G.; Cordaro, M. Do Secondary Electrostatic Interactions Influence Multiple Dihydrogen Bonds? AA−DD Array on an Amine-Borane Aza-Coronand: Theoretical Studies and Synthesis. ChemPhysChem 2021, 22, 593–605. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
Figure 1.
ORTEP diagram of the asymmetric unit of compound 1. The hydrogen interactions are represented by dotted lines.
Figure 1.
ORTEP diagram of the asymmetric unit of compound 1. The hydrogen interactions are represented by dotted lines.

Figure 2.
Molecular packing of compound 1.

Figure 3.
Hydrogen bond motif in compound 1 (a,b) and in compound 2 (c,d).

Figure 4.
General packing in compound 1 (a) and in compound 2 (b).

Figure 5.
Schematic representation of the HTMA fragment in compound 1, showing the bond distances in Angstrom.
Figure 5.
Schematic representation of the HTMA fragment in compound 1, showing the bond distances in Angstrom.

Figure 6.
Non-covalent interaction surfaces regarding compound 2 (A) and compound 1 (B); blue arrows indicate the specific strong hydrogen bonds, while the green arrow highlights the intermolecular electrostatic interactions in the long-range regime.
Figure 6.
Non-covalent interaction surfaces regarding compound 2 (A) and compound 1 (B); blue arrows indicate the specific strong hydrogen bonds, while the green arrow highlights the intermolecular electrostatic interactions in the long-range regime.

Scheme 1.
Energetic diagram of all the four step-by-step protonations of HMTA; for each step, the proton affinity (PA) and the deformation energy (ΔEdef) are displayed.
Scheme 1.
Energetic diagram of all the four step-by-step protonations of HMTA; for each step, the proton affinity (PA) and the deformation energy (ΔEdef) are displayed.

Figure 7.
Electrostatic potential surface (ESP) and the molecular orbitals of HMTA in its neutral form (a), monoprotonated (b), bi protonated (c), and tri protonated (d).
Figure 7.
Electrostatic potential surface (ESP) and the molecular orbitals of HMTA in its neutral form (a), monoprotonated (b), bi protonated (c), and tri protonated (d).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystal data and structure refinement for compound 1.
| Identification Code | Compound 1 |
|---|---|
| Deposition number | 2097042 |
| Empirical formula | C7H10N2O2.5 |
| Formula weight | 162.17 |
| Temperature/K | 296.15 |
| Crystal system | monoclinic |
| Space group | P21/m |
| a/Å | 6.8921(2) |
| b/Å | 6.9032(2) |
| c/Å | 16.5075(4) |
| α/° | 90 |
| β/° | 101.9540(10) |
| γ/° | 90 |
| Volume/Å3 | 768.35(4) |
| Z | 4 |
| ρcalc g/cm3 | 1.402 |
| μ/mm−1 | 0.108 |
| F(000) | 344.0 |
| Crystal size/mm3 | 0.48 × 0.35 × 0.18 |
| Radiation | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 6 to 54 |
| Index ranges | −8 ≤ h ≤ 8, −8 ≤ k ≤ 8, −21 ≤ l ≤ 21 |
| Reflections collected | 27297 |
| Independent reflections | 1805 [Rint = 0.0272, Rsigma = 0.0109] |
| Data/restraints/parameters | 1805/0/135 |
| Goodness-of-fit on F2 | 1.035 |
| Final R indexes [I>=2σ (I)] | R1 = 0.0349, wR2 = 0.0982 |
| Final R indexes [all data] | R1 = 0.0391, wR2 = 0.1037 |
| Largest diff. peak/hole/e Å−3 | 0.21/-0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bella, G.; Nicolò, F.; Bruno, G.; Santoro, A. Intermolecular Forces Driving Hexamethylenetetramine Co-Crystal Formation, a DFT and XRD Analysis. Molecules 2021, 26, 5746. https://doi.org/10.3390/molecules26195746
AMA Style
Bella G, Nicolò F, Bruno G, Santoro A. Intermolecular Forces Driving Hexamethylenetetramine Co-Crystal Formation, a DFT and XRD Analysis. Molecules. 2021; 26(19):5746. https://doi.org/10.3390/molecules26195746
Chicago/Turabian StyleBella, Giovanni, Francesco Nicolò, Giuseppe Bruno, and Antonio Santoro. 2021. "Intermolecular Forces Driving Hexamethylenetetramine Co-Crystal Formation, a DFT and XRD Analysis" Molecules 26, no. 19: 5746. https://doi.org/10.3390/molecules26195746