
Markov State Models and Molecular Dynamics Simulations Provide Understanding of the Nucleotide-Dependent Dimerization-Based Activation of LRRK2 ROC Domain

 ,
,
Abstract
:1. Introduction
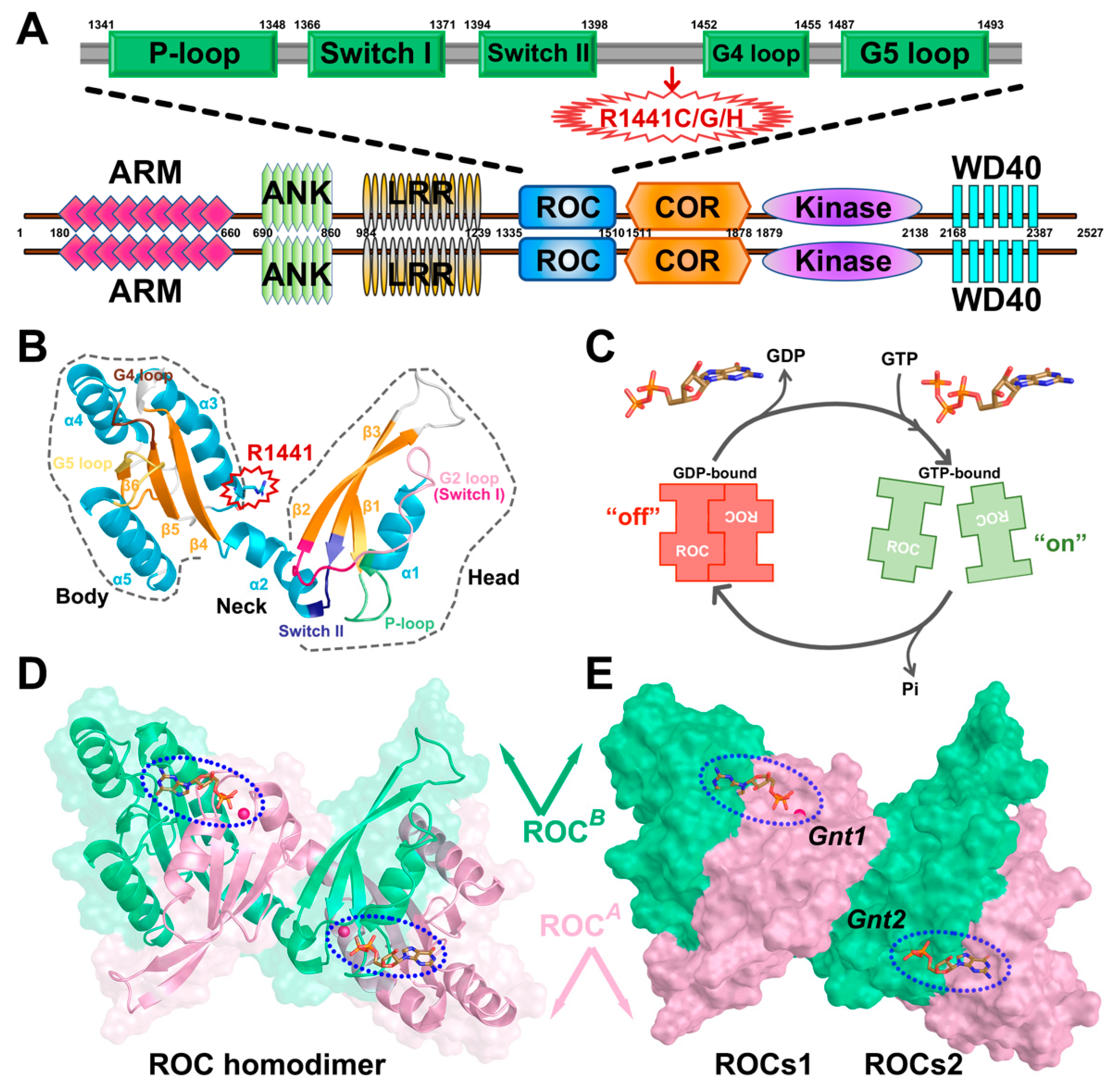
2. Results
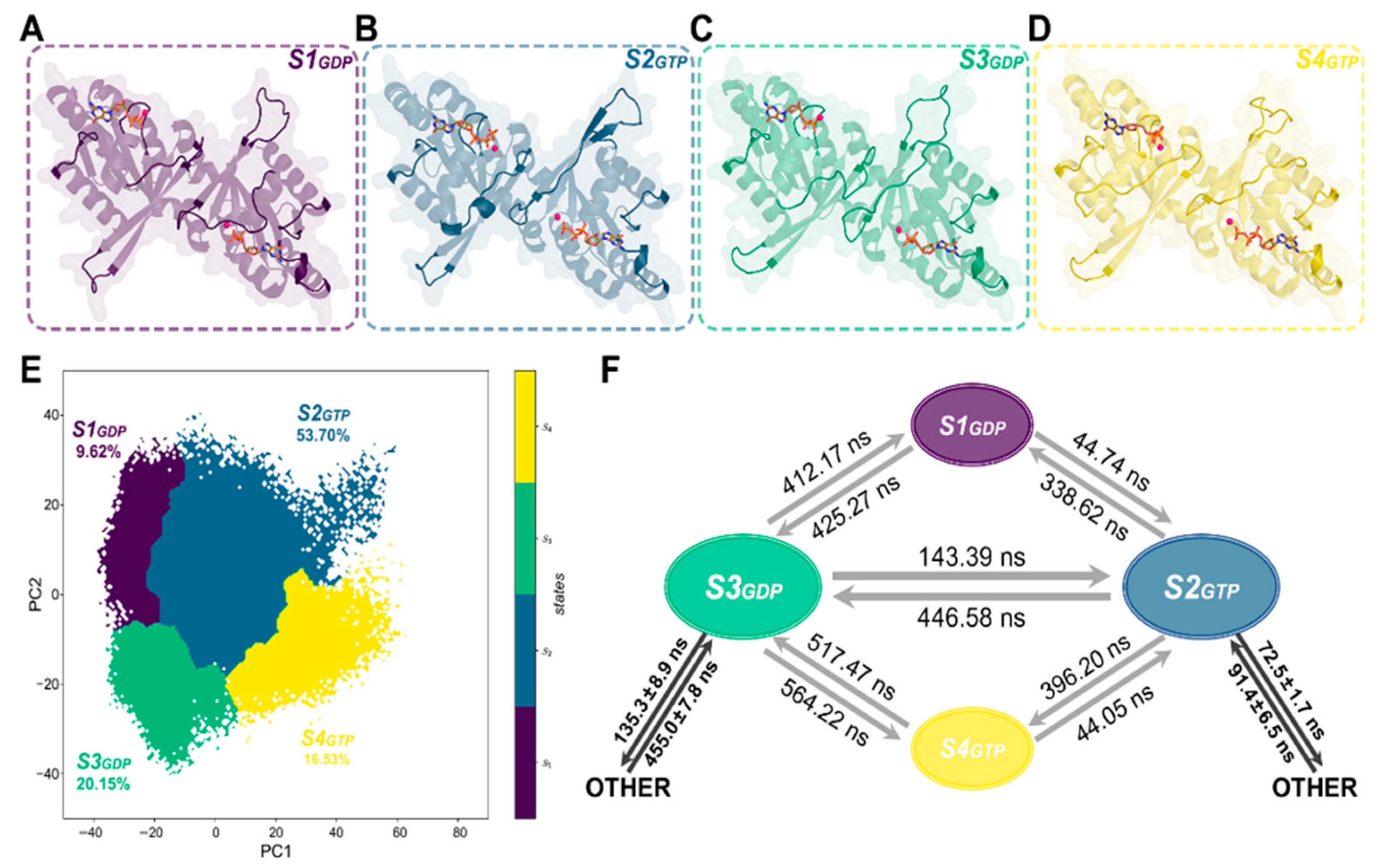
2.1. Construction and Validation of Markov State Models (MSMs)
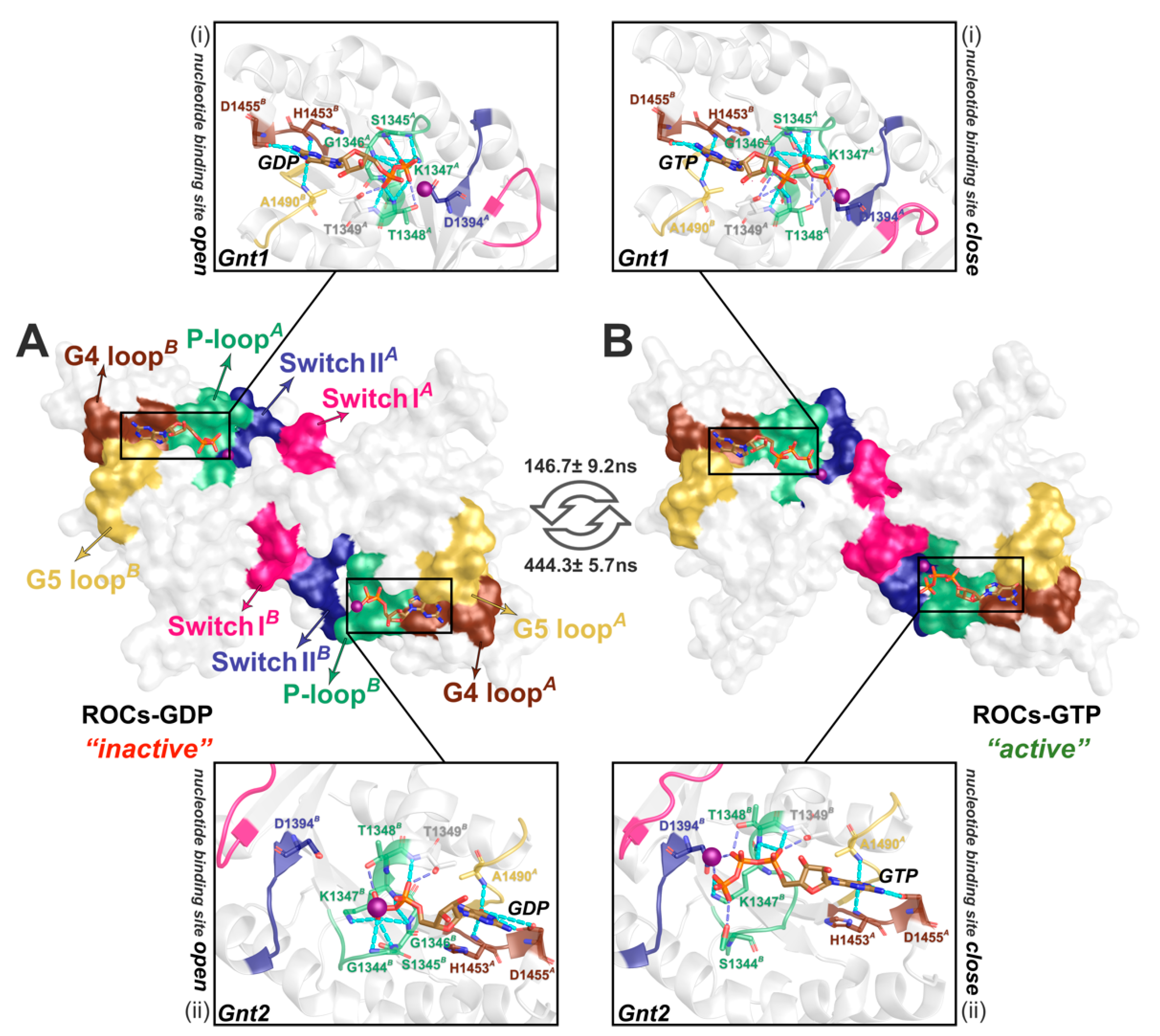
2.2. MSMs Revealed Different Dimerization Extent of ROCs during Nucleotide Turnover
2.3. ROCs Exhibited Classic “Open” to “Closed” Conformational Transition of GTPase
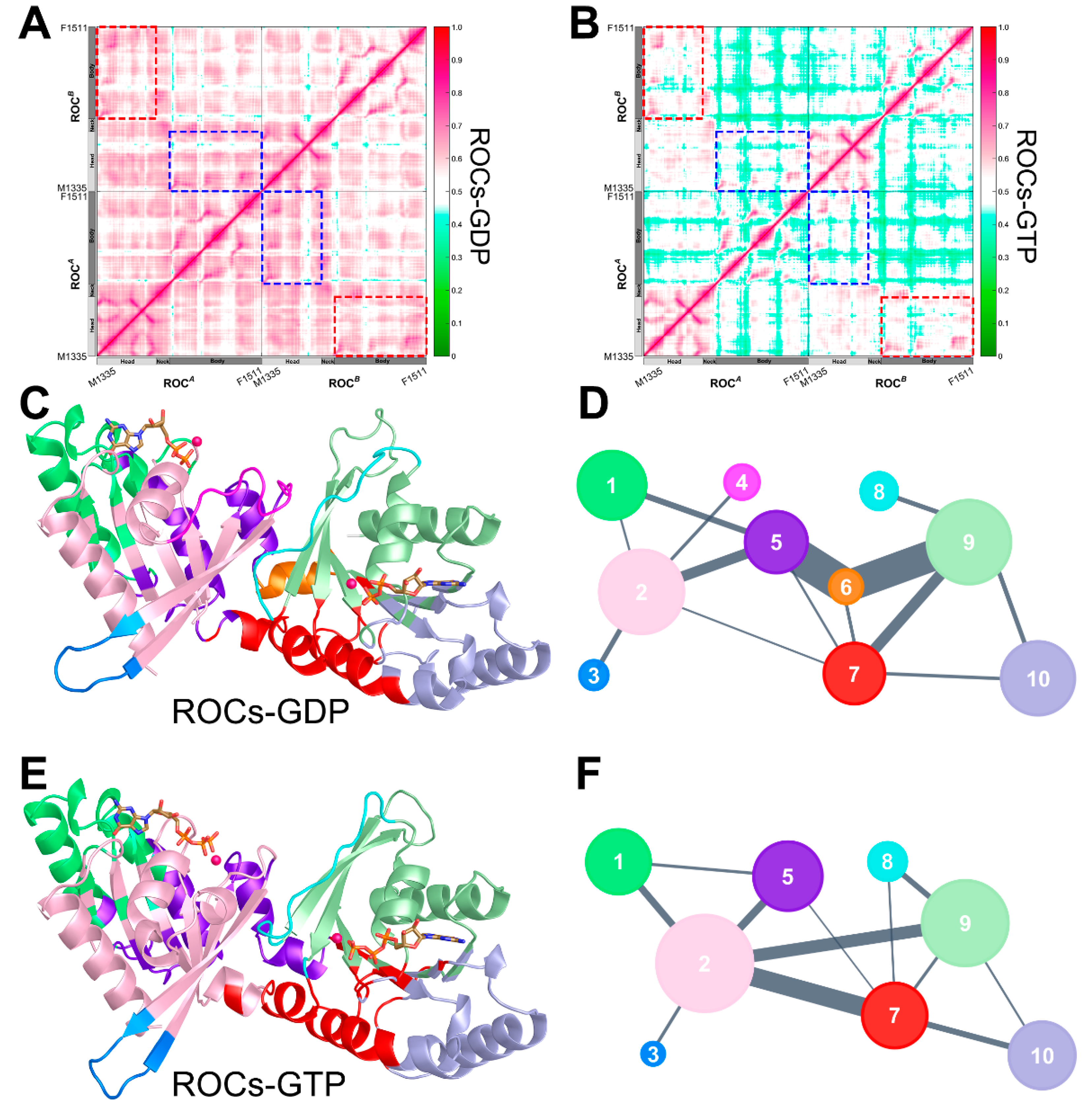
2.4. Nucleotide Turnover Reshaped ROCs Global Structure through Correlated Network
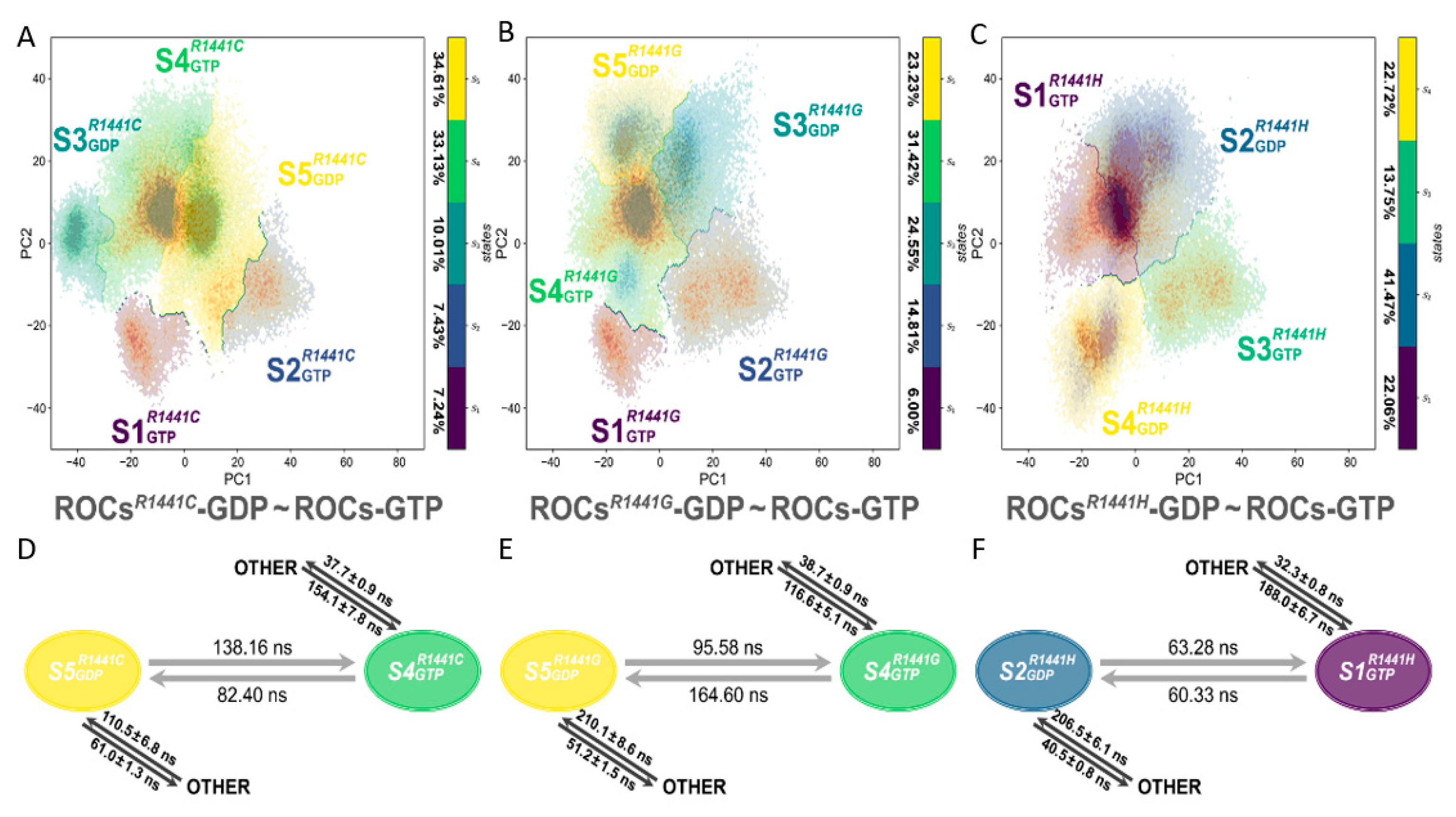
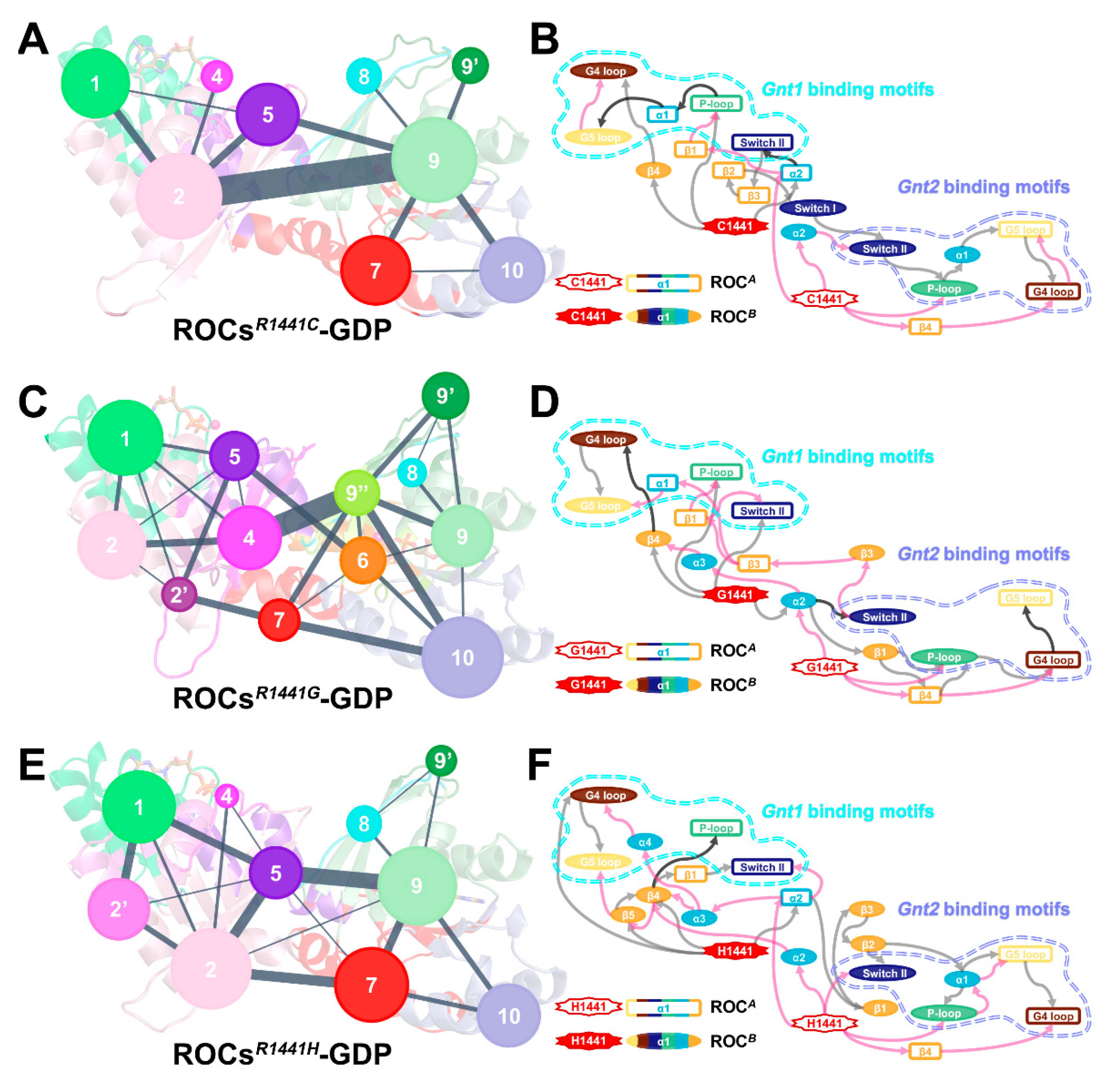
2.5. Molecular Basis and Allostery Underlying Pathogenetic Effects of R1441C/G/H Mutations
3. Discussion
4. Materials and Methods
4.1. Construction of Simulation Systems
4.2. MD Simulations
4.3. Markov State Model Construction and Validation
4.4. Generalized Cross-Correlation Matrix Analysis
4.5. Dynamic Network Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wu, C.X.; Liao, J.; Park, Y.; Reed, X.; Engel, V.A.; Hoang, N.C.; Takagi, Y.; Johnson, S.M.; Wang, M.; Federici, M.; et al. Parkinson’s disease-associated mutations in the GTPase domain of LRRK2 impair its nucleotide-dependent conformational dynamics. J. Biol. Chem. 2019, 294, 5907–5913. [Google Scholar] [CrossRef]
- Petsko, G.A. The next epidemic. Genome Biol. 2006, 7, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, W. Biochemical and molecular features of LRRK2 and its pathophysiological roles in Parkinson’s disease. BMB Rep. 2010, 43, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Halli-Tierney, A.D.; Luker, J.; Carroll, D.G. Parkinson Disease. Am. Fam. Physician 2020, 102, 679–691. [Google Scholar] [PubMed]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Muda, K.; Bertinetti, D.; Gesellchen, F.; Hermann, J.S.; Von Zweydorf, F.; Geerlof, A.; Jacob, A.; Ueffing, M.; Gloeckner, C.J.; Herberg, F.W. Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14-3-3. Proc. Natl. Acad. Sci. USA 2014, 111, E34–E43. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ye, M.; Wang, Y.; Qiu, M.; Fu, T.; Zhang, J.; Zhou, B.; Lu, S. How Parkinson’s disease-related mutations disrupt the dimerization of WD40 domain in LRRK2: A comparative molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2020, 22, 20421–20433. [Google Scholar] [CrossRef]
- Gasser, T. Molecular pathogenesis of Parkinson disease: Insights from genetic studies. Expert Rev. Mol. Med. 2009, 11, e22. [Google Scholar] [CrossRef]
- Gasser, T. Mendelian forms of Parkinson’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2009, 1792, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.P.T.; Moore, D.J. Understanding the GTPase activity of LRRK2: Regulation, function, and neurotoxicity. In Advances in Neurobiology; Spinger: Berlin, Germany, 2017; Volume 14, pp. 71–88. [Google Scholar]
- Marín, I.; Egmond, W.N.; Haastert, P.J.M. The Roco protein family: A functional perspective. FASEB J. 2008, 22, 3103–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosgraaf, L.; Van Haastert, P.J.M. Roc, a Ras/GTPase domain in complex proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2003, 1643, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Fan, Y.; Ru, H.; Wang, L.; Magupalli, V.G.; Taylor, S.S.; Alessi, D.R.; Wu, H. Crystal structure of the WD40 domain dimer of LRRK2. Proc. Natl. Acad. Sci. USA 2019, 116, 1579–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rideout, H.J.; Stefanis, L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson’s disease. Neurochem. Res. 2014, 39, 576–592. [Google Scholar] [CrossRef]
- Schmidt, S.H.; Weng, J.H.; Aoto, P.C.; Boassa, D.; Mathea, S.; Silletti, S.; Hu, J.; Wallbott, M.; Komives, E.A.; Knapp, S.; et al. Conformation and dynamics of the kinase domain drive subcellular location and activation of LRRK2. Proc. Natl. Acad. Sci. USA 2021, 118, e2100844118. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ji, D.; Lei, C.; Chen, Y.; Qiu, Y.; Li, X.; Li, M.; Ni, D.; Pu, J.; Zhang, J.; et al. Mechanistic insights into the effect of phosphorylation on Ras conformational dynamics and its interactions with cell signaling proteins. Comput. Struct. Biotechnol. J. 2021, 19, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ni, D.; Wang, C.; He, X.; Lin, H.; Wang, Z.; Zhang, J. Deactivation Pathway of Ras GTPase Underlies Conformational Substates as Targets for Drug Design. ACS Catal. 2019, 9, 7188–7196. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Comparative effects of oncogenic mutations G12C, G12V, G13D, and Q61H on local conformations and dynamics of K-Ras. Comput. Struct. Biotechnol. J. 2020, 18, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Trancikova, A.; Civiero, L.; Glauser, L.; Bubacco, L.; Greggio, E.; Moore, D.J. GTPase activity regulates kinase activity and cellular phenotypes of parkinson’s disease-associated LRRK2. Hum. Mol. Genet. 2013, 22, 1140–1156. [Google Scholar] [CrossRef] [Green Version]
- Ito, G.; Okai, T.; Fujino, G.; Takeda, K.; Ichijo, H.; Katada, T.; Iwatsubo, T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson’s disease. Biochemistry 2007, 46, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornejo-Olivas, M.; Torres, L.; Velit-Salazar, M.R.; Inca-Martinez, M.; Mazzetti, P.; Cosentino, C.; Micheli, F.; Perandones, C.; Dieguez, E.; Raggio, V.; et al. Variable frequency of LRRK2 variants in the Latin American research consortium on the genetics of Parkinson’s disease (LARGE-PD), a case of ancestry. NPJ Parkinson’s Dis. 2017, 3, 19. [Google Scholar] [CrossRef]
- Ross, O.A.; Whittle, A.J.; Cobb, S.A.; Hulihan, M.M.; Lincoln, S.J.; Toft, M.; Farrer, M.J.; Dickson, D.W. Lrrk2 R1441 substitution and progressive supranuclear palsy. Neuropathol. Appl. Neurobiol. 2006, 32, 23–25. [Google Scholar] [CrossRef]
- Weng, Y.H.; Chen, C.Y.; Lin, K.J.; Chen, Y.L.; Yeh, T.H.; Hsiao, I.T.; Chen, I.J.; Lu, C.S.; Wang, H.L. (R1441C) LRRK2 induces the degeneration of SN dopaminergic neurons and alters the expression of genes regulating neuronal survival in a transgenic mouse model. Exp. Neurol. 2016, 275, 104–115. [Google Scholar] [CrossRef]
- Liao, J.; Wu, C.X.; Burlak, C.; Zhang, S.; Sahm, H.; Wang, M.; Zhang, Z.Y.; Vogel, K.W.; Federici, M.; Riddle, S.M.; et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. USA 2014, 111, 4055–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyaert, E.; Wauters, L.; Guaitoli, G.; Konijnenberg, A.; Leemans, M.; Terheyden, S.; Petrovic, A.; Gallardo, R.; Nederveen-Schippers, L.M.; Athanasopoulos, P.S.; et al. A homologue of the Parkinson’s disease-associated protein LRRK2 undergoes a monomer-dimer transition during GTP turnover. Nat. Commun. 2017, 8, 1008. [Google Scholar] [CrossRef] [PubMed]
- Gasper, R.; Meyer, S.; Gotthardt, K.; Sirajuddin, M.; Wittinghofer, A. It takes two to tango: Regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 2009, 10, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Nixon-Abell, J.; Berwick, D.C.; Harvey, K. L’RRK de Triomphe: A solution for LRRK2 GTPase activity? Biochem. Soc. Trans. 2016, 44, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation Mechanism of the β2-Adrenergic Receptor. Biophys. J. 2012, 102, 239a. [Google Scholar] [CrossRef] [Green Version]
- Ni, D.; Li, X.; He, X.; Zhang, H.; Zhang, J.; Lu, S. Drugging K-RasG12C through covalent inhibitors: Mission possible? Pharmacol. Ther. 2019, 202, 1–17. [Google Scholar] [CrossRef]
- Pal, S.; Kumar, V.; Kundu, B.; Bhattacharya, D.; Preethy, N.; Reddy, M.P.; Talukdar, A. Ligand-based pharmacophore modeling, virtual screening and molecular docking studies for discovery of potential topoisomerase I inhibitors. Comput. Struct. Biotechnol. J. 2019, 17, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Shukla, D.; Pande, V.S.; Roux, B. Transition path theory analysis of c-Src kinase activation. Proc. Natl. Acad. Sci. USA 2016, 113, 9193–9198. [Google Scholar] [CrossRef] [Green Version]
- Long, C.; Chao, E.; Da, L.-T.; Yu, J. A viral T7 polymerse ratcheting along DNA with fidelity control. Comput. Struct. Biotechnol. J. 2019, 17, 638–644. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 2021, 12, 4721. [Google Scholar] [CrossRef]
- Kokkonen, P.; Slanska, M.; Dockalova, V.; Pinto, G.P.; Sánchez-Carnerero, E.M.; Damborsky, J.; Klán, P.; Prokop, Z.; Bednar, D. The impact of tunnel mutations on enzymatic catalysis depends on the tunnel-substrate complementary and the rate-limiting step. Comput. Struct. Biotechnol. J. 2020, 18, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA: Protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.H.; Noé, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Prinz, J.H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schtte, C.; Noé, F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134, 174105. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Noé, F. Variational Approach for Learning Markov Processes from Time Series Data. J. Nonlinear Sci. 2020, 30, 23–66. [Google Scholar] [CrossRef] [Green Version]
- Peppas, N.A.; Robinson, J.R. Bioadhesives for optimization of drug delivery. J. Drug Target. 1995, 3, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Röblitz, S.; Weber, M. Fuzzy spectral clustering by PCCA+: Application to Markov state models and data classification. Adv. Data Anal. Classif. 2013, 7, 147–179. [Google Scholar] [CrossRef]
- Weinan, E.; Vanden-Eijnden, E. Towards a theory of transition paths. J. Stat. Phys. 2006, 123, 503–523. [Google Scholar]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, S.; Zhou, Q.; Zhang, C.; Gao, Y.; Wang, H.; Li, Z.; Wu, D.; Wu, Y.; Huang, Y.Y.; et al. Disocvery of highly selective and orally available benzimidazole-based phosphodiesterase 10 inhibitors with improved solubility and pharmacokinetic properties for treatment of pulmonary arterial hypertension. Acta Pharm. Sin. B 2020, 10, 2339–2347. [Google Scholar] [CrossRef]
- Zeb, A.; Son, M.; Yoon, S.; Kim, J.H.; Park, S.J.; Lee, K.W. Computational simulations identified two candiate inhibitors of Cdk5/p25 to abrogate Tau-assocaited neurological disorders. Comput. Struct. Biotechnol. J. 2019, 17, 579–590. [Google Scholar] [CrossRef]
- Stafa, K.; Trancikova, A.; Webber, P.J.; Glauser, L.; West, A.B.; Moore, D.J. GTPase activity and neuronal toxicity of parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012, 8, e1002526. [Google Scholar] [CrossRef]
- Lange, O.F.; Grubmüller, H. Generalized correlation for biomolecular dynamics. Proteins Struct. Funct. Bioinform. 2006, 62, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chen, Y.; Ni, D.; Huang, Z.; Wei, J.; Feng, L.; Su, J.-C.; Wei, Y.; Ning, S.; Yang, X.; et al. Targeting a cryptic allosteric site of SIRT6 with small-molecule inhibitors that inhibit the migration of pancreatic cancer cells. Acta Pharm. Sin. B 2021, in press. [Google Scholar] [CrossRef]
- Tong, Y.; Pisani, A.; Martella, G.; Karouani, M.; Yamaguchi, H.; Pothos, E.N.; Shen, J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14622–14627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Wu, C.; Park, Y.; Long, X.; Hoang, Q.Q.; Liao, J. The Parkinson’s disease–associated mutation N1437H impairs conformational dynamics in the G domain of LRRK2. FASEB J. 2019, 33, 4814–4823. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, B.K.; Ho, F.Y.; Vetter, I.R.; Van Haastert, P.J.M.; Wittinghofer, A.; Kortholt, A. Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 10322–10327. [Google Scholar] [CrossRef] [Green Version]
- Del Sol, A.; Fujihashi, H.; Amoros, D.; Nussinov, R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006, 2, 2006-0019. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Vishveshwara, S. A study of communication pathways in methionyl-tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 15711–15716. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Zhu, Y.; Long, J.; Ye, F.; Hu, G. Both intra and inter-domain interactions define the intrinsic dynamics and allosteric mechanism in DNMT1s. Comput. Struct. Biotechnol. J. 2020, 23, 749–764. [Google Scholar] [CrossRef]
- Xiong, Y.; Yuan, C.; Chen, R.; Dawson, T.M.; Dawson, V.L. ArfGAP1 is a GTPase activating protein for LRRK2: Reciprocal regulation of ArfGAP1 by LRRK2. J. Neurosci. 2012, 32, 3877–3886. [Google Scholar] [CrossRef] [Green Version]
- Studio, D. Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Accelrys Softw Inc.: San Diego, CA, USA, 2015; pp. 98–104. [Google Scholar]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. Amber18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Lu, S.; Chen, Y.; Wei, J.; Zhao, M.; Ni, D.; He, X.; Zhang, J. Mechanism of allosteric activation of SIRT6 revealed by the action of rationally designed activators. Acta Pharm. Sin. B 2021, 11, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Augustyn, A.; Johansson, A.J.; Borowski, T. Mechanism of sulfate activation catalyzed by ATP sulfurlase-Magnesium inhibits the activity. Comput. Struct. Biotechnol. J. 2019, 17, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, G.; Trstanova, Z. Langevin dynamics with general kinetic energies. Multiscale Model. Simul. 2018, 16, 777–806. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Palermo, G.; Ricci, C.G.; Fernando, A.; Basak, R.; Jinek, M.; Rivalta, I.; Batista, V.S.; McCammon, J.A. Protospacer Adjacent Motif-Induced Allostery Activates CRISPR-Cas9. J. Am. Chem. Soc. 2017, 139, 16028–16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; McCammon, J.A. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc. Natl. Acad. Sci. USA 2017, 114, 7260–7265. [Google Scholar] [CrossRef] [Green Version]
- Abramyan, A.M.; Yano, H.; Xu, M.; Liu, L.; Naing, S.; Fant, A.D.; Shi, L. The Glu102 mutation disrupts higher-order oligomerization of the sigma 1 receptor. Comput. Struct. Biotechnol. J. 2020, 18, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ye, Z.; Su, Y.; Ouyang, D. Predicting complexation performance between cycoldextrins and guest molecules by integrated machine learning and molecular modeling techniques. Acta Pharm. Sin. B 2019, 9, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, G.; Paul, F.; Giorgino, T.; De Fabritiis, G.; Noé, F. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013, 139, 015102. [Google Scholar] [CrossRef] [PubMed]
- Molgedey, L.; Schuster, H.G. Separation of a mixture of independent signals using time delayed correlations. Phys. Rev. Lett. 1994, 72, 3634–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabar, M.R.R. Introduction to Stochastic Processes. In Understanding Complex Systems; Elsevier: Amsterdam, The Netherlands, 2019; pp. 9–18. [Google Scholar]
- Metzner, P.; Schütte, C.; Vanden-Eijnden, E. Transition Path Theory for Markov Jump Processes. Multiscale Model. Simul. 2009, 7, 1192–1219. [Google Scholar] [CrossRef]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Mian, K.H. Interactive Dynamic Network Analysis. Dyn. Soc. Netw. Model. Anal. Work. Summ. Pap. 1982, 117–119. [Google Scholar]
- Hougardy, S. The Floyd-Warshall algorithm on graphs with negative cycles. Inf. Process. Lett. 2010, 110, 279–281. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Representative | Binding Free Energy | Standard Deviations |
|---|---|---|---|
| ROCs-GDP | S3GDP | −296.39 | 19.71 |
| ROCs-GTP | S2GTP | −285.09 | 23.52 |
| ROCsR1441C-GDP | S5GDPR1441C | −276.31 | 18.02 |
| ROCsR1441G-GDP | S5GDPR144G | −274.93 | 17.48 |
| ROCsR1441H-GDP | S2GDPR144H | −274.98 | 17.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Qi, Z.; Ni, D.; Lu, S.; Chen, L.; Chen, X. Markov State Models and Molecular Dynamics Simulations Provide Understanding of the Nucleotide-Dependent Dimerization-Based Activation of LRRK2 ROC Domain. Molecules 2021, 26, 5647. https://doi.org/10.3390/molecules26185647
Li X, Qi Z, Ni D, Lu S, Chen L, Chen X. Markov State Models and Molecular Dynamics Simulations Provide Understanding of the Nucleotide-Dependent Dimerization-Based Activation of LRRK2 ROC Domain. Molecules. 2021; 26(18):5647. https://doi.org/10.3390/molecules26185647
Chicago/Turabian StyleLi, Xinyi, Zengxin Qi, Duan Ni, Shaoyong Lu, Liang Chen, and Xiangyu Chen. 2021. "Markov State Models and Molecular Dynamics Simulations Provide Understanding of the Nucleotide-Dependent Dimerization-Based Activation of LRRK2 ROC Domain" Molecules 26, no. 18: 5647. https://doi.org/10.3390/molecules26185647