
Novel Bradykinin Receptor Inhibitors Inhibit Proliferation and Promote the Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the ERK Pathway


Abstract
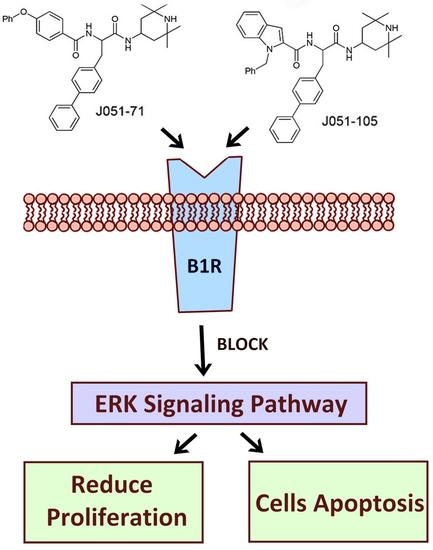
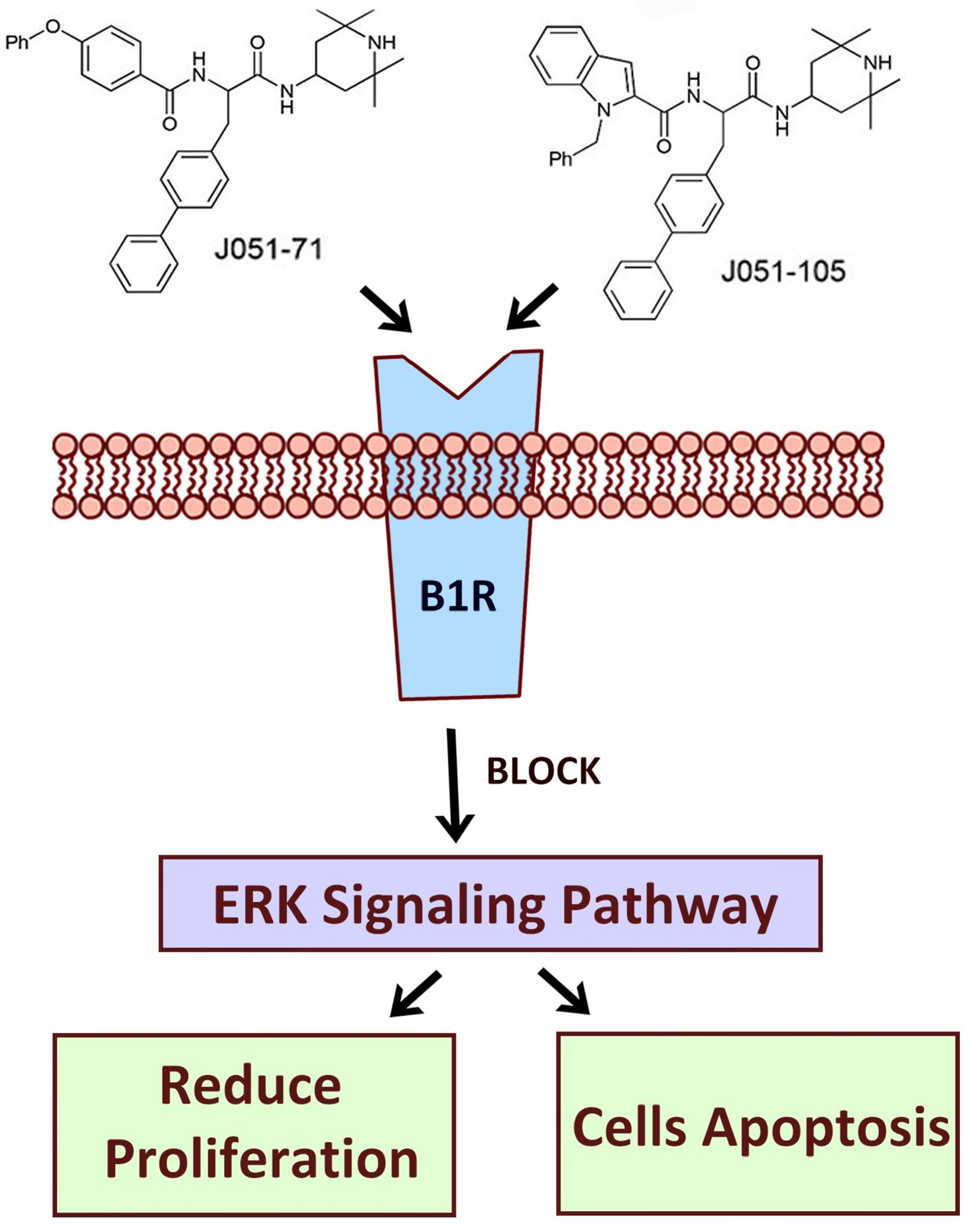
:
1. Introduction
2. Results
2.1. Design and Synthesis of BK Receptor Inhibitors
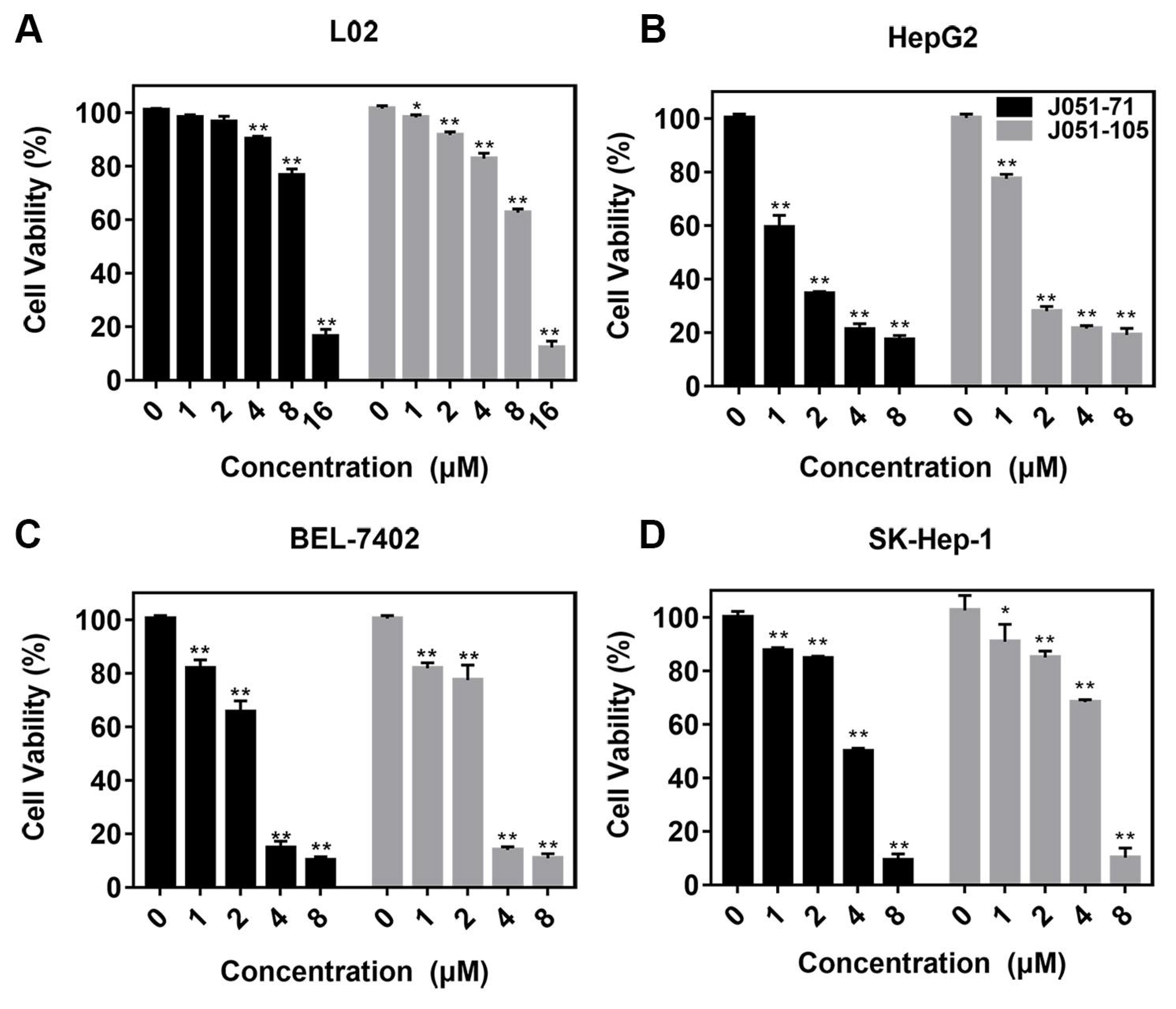
2.2. Anticancer Activity of BK Receptor Inhibitors
2.3. BK Receptor Inhibitors Inhibit the Cellular Activity of Primary HCC Cells
2.4. BK Receptor Inhibitors Inhibit the Proliferation of HCC Cells
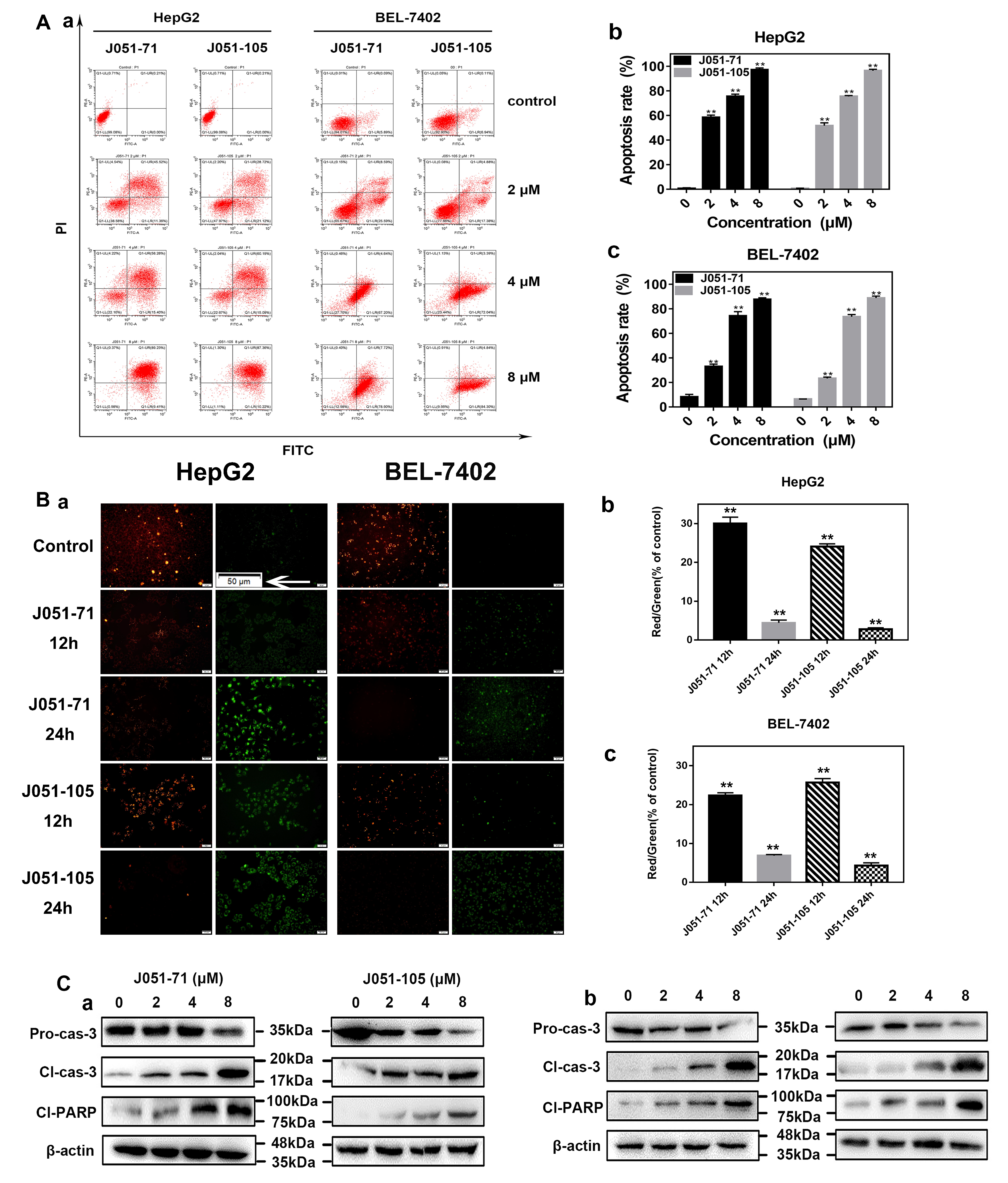
2.5. BK Receptor Inhibitors Promote the Apoptosis of HCC Cells
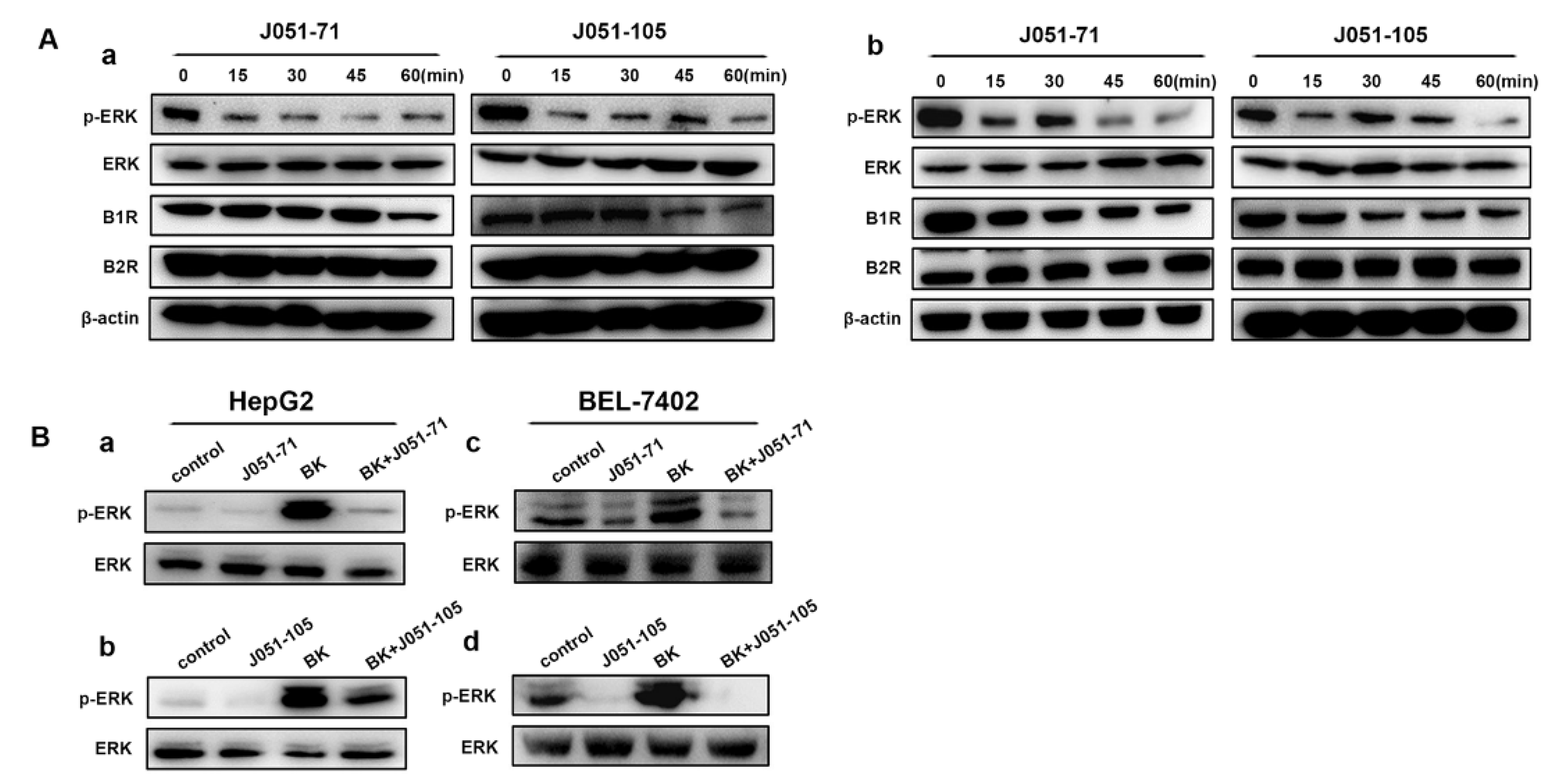
2.6. BK Receptor Inhibitors Reduce the Proliferation of HCC Cells by Inhibiting the ERK Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of BK Receptor Inhibitors
4.3. Cell Culture, Cell Viability, and Proliferation Assays
4.4. Colony Formation
4.5. Annexin V/PI Double Staining
4.6. Measurement of Mitochondrial Membrane Potentials
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| HCC | Hepatocellular carcinoma |
| BK | bradykinin |
| GPCRs | G-protein-coupled receptors |
| B1R | B1 receptor |
| B2R | B2 receptor |
| KKS | kinin-kallikrein system |
| TCE | trichloroethylene |
| MAPK | mitogen-activated protein kinase |
| IL | interleukin |
| TNF | tumor necrosis factor |
| BOC | Boc-4-phenyl-Phe-OH |
| HATU | hexafluorophosphate |
| DIPEA | diisopropylethylamine |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| MeOH | methanol |
| DCM | dichloromethane |
| EtOAc | ethyl acetate |
| PE | petroleum ether |
| IBCB | Institute of Biochemistry and Cell Biology |
| NMR | nuclear magnetic resonance |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | fetal bovine serum |
| PI | propidium iodide |
References
- Shi, J.; Lai, E.C.; Li, N.; Guo, W.X.; Xue, J.; Lau, W.Y.; Wu, M.C.; Cheng, S.Q. Surgical treatment of hepatocellular carcinoma with portal vein tumor thrombus. Ann. Surg. Oncol. 2010, 7, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Nagano, H.; Ota, H.; Morimoto, O.; Nakamura, M.; Wada, H.; Node, T.; Damdinsuren, B.; Marubashi, S.; Miyamoto, A.; et al. Patterns and clinicopathologic features of extrahepatic recurrence of hepatocellular carcinoma after curative resection. Surgery 2007, 141, 196–202. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, R.; Pandey, A.K. Hepatocellular Carcinoma: Causes, Mechanism of Progression and Biomarkers. Curr. Chem. Genom. Transl. Med. 2018, 12, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Hartke, J.; Johnson, M.; Ghabril, M. The diagnosis and treatment of hepatocellular carcinoma. Semin. Diagn. Pathol. 2017, 34, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Gerolami, R.; Caparello, C.; et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J. Hepatol. 2018, 69, 353–358. [Google Scholar] [CrossRef]
- Ohri, N.; Kaubisch, A.; Garg, M.; Guha, C. Targeted Therapy for Hepatocellular Carcinoma. Semin. Radiat. Oncol. 2016, 26, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Oncogenic Signal and Tumor Microenvironment in Hepatocellular Carcinoma. Oncology 2017, 93, 160–164. [Google Scholar] [CrossRef]
- Costa, P.L.N.; Sirois, P.; Tannock, I.F.; Chammas, R. The role of kinin receptors in cancer and therapeutic opportunities. Cancer Lett. 2014, 345, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Neto, C.M.; Dillenburg-Pilla, P.; Heinrich, T.A.; Parreiras-e-Silva, L.T.; Pereira, M.G.; Reis, R.I.; Souza, P.P. Participation of kallikrein-kinin system in different pathologies. Int. Immunopharmacol. 2008, 8, 135–142. [Google Scholar] [CrossRef]
- Calixto, J.B.; Medeiros, R.; Fernandes, E.S.; Ferreira, J.; Cabrini, D.A.; Campos, M.M. Kinin B1 receptors: Key G-protein-coupled receptors and their role in inflammatory and painful processes. Br. J. Pharmacol. 2004, 143, 803–818. [Google Scholar] [CrossRef] [Green Version]
- Kuhr, F.; Lowry, J.; Zhang, Y.; Brovkovych, V.; Skidgel, R.A. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 2010, 44, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenfeld, P.; Millan, C.; Matus, C.E.; Figueroa, J.E.; Burgos, R.A.; Nualart, F.; Bhoola, K.D.; Figueroa, C.D. Activation of kinin B1 receptors induces chemotaxis of human neutrophils. J. Leukoc. Biol. 2006, 80, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, J.X.; Ye, L.P.; Li, S.L.; Wang, F.; Zha, W.S.; Shen, T.; Wu, C.; Zhu, Q.X. Plasma Kallikrein-Kinin system mediates immune-mediated renal injury in trichloroethylene-sensitized mice. J. Immunotoxicol. 2016, 13, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, N.; Yang, L.; Zang, D.; Yang, P.; Wang, H.; Shen, T.; Zhu, Q.X. Role of selective blocking of bradykinin B1 receptor in attenuating immune liver injury in trichloroethylene-sensitized mice. Cytokine 2018, 108, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, N.; Yang, L.; Xie, H.; Yang, Y.; Wang, H.; Shen, T.; Zhu, Q. Bradykinin contributes to immune liver injury via B2R receptor-mediated pathways in trichloroethylene sensitized mice: A role in Kupffer cell activation. Toxicology 2019, 415, 37–48. [Google Scholar] [CrossRef]
- Wang, X.; Yang, C.; Zhang, Q.; Wang, C.; Zhou, X.; Zhang, X.; Liu, S. In vitro anticancer effects of esculetin against human leukemia cell lines involves apoptotic cell death, autophagy, G0/G1 cell cycle arrest and modulation of Raf/MEK/ERK signalling pathway. J. BUON 2019, 24, 1686–1691. [Google Scholar]
- Meng, X.; Shi, Y.; Xiang, X.; Li, C.; Ge, X.; Pan, K.; Liang, Y. Influence of miR-101 on proliferation of liver cancer cells through the MAPK/ERK signaling pathway. Oncol. Lett. 2020, 19, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Li, Q.; Zhou, C.; Zhao, Y.; Li, R.; Zhang, Y. Demethyleneberberine attenuates concanavalin A-induced autoimmune hepatitis in mice through inhibition of NF-κB and MAPK signaling. Int. Immunopharmacol. 2020, 80, 106137. [Google Scholar] [CrossRef]
- Cusimano, A.; Foderà, D.; D’Alessandro, N.; Lampiasi, N.; Azzolina, A.; Montalto, G.; Cervello, M. Potentiation of the antitumor effects of both selective cyclooxygenase-1 and cyclooxygenase-2 inhibitors in human hepatic cancer cells by inhibition of the MEK/ERK pathway. Cancer Biol. Ther. 2007, 6, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.M.; Gera, L.; Chan, D.C.; Bunn, P.A., Jr.; York, E.J.; Simkeviciene, V.; Helfrich, B. Bradykinin-related compounds as new drugs for cancer and inflammation. Can. J. Physiol. Pharmacol. 2002, 80, 275–280. [Google Scholar] [CrossRef]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio-Protocol 2019, 9, e3128. [Google Scholar] [CrossRef]
- Whalley, E.T.; Figueroa, C.D.; Gera, L.; Bhoola, K.D. Discovery and therapeutic potential of kinin receptor antagonists. Expert Opin. Drug Discov. 2012, 7, 1129–1148. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M.; Gera, L.; Chan, D.C.; York, E.J.; Simkeviciene, V.; Bunn, P.A., Jr.; Taraseviciene-Stewart, L. Combination cancer chemotherapy with one compound: Pluripotent bradykinin antagonists. Peptides 2005, 26, 1288–1291. [Google Scholar] [CrossRef]
- Barth, M.; Bondoux, M.; Luccarini, J.M.; Peyrou, V.; Dodey, P.; Pruneau, D.; Massardier, C.; Paquet, J.L. From bradykinin B2 receptor antagonists to orally active and selective bradykinin B1 receptor antagonists. J. Med. Chem. 2012, 55, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Avdieiev, S.; Gera, L.; Havrylyuk, D.; Hodges, R.S.; Lesyk, R.; Ribrag, V.; Vassetzkyf, Y.; Kavsan, V. Bradykinin antagonists and thiazolidinone derivatives as new potential anti-cancer compounds. Bioorg. Med. Chem. 2014, 22, 3815–3823. [Google Scholar] [CrossRef] [PubMed]
- Jutras, S.; Bachvarova, M.; Keita, M.; Bascands, J.L.; Mes-Masson, A.M.; Stewart, J.M.; Bachvarov, D. Strong cytotoxic effect of the bradykinin antagonist BKM-570 in ovarian cancer cells-analysis of the molecular mechanisms of its antiproliferative action. FEBS J. 2001, 277, 5146–5160. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, C. Regulation of MAP kinase activity by peptide receptor signalling pathway: Paradigms of multiplicity. Cell Signal. 2010, 13, 777–785. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | J051-71 (μM) | J051-105 (μM) | Paclitaxel (μM) | Cisplatin (μM) |
|---|---|---|---|---|
| A549 | 1.28 | 2.33 | 0.90 | 9.38 |
| SGC-7901 | 5.07 | 5.49 | 9.05 | 12.4 |
| MCF-7 | 2.20 | 2.47 | <2.0 | 8.20 |
| HeLa | 5.75 | 5.00 | 12.30 | 3.41 |
| MG63 | 3.22 | 3.46 | <2.0 | 23.17 |
| Bx-PC3 | 2.11 | 2.99 | 15.27 | 20.83 |
| Cell Line | J051-71 (μM) | J051-105 (μM) |
|---|---|---|
| L02 | 9.53 | 10.33 |
| HepG2 | 1.38 | 1.53 |
| BEL-7402 | 2.53 | 2.76 |
| SK-Hep-1 | 4.78 | 5.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, B.; Huang, Y.; Yao, W.; Tao, F.; Chen, Y. Novel Bradykinin Receptor Inhibitors Inhibit Proliferation and Promote the Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the ERK Pathway. Molecules 2021, 26, 3915. https://doi.org/10.3390/molecules26133915
Wang Y, Zhang B, Huang Y, Yao W, Tao F, Chen Y. Novel Bradykinin Receptor Inhibitors Inhibit Proliferation and Promote the Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the ERK Pathway. Molecules. 2021; 26(13):3915. https://doi.org/10.3390/molecules26133915
Chicago/Turabian StyleWang, Yiou, Bingxue Zhang, Yibing Huang, Wenjun Yao, Fei Tao, and Yuxin Chen. 2021. "Novel Bradykinin Receptor Inhibitors Inhibit Proliferation and Promote the Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the ERK Pathway" Molecules 26, no. 13: 3915. https://doi.org/10.3390/molecules26133915