
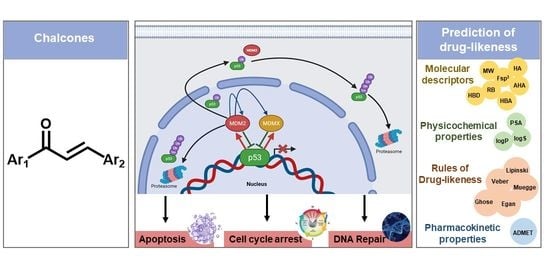
Chalcones as Promising Antitumor Agents by Targeting the p53 Pathway: An Overview and New Insights in Drug-Likeness

 , , , and
, , , and
Abstract
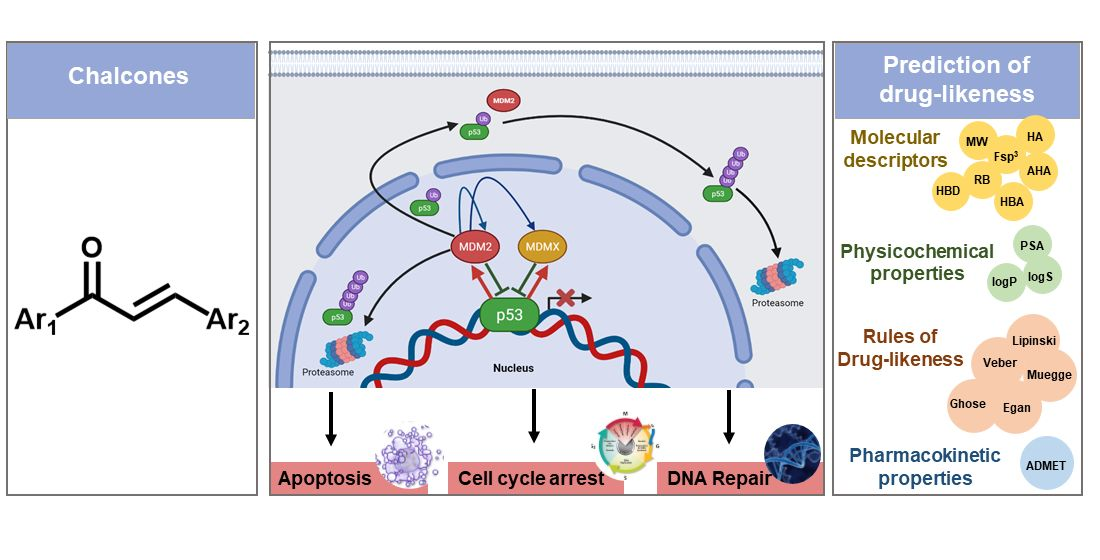
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
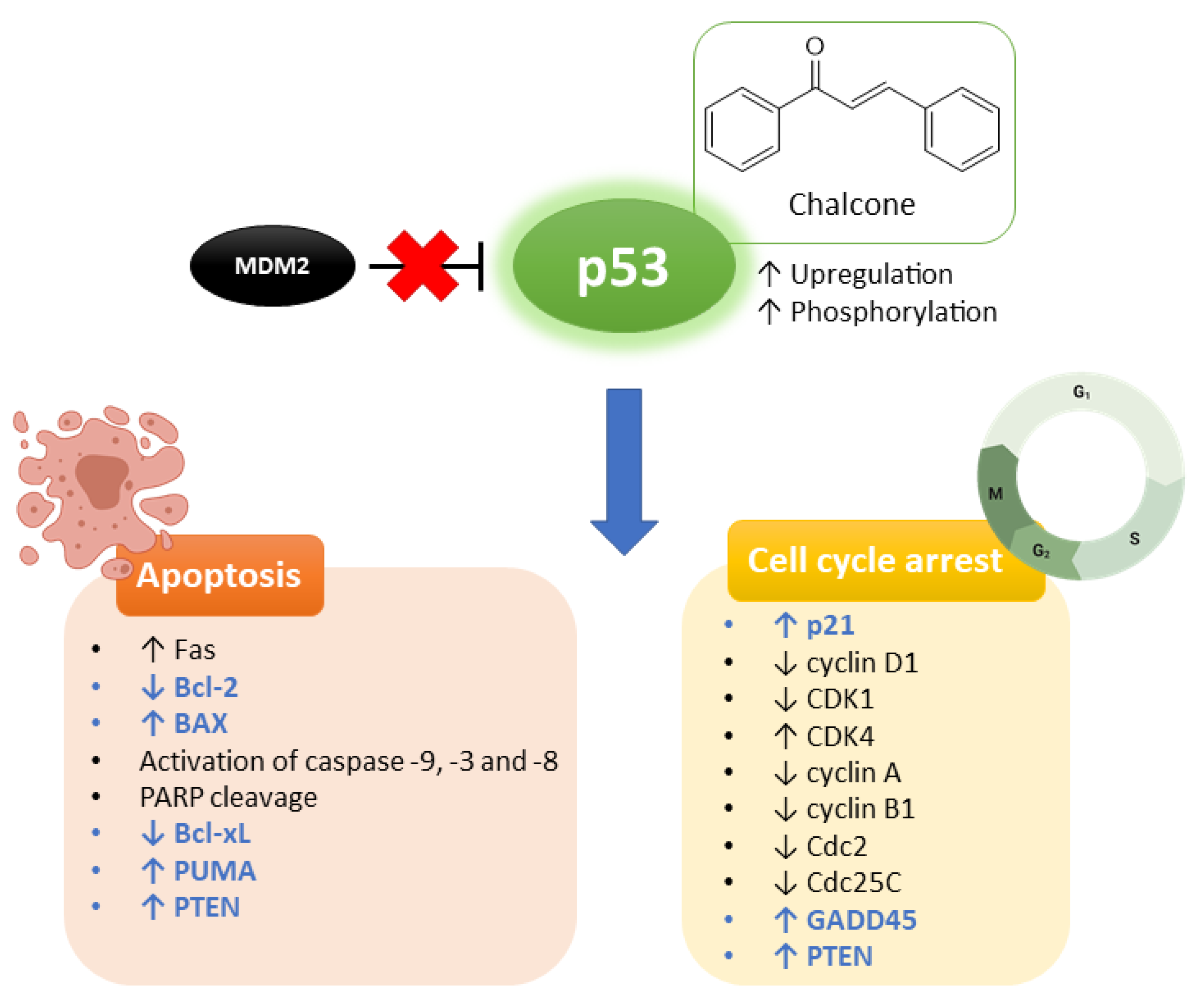
1. Introduction
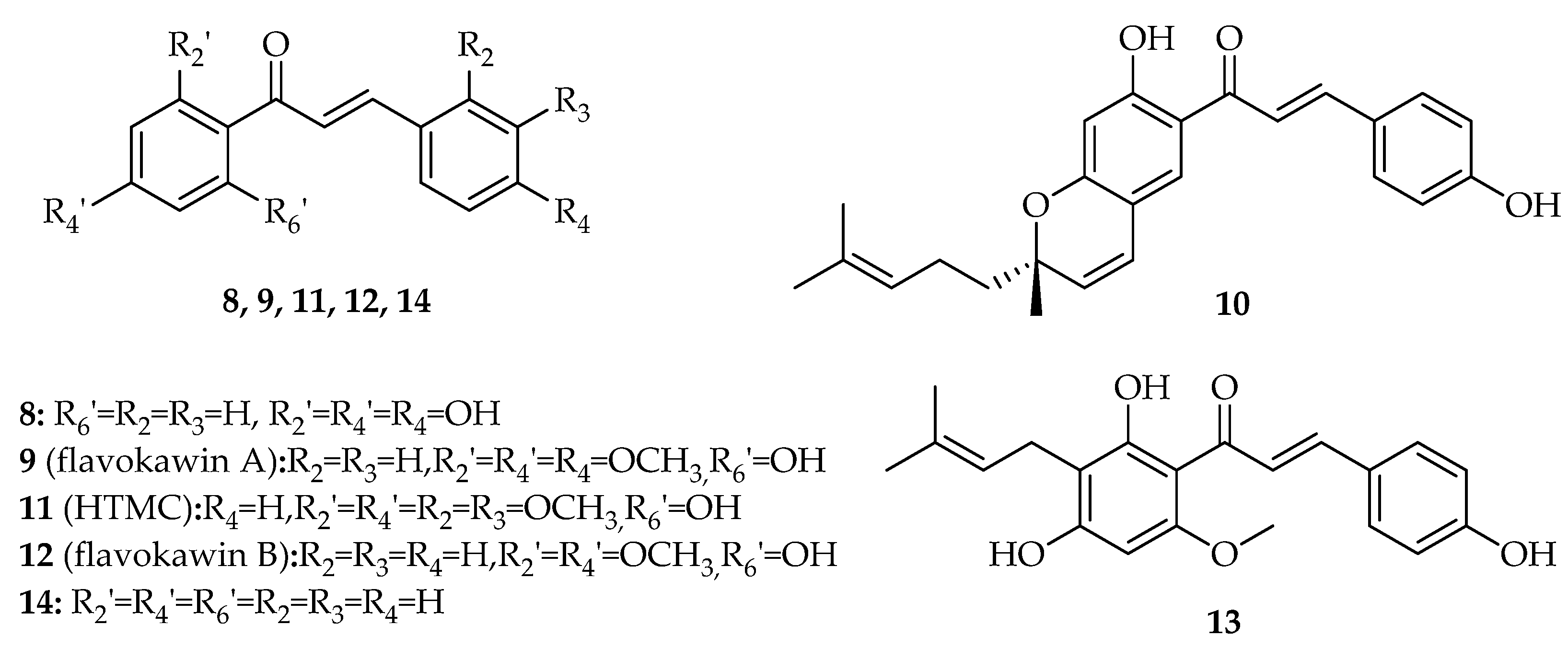
2. Interference with p53 Pathway by Natural Chalcones
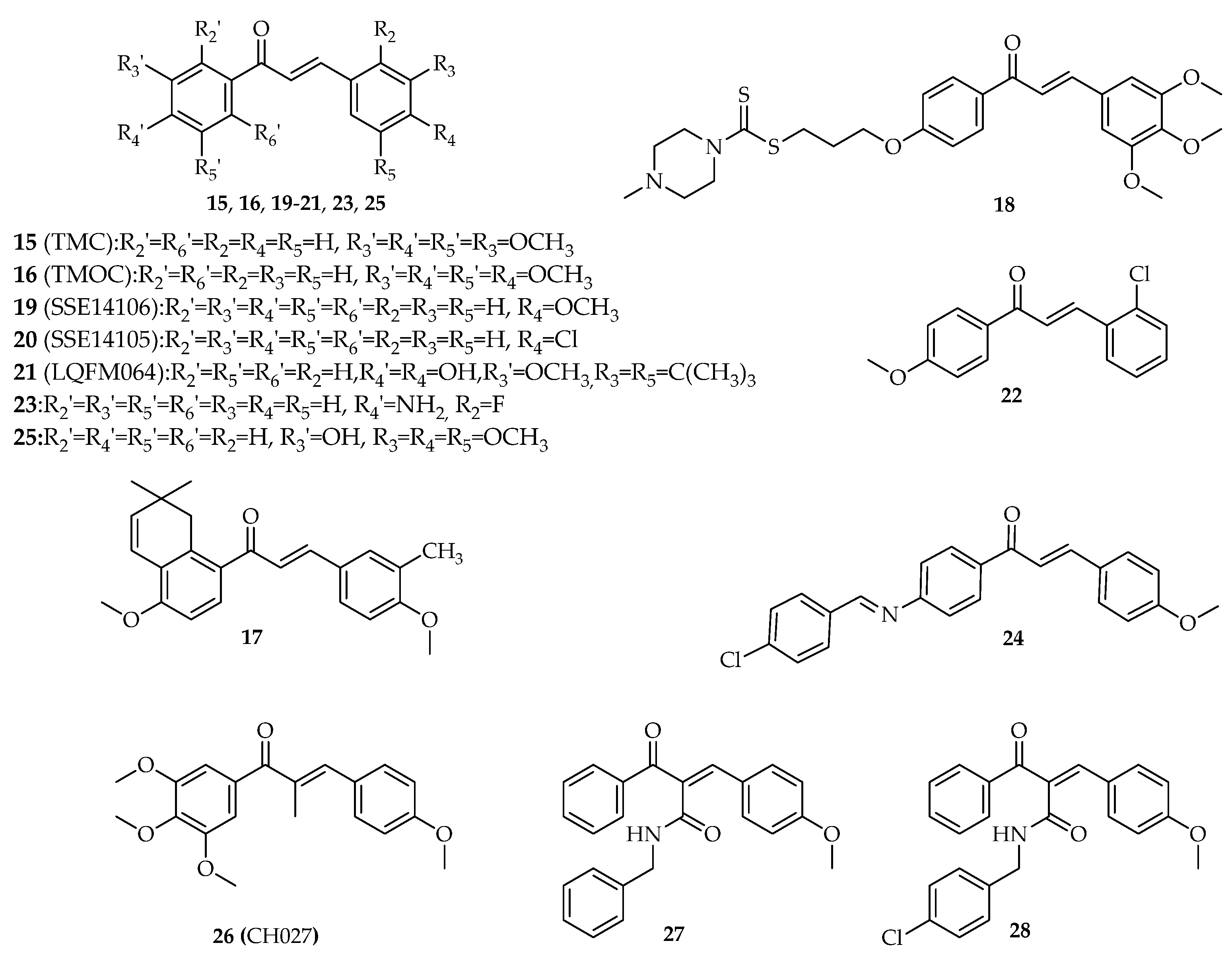
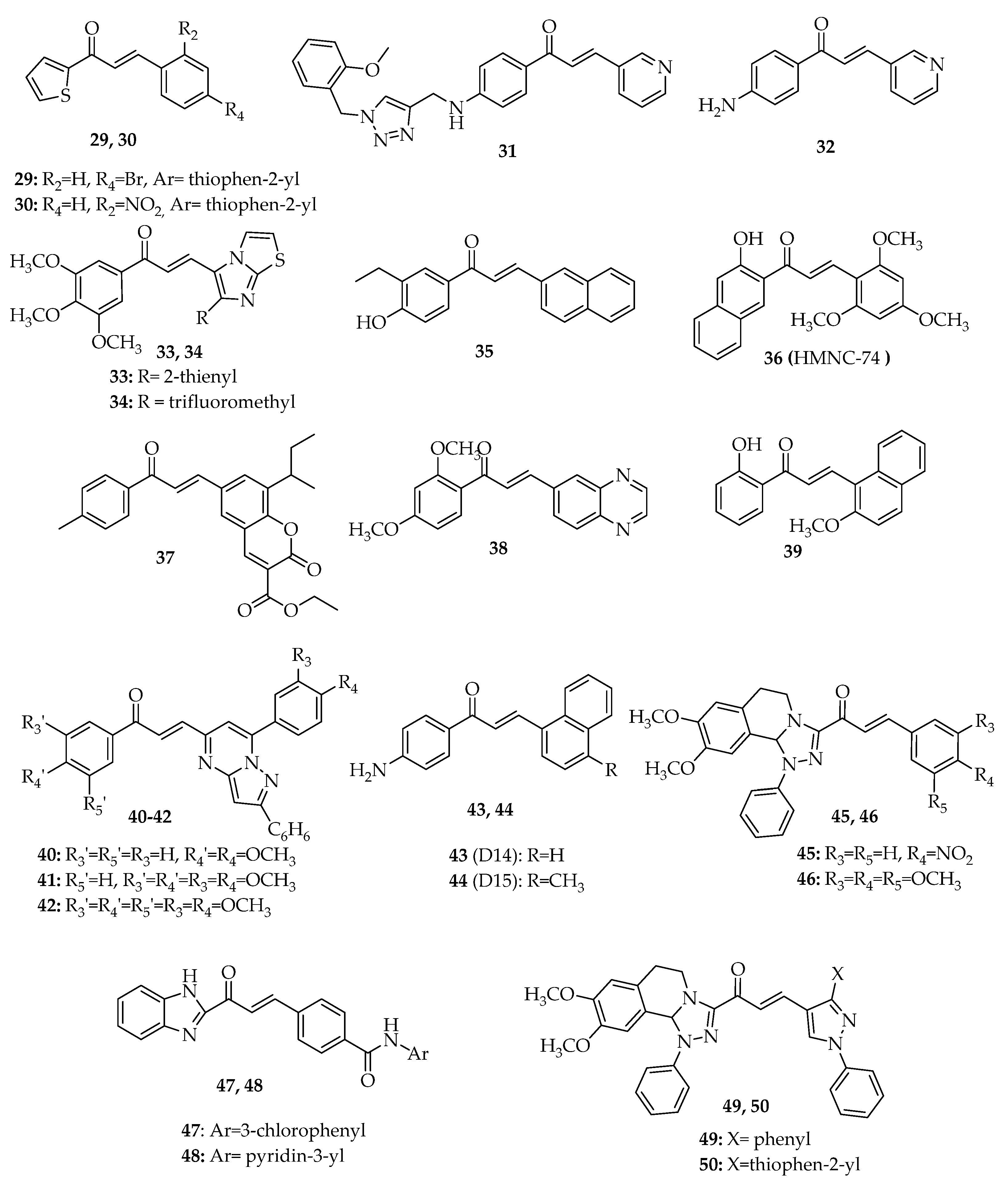
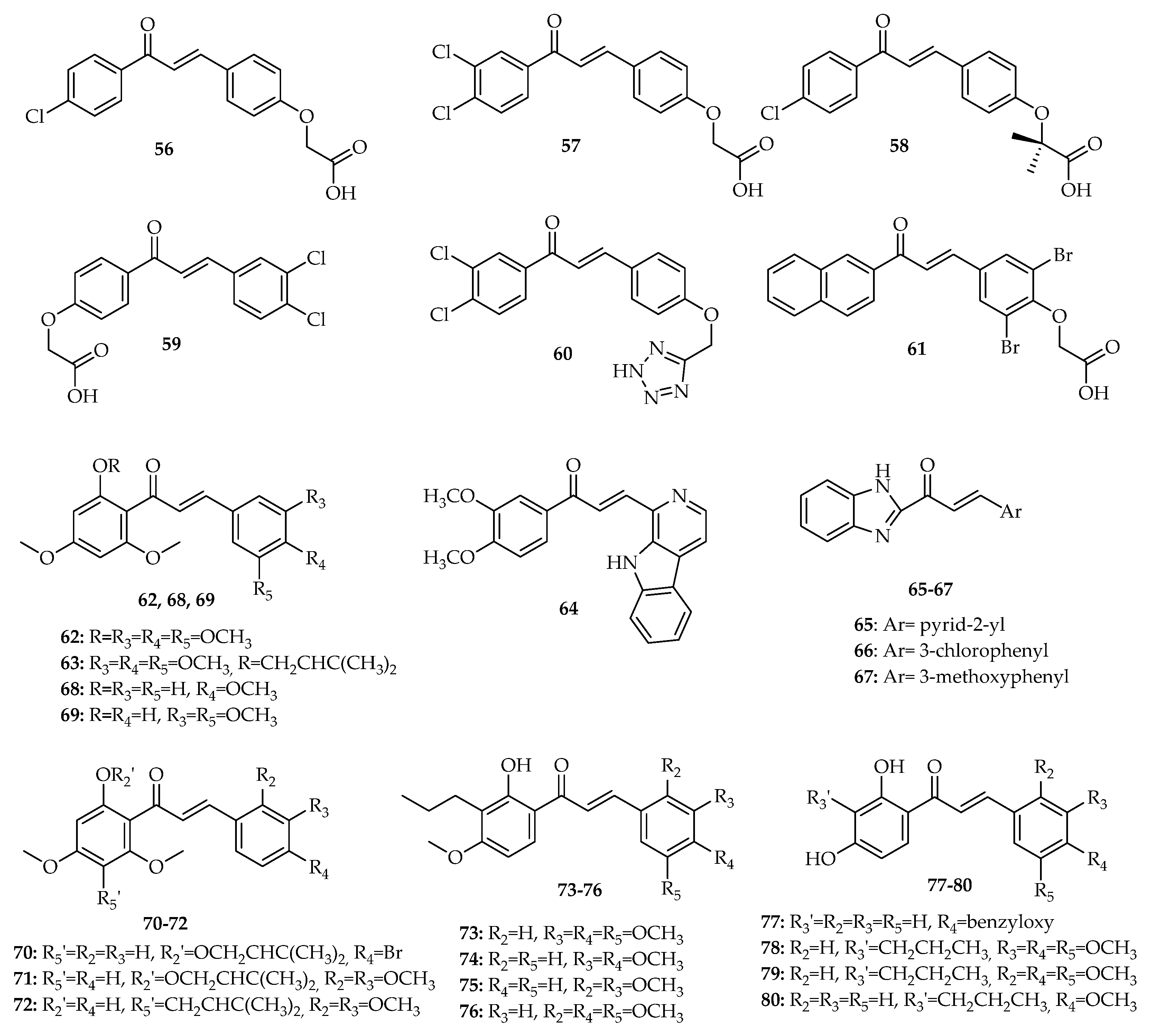
3. Interference with p53 Pathway by Synthetic Chalcones
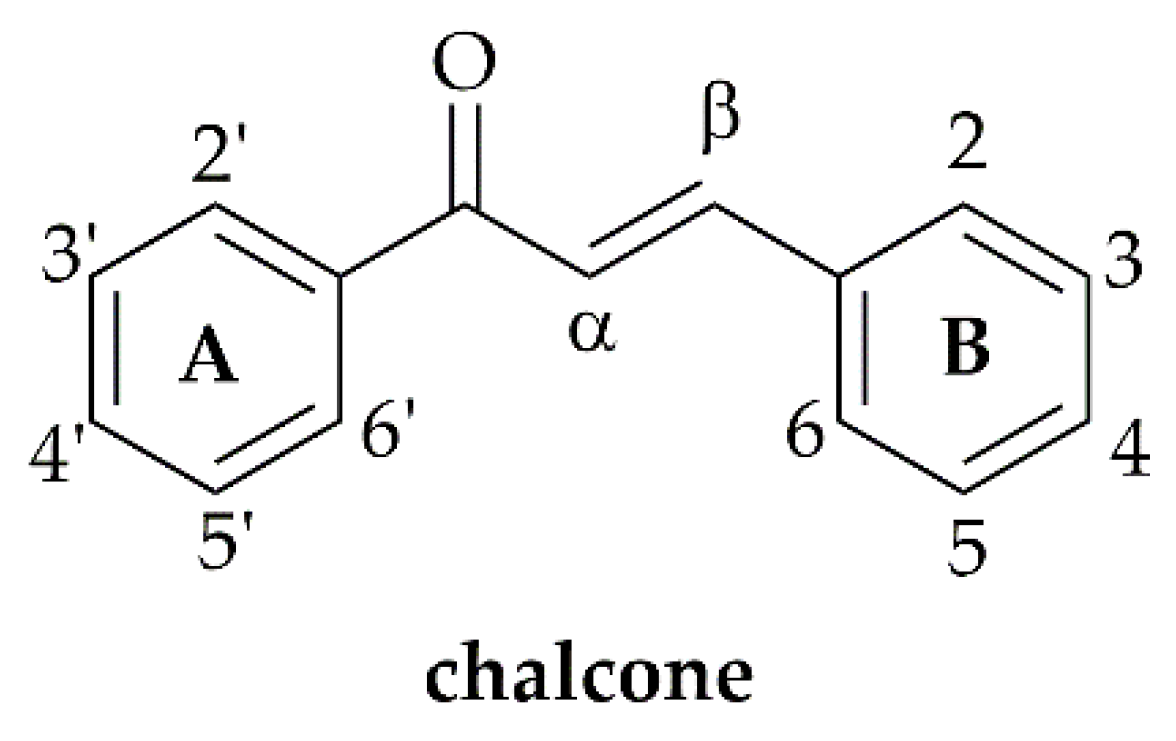
3.1. Chalcones with Phenyl Groups
3.1.1. α,β-Non-Substituted Chalcones
3.1.2. α-Substituted Chalcones
3.2. Chalcones with Other Aryl Groups
3.2.1. Chalcones with Simple Aryl Groups
3.2.2. Chalcones with Fused Aryl Groups
3.3. Chalcone Analogues
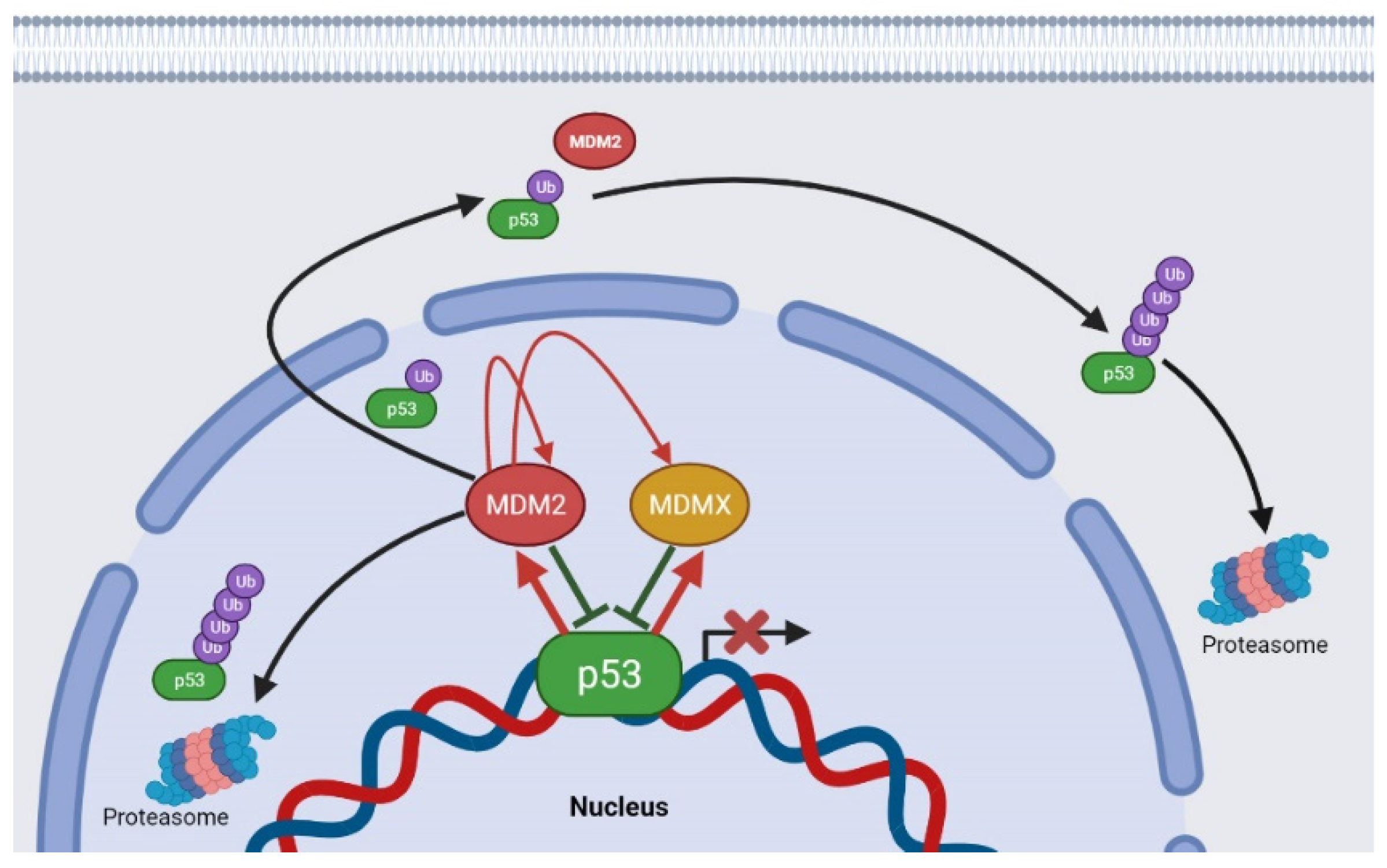
4. Chalcones as Disruptors of the p53–MDM2 Interaction
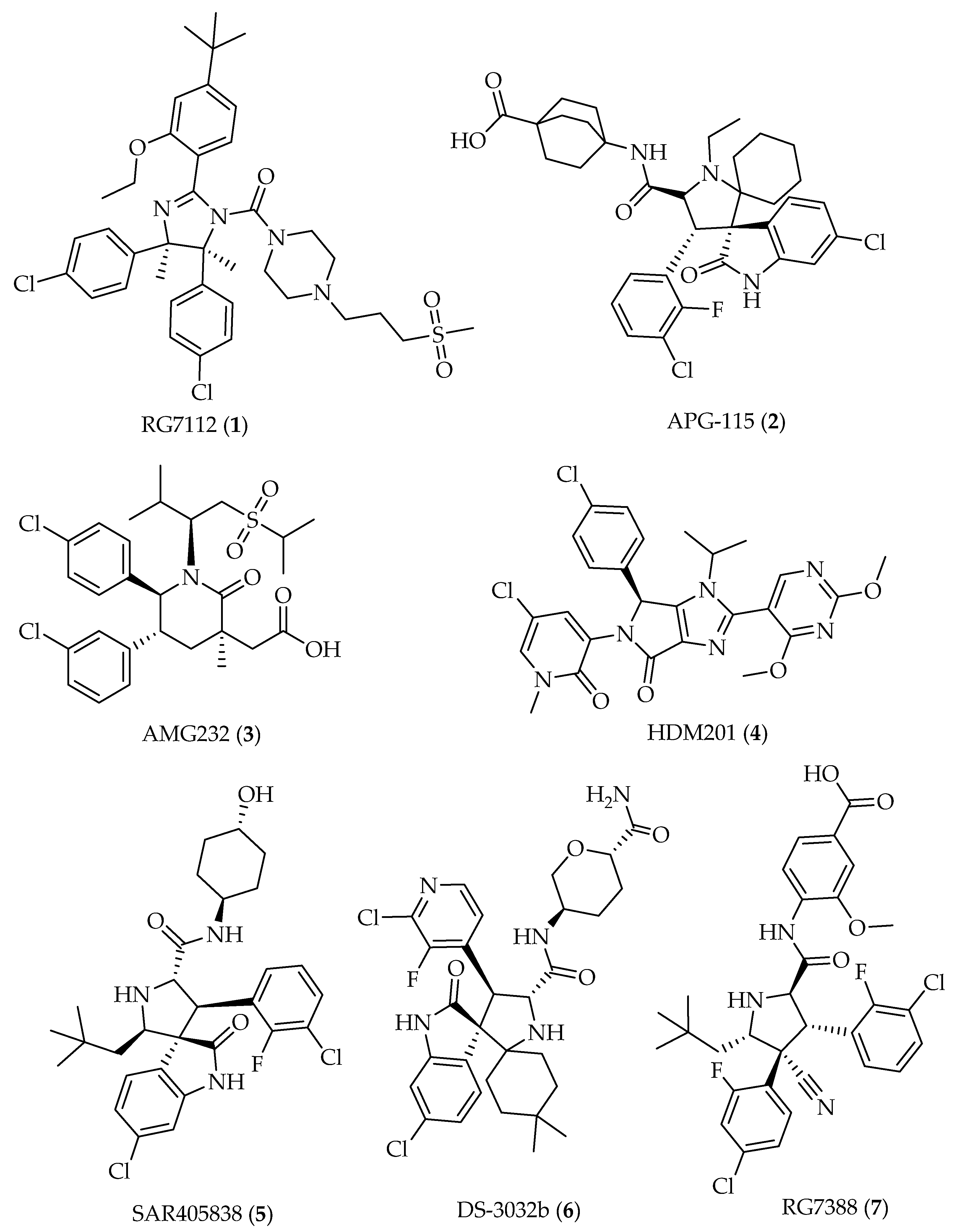
5. Prediction of Drug-likeness and Comparison with Reported Compounds Targeting MDM2 in Clinical Trials
5.1. Molecular Descriptors and Physicochemical Properties
5.2. Rule of Drug-Likeness
5.3. Pharmacokinetic Properties
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADME | Absorption, distribution, metabolism, excretion |
| AHA | Aromatic heavy atoms |
| Akt | Protein kinase B |
| Bak | Bcl-2 antagonist/Killer |
| Bax | Bcl-2-associated X protein |
| BBB | Blood–brain barrier |
| Bcl-2 | B cell lymphoma 2 |
| Bid | BH3-interacting domain death agonist |
| CNS | Central nervous system |
| Co-IP | Co-immunoprecipitation |
| DUB | Deubiquitinating |
| ELISA | Enzyme-linked immunosorbent assay |
| EMSA | Electrophoretic gel mobility shift assay |
| EMT | Epithelial–mesenchymal transition |
| Far | Fraction of aromatic heavy atoms |
| Fsp3 | Fraction of carbon sp3 |
| HA | Heavy atoms |
| HBA | Hydrogen bond acceptors |
| HBD | Hydrogen bond donors |
| IC50 | Half-maximal inhibitory concentration |
| MDM2 | Murine double minute 2 |
| MDMX | Murine double minute X |
| MW | Molecular weight |
| MW irradiation | Microwave irradiation |
| NF-kB | Nuclear factor kapa B |
| PARP | Poly ADP ribose polymerase |
| p-mTOR | Phosphorylated mTOR |
| PSA | Polar surface area |
| PTEN | Phosphatase and tensin homolog |
| PUMA | p53 upregulated modulator of apoptosis |
| mFasL | Membrane-bound Fas ligand |
| RB | Rotatable bonds |
| ROS | Reactive oxygen species |
| RT-PCR | Real-time polymerase chain reaction |
| SAR | Structure–activity relationship |
| wt | Wild-type |
References
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K. Therapeutic potential of chalcones as cardiovascular agents. Life Sci. 2016, 148, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Kontogiorgis, C.; Mantzanidou, M.; Hadjipavlou-Litina, D. Chalcones and their potential role in inflammation. Mini Rev. Med. Chem. 2008, 8, 1224–1242. [Google Scholar] [CrossRef] [PubMed]
- Boumendjel, A.; Ronot, X.; Boutonnat, J. Chalcones derivatives acting as cell cycle blockers: Potential anti-cancer drugs? Curr. Drug Targets 2009, 10, 363–371. [Google Scholar] [PubMed]
- Go, M.; Wu, X.; Liu, X. Chalcones: An update on cytotoxic and chemoprotective properties. Curr. Med. Chem. 2005, 12, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Chalcones and their therapeutic targets for the management of diabetes: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2015, 92, 839–865. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Chalcone scaffolds as anti-infective agents: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 101, 496–524. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Hong, B.; van den Heuvel, P.J.; Prabhu, V.; Zhang, S.; El-Deiry, W. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Sane, S.; Rezvani, K. Essential roles of E3 ubiquitin ligases in p53 regulation. Int. J. Mol. Sci. 2017, 18, 442. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.; Wu, L.; Levine, A. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [Green Version]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.-M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88. [Google Scholar]
- Liu, Y.; Wang, X.; Wang, G.; Yang, Y.; Yuan, Y.; Ouyang, L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 2019, 176, 92–104. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.-Y. Targeting the MDM2–p53 protein–protein interaction for new cancer therapy: Progress and challenges. Cold Spring Harb. Perspect Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D.; McEachern, D.; Przybranowski, S.; Li, X.; Luo, R.; Wen, B. Discovery of 4-((3′ R, 4′ S, 5′ R)-6 ″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2 ″-oxodispiro [cyclohexane-1, 2′-pyrrolidine-3′, 3 ″-indoline]-5′-carboxamido) bicyclo [2.2. 2] octane-1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J. Med. Chem. 2017, 60, 2819–2839. [Google Scholar]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2–p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment: Miniperspective. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.D.; Tang, Q.; Kong, Y.; Rong, T.; Wang, Q.; Li, N.; Fang, X.; Gu, J.; Xiong, D.; Yin, Y. MDM2 inhibitor APG-115 exerts potent antitumor activity and synergizes with standard-of-care agents in preclinical acute myeloid leukemia models. Cell Death Discov. 2021, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G. Dose and schedule determine distinct molecular mechanisms underlying the efficacy of the p53–MDM2 inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Blotner, S.; Chen, L.-C.; Ferlini, C.; Zhi, J. Phase 1 summary of plasma concentration–QTc analysis for idasanutlin, an MDM2 antagonist, in patients with advanced solid tumors and AML. Cancer Chemother. Pharmac. 2018, 81, 597–607. [Google Scholar] [CrossRef]
- Gounder, M.M.; Bauer, T.M.; Schwartz, G.K.; Masters, T.; Carvajal, R.D.; Song, S.; Kumar, P.; Gajee, R.; Zernovak, O.; Rosen, M.M.; et al. A phase 1 study of the MDM2 inhibitor DS-3032b in patients (pts) with advanced solid tumors and lymphomas. J. Clin. Oncol. 2016, 34 (Suppl. 15), 2581. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Kuo, P.L.; Chiang, L.C.; Lin, C.C. Isoliquiritigenin inhibits the proliferation and induces the apoptosis of human non-small cell lung cancer A549 cells. Clin. Exp. Pharmacol. Physiol. 2004, 31, 414–418. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Kuo, P.-L.; Lin, C.-C. Isoliquiritigenin induces apoptosis and cell cycle arrest through p53-dependent pathway in Hep G2 cells. Life Sci. 2005, 77, 279–292. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Chia, C.C.; Chen, P.J.; Huang, S.E.; Huang, S.C.; Kuo, P.L. Shallot and licorice constituent isoliquiritigenin arrests cell cycle progression and induces apoptosis through the induction of ATM/p53 and initiation of the mitochondrial system in human cervical carcinoma HeLa cells. Mol. Nutr. Food Res. 2009, 53, 826–835. [Google Scholar] [CrossRef]
- Kim, D.-H.; Park, J.E.; Chae, I.G.; Park, G.; Lee, S.; Chun, K.-S. Isoliquiritigenin inhibits the proliferation of human renal carcinoma Caki cells through the ROS-mediated regulation of the Jak2/STAT3 pathway. Oncol. Rep. 2017, 38, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Simoneau, A.R.; Xie, J.; Shahandeh, B.; Zi, X. Effects of the kava chalcone flavokawain A differ in bladder cancer cells with wild-type versus mutant p53. Cancer Prev. Res. 2008, 1, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, S.-C.; Hsu, C.-L.; Yu, Y.-S.; Yen, G.-C. Cytotoxic effects of new geranyl chalcone derivatives isolated from the leaves of Artocarpus communis in SW 872 human liposarcoma cells. J. Agric. Food. Chem. 2008, 56, 8859–8868. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.K.; Kao, T.-Y.; Ko, J.-L.; Tzeng, Y.-M. Chalcone HTMC causes in vitro selective cytotoxicity, cell-cycle G1 phase arrest through p53-dependent pathway in human lung adenocarcinoma A549 cells, and in vivo tumor growth suppression. Bioorg. Med. Chem. Lett. 2010, 20, 6508–6512. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Lin, W.-H.; Wang, S.-Y.; Chen, C.-S.; Liao, J.-W.; Chang, H.-W.; Chen, S.-C.; Lin, K.-Y.; Wang, L.; Yang, H.-L. Flavokawain B inhibits growth of human squamous carcinoma cells: Involvement of apoptosis and cell cycle dysregulation in vitro and in vivo. J. Nutr. Biochem. 2012, 23, 368–378. [Google Scholar] [CrossRef]
- Sławińska-Brych, A.; Zdzisińska, B.; Dmoszyńska-Graniczka, M.; Jeleniewicz, W.; Kurzepa, J.; Gagoś, M.; Stepulak, A. Xanthohumol inhibits the extracellular signal regulated kinase (ERK) signalling pathway and suppresses cell growth of lung adenocarcinoma cells. Toxicology 2016, 357, 65–73. [Google Scholar] [CrossRef]
- Silva, G.; Marins, M.; Fachin, A.L.; Lee, S.H.; Baek, S.J. Anti-cancer activity of trans-chalcone in osteosarcoma: Involvement of Sp1 and p53. Mol. Carcinog. 2016, 55, 1438–1448. [Google Scholar] [CrossRef]
- Silva, G.; Marins, M.; Chaichanasak, N.; Yoon, Y.; Fachin, A.L.; Pinhanelli, V.C.; Regasini, L.O.; dos Santos, M.B.; Ayusso, G.M.; de Carvalho Marques, B. Trans-chalcone increases p53 activity via DNAJB1/HSP40 induction and CRM1 inhibition. PLoS ONE 2018, 13, e0202263. [Google Scholar] [CrossRef]
- da Silva Siqueira, E.; Concato, V.M.; Tomiotto-Pellissier, F.; Silva, T.F.; da Silva Bortoleti, B.T.; Gonçalves, M.D.; Costa, I.N.; Junior, W.A.V.; Pavanelli, W.R.; Panis, C. Trans-chalcone induces death by autophagy mediated by p53 up-regulation and β-catenin down-regulation on human hepatocellular carcinoma HuH7. 5 cell line. Phytomedicine 2021, 80, 153373. [Google Scholar] [CrossRef]
- Lai, C.-K.; Rao, Y.K.; Chang, K.-R.; Lin, C.-W.; Su, H.-L.; Chang, C.-S.; Lai, C.-H.; Tzeng, Y.-M. 3, 3′, 4′, 5′-Tetramethoxychalcone inhibits human oral cancer cell proliferation and migration via p53-mediated mitochondrial-dependent apoptosis. Anticancer Res. 2014, 34, 1811–1819. [Google Scholar]
- Qi, Z.; Liu, M.; Liu, Y.; Zhang, M.; Yang, G. Tetramethoxychalcone, a chalcone derivative, suppresses proliferation, blocks cell cycle progression, and induces apoptosis of human ovarian cancer cells. PLoS ONE 2014, 9, e106206. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, Z.; Wen, J.; Ma, F.; Wang, F.; Yu, K.; Tang, M.; Wu, W.; Dong, Y.; Cheng, X. SKLB-M8 induces apoptosis through the AKT/mTOR signaling pathway in melanoma models and inhibits angiogenesis with decrease of ERK1/2 phosphorylation. J. Pharmacol. Sci. 2014, 126, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.-J.; Zhang, S.-Y.; Liu, Y.-C.; Zhang, L.; Liu, J.-J.; Song, J.; Zhao, R.-H.; Li, F.; Sun, H.-H.; Liu, H.-M. Design, synthesis and antiproliferative activity studies of novel dithiocarbamate–chalcone derivates. Bioorg. Med. Chem. Lett. 2016, 26, 3918–3922. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, S.; Khan, S.; Bilal, A.; Manzoor, S.; Abdullah, M.; Emwas, A.-H.; Sioud, S.; Gao, X.; Chotana, G.A.; Faisal, A. Synthesis and evaluation of modified chalcone based p53 stabilizing agents. Bioorg. Med. Chem. Lett. 2017, 27, 4101–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, B.L.S.; da Silva, A.C.G.; de Ávila, R.I.; Cortez, A.P.; Luzin, R.M.; Lião, L.M.; de Souza Gil, E.; Sanz, G.; Vaz, B.G.; Sabino, J.R. A novel chalcone derivative, LQFM064, induces breast cancer cells death via p53, p21, KIT and PDGFRA. Eur. J. Pharm. Sci. 2017, 107, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Wang, X.; Dong, L.; Sun, P. Curcumin derivative L6H4 inhibits proliferation and invasion of gastric cancer cell line BGC-823. J. Cell. Biochem. 2019, 120, 1011–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, M.B.; Bertholin Anselmo, D.; de Oliveira, J.G.; Jardim-Perassi, B.V.; Alves Monteiro, D.; Silva, G.; Gomes, E.; Lucia Fachin, A.; Marins, M.; de Campos Zuccari, D.A.P. Antiproliferative activity and p53 upregulation effects of chalcones on human breast cancer cells. J. Enzym. Inhib. Med. Chem. 2019, 34, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.S.; Hussein, R.A.; El-Sayed, W.M. Substitution at phenyl rings of chalcone and schiff base moieties accounts for their antiproliferative activity. Anticancer Agents Med. Chem. 2019, 19, 620–626. [Google Scholar] [CrossRef]
- Li, K.; Zhao, S.; Long, J.; Su, J.; Wu, L.; Tao, J.; Zhou, J.; Zhang, J.; Chen, X.; Peng, C. A novel chalcone derivative has antitumor activity in melanoma by inducing DNA damage through the upregulation of ROS products. Cancer Cell Int. 2020, 20, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Srinivasan, B.; Xing, C.; Lü, J. A new chalcone derivative (E)-3-(4-methoxyphenyl)-2-methyl-1-(3, 4, 5-trimethoxyphenyl) prop-2-en-1-one suppresses prostate cancer involving p53-mediated cell cycle arrests and apoptosis. Anticancer Res. 2012, 32, 3689–3698. [Google Scholar]
- Riaz, S.; Iqbal, M.; Ullah, R.; Zahra, R.; Chotana, G.A.; Faisal, A.; Saleem, R.S.Z. Synthesis and evaluation of novel α-substituted chalcones with potent anti-cancer activities and ability to overcome multidrug resistance. Bioorg. Chem. 2019, 87, 123–135. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcelos, A.; Campos, V.F.; Nedel, F.; Seixas, F.K.; Dellagostin, O.A.; Smith, K.R.; de Pereira, C.M.P.; Stefanello, F.M.; Collares, T.; Barschak, A.G. Cytotoxic and apoptotic effects of chalcone derivatives of 2-acetyl thiophene on human colon adenocarcinoma cells. Cell Biochem. Funct. 2013, 31, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-J.; Zhang, S.-Y.; Liu, Y.-C.; Yue, X.-X.; Liu, J.-J.; Song, J.; Zhao, R.-H.; Li, F.; Sun, H.-H.; Zhang, Y.-B. Design, synthesis and antiproliferative activity studies of 1, 2, 3-triazole–chalcones. MedChemComm 2016, 7, 1664–1671. [Google Scholar] [CrossRef]
- Arshad, L.; Haque, M.A.; Abbas Bukhari, S.N.; Jantan, I. An overview of structure–activity relationship studies of curcumin analogs as antioxidant and anti-inflammatory agents. Future Med. Chem. 2017, 9, 605–626. [Google Scholar] [CrossRef]
- Kamal, A.; Dastagiri, D.; Ramaiah, M.J.; Reddy, J.S.; Bharathi, E.V.; Srinivas, C.; Pushpavalli, S.; Pal, D.; Pal-Bhadra, M. Synthesis of imidazothiazole–chalcone derivatives as anticancer and apoptosis inducing agents. ChemMedChem 2010, 5, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, F.S.; Chiaradia, L.D.; Licínio, M.A.; De Moraes, A.C.R.; Curta, J.C.; Costa, A.; Mascarello, A.; Creczinsky-Pasa, T.B.; Nunes, R.J.; Yunes, R.A. Induction of apoptosis and cell cycle arrest in L-1210 murine lymphoblastic leukaemia cells by (2E)-3-(2-naphthyl)-1-(3′-methoxy-4′-hydroxy-phenyl)-2-propen-1-one. J. Pharm. Pharmacol. 2010, 62, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Yong, Y.; Lee, J.; Ahn, S.; Jung, K.-Y.; Koh, D.; Lee, Y.H.; Lim, Y. A novel hydroxymethoxynaphthochalcone induces apoptosis through the p53-dependent caspase-mediated pathway in HCT116 human colon cancer cells. J. Korean Soc. Appl. Biol. Chem. 2014, 57, 413–418. [Google Scholar] [CrossRef]
- Lee, J.M.; Lee, M.S.; Koh, D.; Lee, Y.H.; Lim, Y.; Shin, S.Y. A new synthetic 2′-hydroxy-2, 4, 6-trimethoxy-5′, 6′-naphthochalcone induces G2/M cell cycle arrest and apoptosis by disrupting the microtubular network of human colon cancer cells. Cancer Lett. 2014, 354, 348–354. [Google Scholar] [CrossRef]
- Singh, N.; Sarkar, J.; Sashidhara, K.V.; Ali, S.; Sinha, S. Anti-tumour activity of a novel coumarin–chalcone hybrid is mediated through intrinsic apoptotic pathway by inducing PUMA and altering Bax/Bcl-2 ratio. Apoptosis 2014, 19, 1017–1028. [Google Scholar] [CrossRef]
- Loch-Neckel, G.; Bicca, M.A.; Leal, P.C.; Mascarello, A.; Siqueira, J.M.; Calixto, J.B. In vitro and in vivo anti-glioma activity of a chalcone-quinoxaline hybrid. Eur. J. Med. Chem. 2015, 90, 93–100. [Google Scholar] [CrossRef]
- Shin, S.Y.; Ahn, S.; Koh, D.; Lim, Y. p53-dependent and-independent mechanisms are involved in (E)-1-(2-hydroxyphenyl)-3-(2-methoxynaphthalen-1-yl) prop-2-en-1-one (HMP)-induced apoptosis in HCT116 colon cancer cells. Biochem. Biophys. Res. Commun. 2016, 479, 913–919. [Google Scholar] [CrossRef]
- Bagul, C.; Rao, G.K.; Makani, V.K.K.; Tamboli, J.R.; Pal-Bhadra, M.; Kamal, A. Synthesis and biological evaluation of chalcone-linked pyrazolo [1, 5-a] pyrimidines as potential anticancer agents. MedChemComm 2017, 8, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Seba, V.; Silva, G.; Santos, M.; Baek, S.; França, S.; Fachin, A.; Regasini, L.; Marins, M. Chalcone Derivatives 4′-Amino-1-Naphthyl-Chalcone (D14) and 4′-Amino-4-Methyl-1-Naphthyl-Chalcone (D15) Suppress Migration and Invasion of Osteosarcoma Cells Mediated by p53 Regulating EMT-Related Genes. Int. J. Mol. Sci. 2018, 19, 2838. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.F.; Hassaneen, H.M.; Abdelhamid, I.A. Cytotoxicity, molecular modeling, cell cycle arrest, and apoptotic induction induced by novel tetrahydro-[1,2,4] triazolo [3,4-a] isoquinoline chalcones. Eur. J. Med. Chem. 2018, 143, 532–541. [Google Scholar] [CrossRef]
- Wu, L.; Yang, Y.; Wang, Z.; Wu, X.; Su, F.; Li, M.; Jing, X.; Han, C. Design, Synthesis, and Biological Evaluation of Aromatic Amide-Substituted Benzimidazole-Derived Chalcones. The Effect of Upregulating TP53 Protein Expression. Molecules 2020, 25, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.F.; Sroor, F.M.; Ibrahim, N.S.; Salem, G.S.; El-Sayed, H.H.; Mahmoud, M.M.; Wagdy, M.-A.M.; Ahmed, A.M.; Mahmoud, A.-A.T.; Ibrahim, S.S. Novel [l, 2, 4] triazolo [3, 4-a] isoquinoline chalcones as new chemotherapeutic agents: Block IAP tyrosine kinase domain and induce both intrinsic and extrinsic pathways of apoptosis. Investig. New Drugs 2021, 39, 98–110. [Google Scholar] [CrossRef]
- Modzelewska, A.; Pettit, C.; Achanta, G.; Davidson, N.E.; Huang, P.; Khan, S.R. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg. Med. Chem. 2006, 14, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Achanta, G.; Modzelewska, A.; Feng, L.; Khan, S.R.; Huang, P. A boronic-chalcone derivative exhibits potent anticancer activity through inhibition of the proteasome. Mol. Pharmacol. 2006, 70, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Issaenko, O.A.; Amerik, A.Y. Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle 2012, 11, 1804–1817. [Google Scholar] [CrossRef] [Green Version]
- Selvendiran, K.; Tong, L.; Vishwanath, S.; Bratasz, A.; Trigg, N.J.; Kutala, V.K.; Hideg, K.; Kuppusamy, P. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J. Biol. Chem. 2007, 282, 28609–28618. [Google Scholar] [CrossRef] [Green Version]
- Selvendiran, K.; Tong, L.; Bratasz, A.; Kuppusamy, M.L.; Ahmed, S.; Ravi, Y.; Trigg, N.J.; Rivera, B.K.; Kálai, T.; Hideg, K. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol. Cancer Ther. 2010, 9, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Anchoori, R.K.; Khan, S.R.; Sueblinvong, T.; Felthauser, A.; Iizuka, Y.; Gavioli, R.; Destro, F.; Vogel, R.I.; Peng, S.; Roden, R.B. Stressing the ubiquitin-proteasome system without 20S proteolytic inhibition selectively kills cervical cancer cells. PLoS ONE 2011, 6, e23888. [Google Scholar] [CrossRef]
- Ma, Y.-C.; Wang, Z.-X.; Jin, S.-J.; Zhang, Y.-X.; Hu, G.-Q.; Cui, D.-T.; Wang, J.-S.; Wang, M.; Wang, F.-Q.; Zhao, Z.-J. Dual Inhibition of Topoisomerase II and Tyrosine Kinases by the Novel Bis-Fluoroquinolone Chalcone-Like Derivative HMNE3 in Human Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0162821. [Google Scholar] [CrossRef] [Green Version]
- Stoll, R.; Renner, C.; Hansen, S.; Palme, S.; Klein, C.; Belling, A.; Zeslawski, W.; Kamionka, M.; Rehm, T.; Mühlhahn, P. Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry 2001, 40, 336–344. [Google Scholar] [CrossRef]
- Leão, M.; Soares, J.; Gomes, S.; Raimundo, L.; Ramos, H.; Bessa, C.; Queiroz, G.; Domingos, S.; Pinto, M.; Inga, A. Enhanced cytotoxicity of prenylated chalcone against tumour cells via disruption of the p53–MDM2 interaction. Life Sci. 2015, 142, 60–65. [Google Scholar] [CrossRef]
- Singh, A.K.; Chauhan, S.S.; Singh, S.K.; Verma, V.V.; Singh, A.; Arya, R.K.; Maheshwari, S.; Akhtar, M.S.; Sarkar, J.; Rangnekar, V.M. Dual targeting of MDM2 with a novel small-molecule inhibitor overcomes TRAIL resistance in cancer. Carcinogenesis 2016, 37, 1027–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.-T.; Jiang, Z.; Shen, J.-J.; Yi, H.; Zhan, Y.-C.; Sha, M.-Q.; Wang, Z.; Xue, S.-T.; Li, Z.-R. Design, synthesis and biological evaluation of novel benzimidazole-2-substituted phenyl or pyridine propyl ketene derivatives as antitumour agents. Eur. J. Med. Chem. 2016, 114, 328–336. [Google Scholar] [CrossRef]
- Brandão, P.; Loureiro, J.B.; Carvalho, S.; Hamadou, M.H.; Cravo, S.; Moreira, J.; Pereira, D.; Palmeira, A.; Pinto, M.; Saraiva, L. Targeting the MDM2-p53 protein-protein interaction with prenylchalcones: Synthesis of a small library and evaluation of potential antitumor activity. Eur. J. Med. Chem. 2018, 156, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.; Lima, R.T.; Palmeira, A.; Seca, H.; Soares, J.; Gomes, S.; Raimundo, L.; Maciel, C.; Pinto, M.; Sousa, E. Design and synthesis of new inhibitors of p53–MDM2 interaction with a chalcone scaffold. Arab. J. Chem. 2019, 12, 4150–4161. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y. Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill, K.L.J.; Garnett, J.; Meaux, I.; Ma, X.; Creighton, C.J.; Bolshakov, S.; Barriere, C.; Debussche, L.; Lazar, A.J.; Prudner, B.C. SAR405838: A novel and potent inhibitor of the MDM2: p53 axis for the treatment of dedifferentiated liposarcoma. Clin. Cancer Res. 2016, 22, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Nakamaru, K.; Seki, T.; Tazaki, K.; Tse, A. Abstract B5: Preclinical characterization of a novel orally-available MDM2 inhibitor DS-3032b: Anti-tumor profile and predictive biomarkers for sensitivity. In Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Boston, MA, USA, 5–9 November 2015. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bade, R.; Chan, H.-F.; Reynisson, J. Characteristics of known drug space. Natural products, their derivatives and synthetic drugs. Eur. J. Med. Chem. 2010, 45, 5646–5652. [Google Scholar] [CrossRef]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharmacol. Sci. 1999, 88, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Anwar-Mohamed, A.; El-Kadi, A. P-glycoprotein effects on drugs pharmacokinetics and drug-drug-interactions and their clinical implications. Libyan J. Pharm. Clin. Pharmacol. 2012, 1, 48154. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, J.; Almeida, J.; Saraiva, L.; Cidade, H.; Pinto, M. Chalcones as Promising Antitumor Agents by Targeting the p53 Pathway: An Overview and New Insights in Drug-Likeness. Molecules 2021, 26, 3737. https://doi.org/10.3390/molecules26123737
Moreira J, Almeida J, Saraiva L, Cidade H, Pinto M. Chalcones as Promising Antitumor Agents by Targeting the p53 Pathway: An Overview and New Insights in Drug-Likeness. Molecules. 2021; 26(12):3737. https://doi.org/10.3390/molecules26123737
Chicago/Turabian StyleMoreira, Joana, Joana Almeida, Lucília Saraiva, Honorina Cidade, and Madalena Pinto. 2021. "Chalcones as Promising Antitumor Agents by Targeting the p53 Pathway: An Overview and New Insights in Drug-Likeness" Molecules 26, no. 12: 3737. https://doi.org/10.3390/molecules26123737