
Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors

 ,
,  and
and
Abstract
:1. Introduction
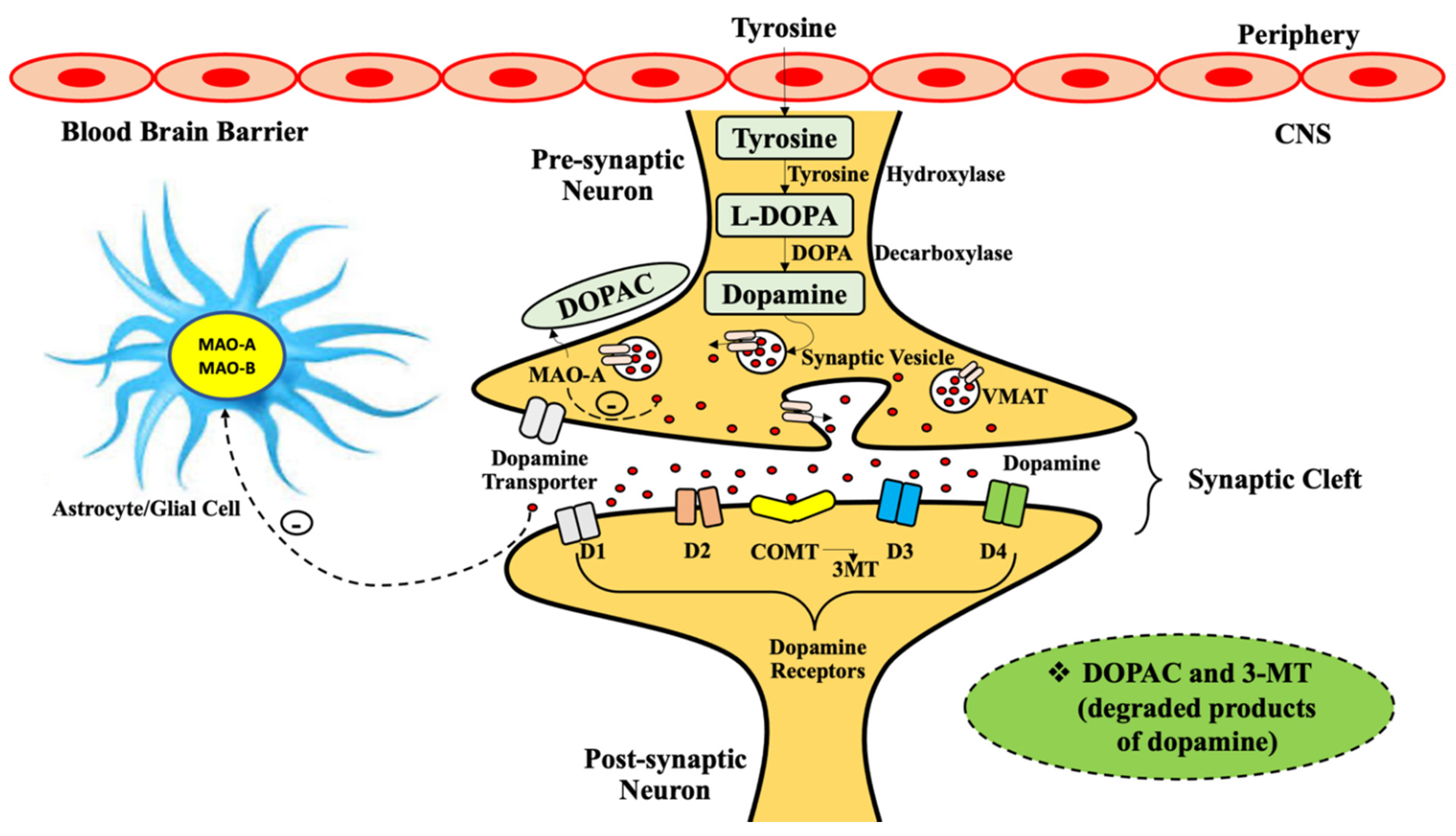
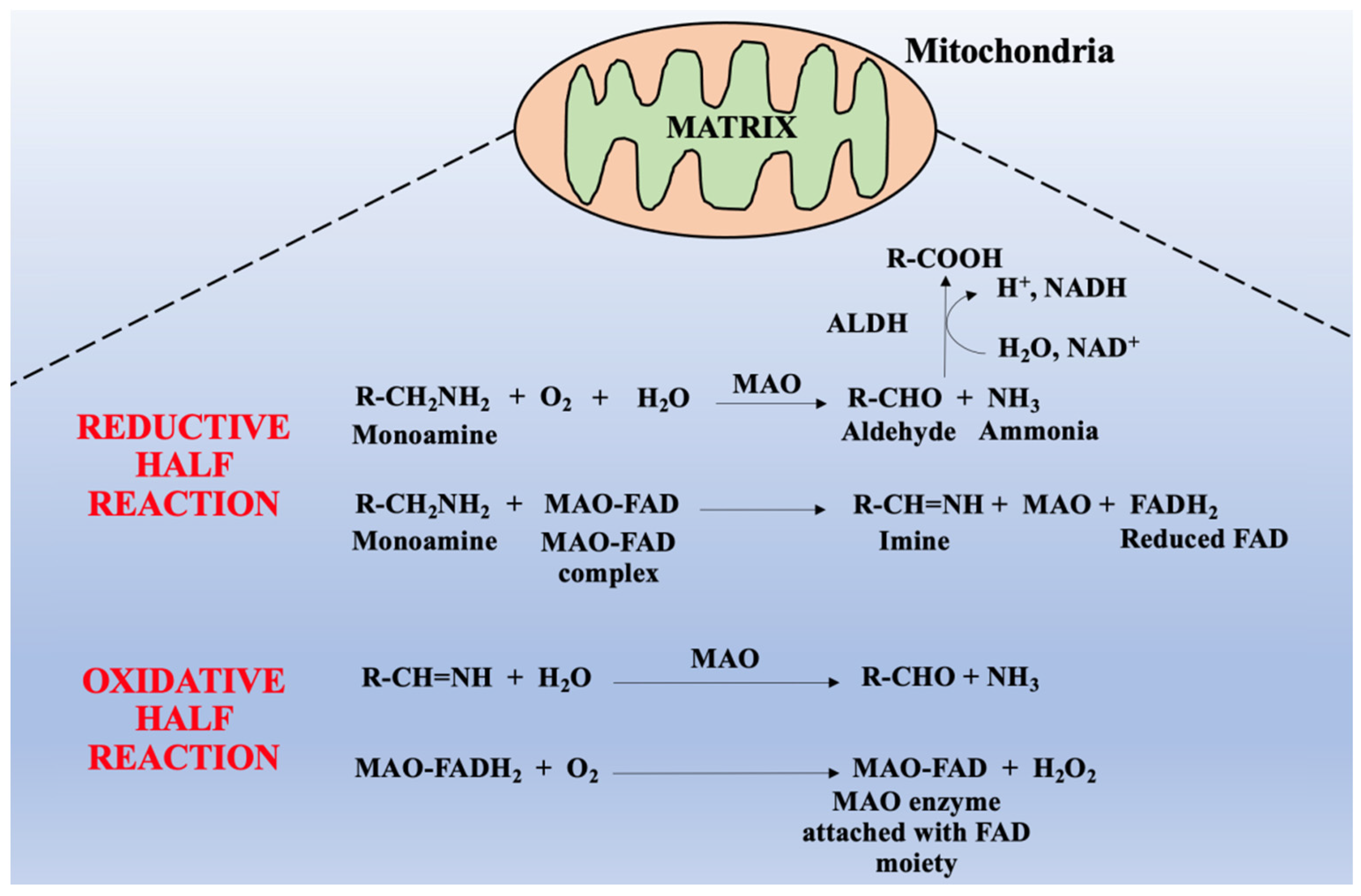
2. General Physiology of Monoamine Oxidases
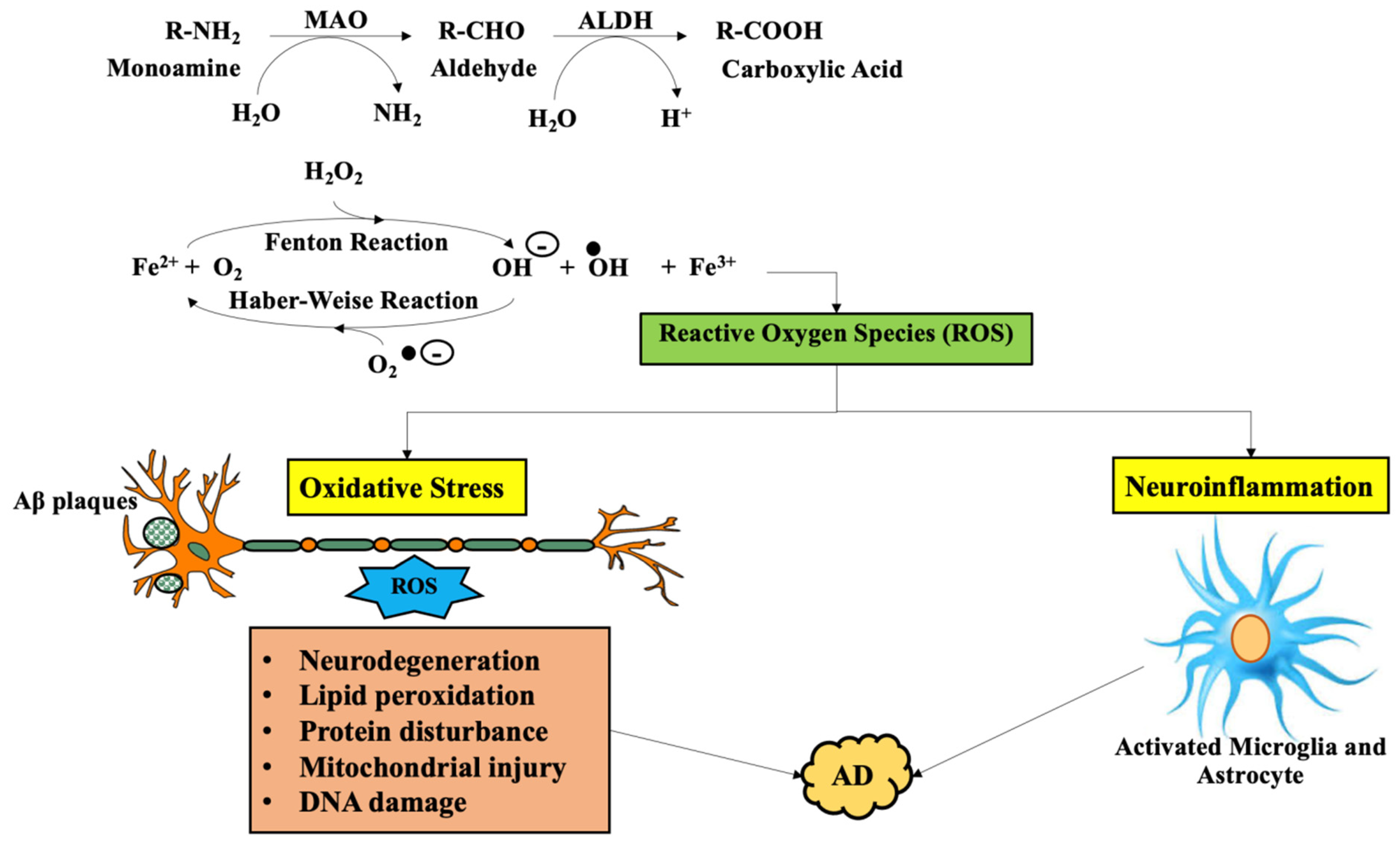
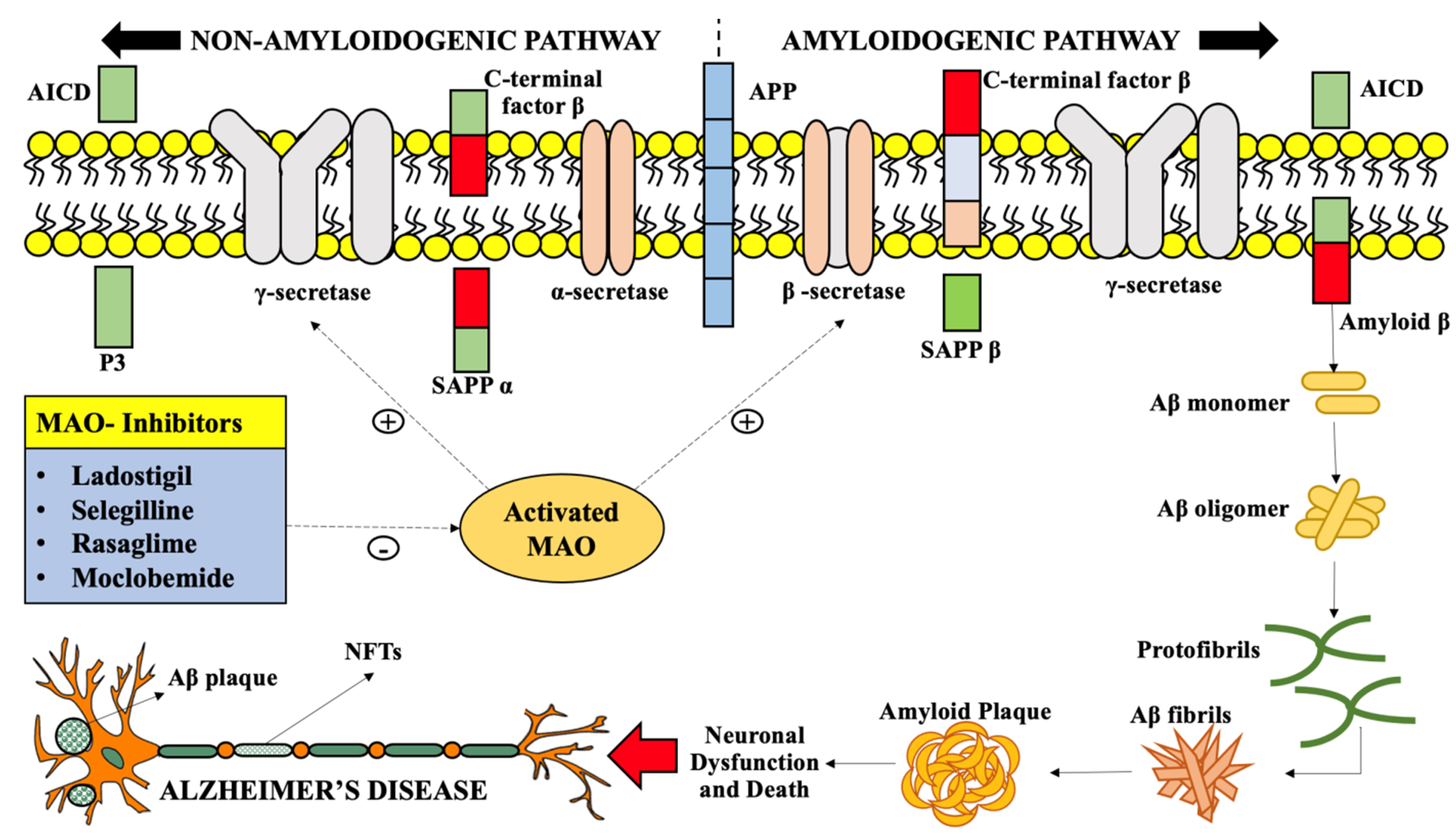
3. Involvement of Monoamine Oxidase in Alzheimer’s Disease
4. Inhibition of MAO-A and MAO-B as a Therapeutic Approach in Alzheimer’s Disease Treatment
4.1. Sequestration of Reactive Aldehydes
4.2. Incline in GABA Levels
4.3. Inhibition of Primary Amine Oxidase
4.4. Miscellaneous Neuroprotective Actions of Monoamine Oxidase Inhibitors
5. Therapeutic Potential of Monoamine Oxidase Inhibitors in Multiple Neurodegenerative Diseases
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332–384. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report; Alzheimer’s Disease International: London, UK, 2013. [Google Scholar]
- Wyss-Coray, T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat. Med. 2006, 12, 1005–1015. [Google Scholar] [PubMed]
- Avila-Muñoz, E.; Arias, C. When astrocytes become harmful: Functional and inflammatory responses that contribute to Alzheimer’s disease. Ageing Res. Rev. 2014, 18, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meraz Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.; Kesler, N. Monoamine oxidase and mitochondrial respiration. J. Neurochem. 1999, 73, 2310–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease. Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, T. Monoamine oxidase-B inhibitors in the treatment of Alzheimers disease. Neurobiol. Aging 2000, 21, 343–348. [Google Scholar] [CrossRef]
- Youdim, M.; Riederer, P. The relevance of glial monoamine oxidase-B and polyamines to the action of selegiline in parkinson’s disease. Mov. Disord. 1993, 8, S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, L.; Saetre, P.; Balciuniene, J.; Castensson, A.; Cairns, N.; Jazin, E.E. Increased monoamine oxidase messenger RNA expression levels in frontal cortex of Alzheimer’s disease patients. Neurosci. Lett. 2002, 326, 56–60. [Google Scholar] [CrossRef]
- Gulyás, B.; Pavlova, E.; Kása, P.; Gulya, K.; Bakota, L.; Várszegi, S.; Keller, É.; Horváth, M.C.; Nag, S.; Hermecz, I. Activated MAO-B in the brain of Alzheimer patients, demonstrated by (1C)-L-deprenyl using whole hemisphere autoradiography. Neurochem. Int. 2011, 58, 60–68. [Google Scholar] [CrossRef]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219. [Google Scholar]
- Carter, S.F.; Schöll, M.; Almkvist, O.; Wall, A.; Engler, H.; Långström, B.; Nordberg, A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [Green Version]
- Magni, G.; Meibach, R. Lazabemide for the long-term treatment of Alzheimer’s disease. Eur. Neuropsychopharmacol. 1999, 9, 142. [Google Scholar] [CrossRef]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem. 2018, 145, 445–497. [Google Scholar] [CrossRef]
- Ramsay, R.R. Monoamine oxidases: The biochemistry of the proteins as targets in medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2012, 12, 2189–2209. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Hirata, M.; Kagawa, S.; Magata, Y.; Ohmomo, Y.; Temma, T. Synthesis and characterization of novel radiofluorinated probes for positron emission tomography imaging of monoamine oxidase B. J. Label. Compd. Radiopharm. 2019, 62, 580–587. [Google Scholar] [CrossRef]
- McDonald, G.R.; Olivieri, A.; Ramsay, R.R.; Holt, A. On the formation and nature of the imidazoline I2 binding site on human monoamine oxidase-B. Pharmacol. Res. 2010, 62, 475–488. [Google Scholar] [CrossRef]
- Edmondson, E.D. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: Biological implications. Curr. Pharm. Des. 2014, 20, 155–160. [Google Scholar] [CrossRef]
- Fitzpatrick, P.F. Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 2010, 493, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Parada, M.; Fierro, A.; Iturriaga-Vasquez, P.; Cassels, B.K. Monoamine oxidase inhibition in the light of new structural data. Curr. Enzym. Inhib. 2005, 1, 85–95. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Mantha, A.K.; Kumar, V. Recent developments on the structure-activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016, 6, 42660–42683. [Google Scholar] [CrossRef]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [Green Version]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-Dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. USA 2005, 102, 12684–12689. [Google Scholar] [CrossRef] [Green Version]
- Hong, R.; Li, X. Discovery of monoamine oxidase inhibitors by medicinal chemistry approaches. MedChemComm 2019, 10, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Barner, E.L.; Gray, S.L. Donepezil use in Alzheimer disease. Ann. Pharmacother. 1998, 32, 70–77. [Google Scholar] [CrossRef]
- Kennedy, B.; Ziegler, M.; Alford, M.; Hansen, L.; Thal, L.; Masliah, E. Early and persistent alterations in prefrontal cortex MAO A and B in Alzheimer’s disease. J. Neural Transm. 2003, 110, 789–801. [Google Scholar] [CrossRef]
- Burke, W.J.; Li, S.W.; Schmitt, C.A.; Xia, P.; Chung, H.D.; Gillespie, K.N. Accumulation of 3, 4-dihydroxyphenylglycolaldehyde, the neurotoxic monoamine oxidase A metabolite of norepinephrine, in locus ceruleus cell bodies in Alzheimer’s disease: Mechanism of neuron death. Brain Res. 1999, 816, 633–637. [Google Scholar] [CrossRef]
- Chan-Palay, V.; Höchli, M.; Savaskan, E.; Hungerecker, G. Calbindin D-28k and monoamine oxidase A immunoreactive neurons in the nucleus basalis of Meynert in senile dementia of the Alzheimer type and Parkinson’s disease. Dement. Geriatr. Cogn. Disord. 1993, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Danilova, R.; Moskvityna, T.; Obukhova, M.; Belopolskaya, M.; Ashmarin, I. Pargyline conjugate-induced long-term activation of monoamine oxidase as an immunological model for depression. Neurochem. Res. 1999, 24, 1147–1151. [Google Scholar] [CrossRef]
- Kumagae, Y.; Matsui, Y.; Iwata, N. Deamination of norepinephrine, dopamine, and serotonin by type A monoamine oxidase in discrete regions of the rat brain and inhibition by RS-8359. Jpn. J. Pharmacol. 1991, 55, 121–128. [Google Scholar] [CrossRef]
- Moret, C.; Briley, M. The importance of norepinephrine in depression. Neuropsychiatr. Dis. Treat. 2011, 7, 9. [Google Scholar]
- Grailhe, R.; Cardona, A.; Even, N.; Seif, I.; Changeux, J.-P.; Cloëz-Tayarani, I. Regional changes in the cholinergic system in mice lacking monoamine oxidase A. Brain Res. Bull. 2009, 78, 283–289. [Google Scholar] [CrossRef]
- Schneier, F.R. Pharmacotherapy of social anxiety disorder. Expert Opin. Pharm. 2011, 12, 615–625. [Google Scholar] [CrossRef]
- Dhull, D.K.; Jindal, A.; Dhull, R.K.; Aggarwal, S.; Bhateja, D.; Padi, S.S. Neuroprotective effect of cyclooxygenase inhibitors in ICV-STZ induced sporadic Alzheimer’s disease in rats. J. Mol. Neurosci. 2012, 46, 223–235. [Google Scholar] [CrossRef]
- Muck-Seler, D.; Presecki, P.; Mimica, N.; Mustapic, M.; Pivac, N.; Babic, A.; Nedic, G.; Folnegovic-Smalc, V. Platelet serotonin concentration and monoamine oxidase type B activity in female patients in early, middle and late phase of Alzheimer’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2009, 33, 1226–1231. [Google Scholar] [CrossRef] [Green Version]
- Bar-Am, O.; Amit, T.; Weinreb, O.; Youdim, M.B.; Mandel, S. Propargylamine containing compounds as modulators of proteolytic cleavage of amyloid protein precursor: Involvement of MAPK and PKC activation. J. Alzheimer’s Dis. 2010, 21, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Cochet, M.; Donneger, R.; Cassier, E.; Gaven, F.; Lichtenthaler, S.F.; Marin, P.; Bockaert, J.; Dumuis, A.; Claeysen, S. 5-HT4 receptors constitutively promote the non-amyloidogenic pathway of APP cleavage and interact with ADAM10. ACS Chem. Neurosci. 2013, 4, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Baik, S.H.; Kang, S.; Cho, S.W.; Bae, J.; Cha, M.-Y.; Sailor, M.J.; Mook-Jung, I.; Ahn, K.H. Close correlation of monoamine oxidase activity with progress of Alzheimer’s disease in mice, observed by in vivo two-photon imaging. ACS Cent. Sci. 2016, 2, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglial activation and neuroinflammation in Alzheimer’s disease: A critical examination of recent history. Front. Aging Neurosci. 2010, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- Saura, J.; Luque, J.; Cesura, A.; Da Prada, M.; Chan-Palay, V.; Huber, G.; Löffler, J.; Richards, J. Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience 1994, 62, 15–30. [Google Scholar] [CrossRef]
- Jossan, S.; Gillberg, P.; Gottfries, C.; Karlsson, I.; Oreland, L. Monoamine oxidase B in brains from patients with Alzheimer’s disease: A biochemical and autoradiographical study. Neuroscience 1991, 45, 1–12. [Google Scholar] [CrossRef]
- Novaroli, L.; Reist, M.; Favre, E.; Carotti, A.; Catto, M.; Carrupt, P.-A. Human recombinant monoamine oxidase B as reliable and efficient enzyme source for inhibitor screening. Bioorg. Med. Chem. 2005, 13, 6212–6217. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Inoue, M.; Hromadkova, L.; Teranishi, Y.; Yamamoto, N.G.; Wiehager, B.; Bogdanovic, N.; Winblad, B.; Sandebring-Matton, A.; Frykman, S. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimers Res. Ther. 2017, 9, 1–19. [Google Scholar] [CrossRef]
- Quartey, M.O.; Nyarko, J.N.; Pennington, P.R.; Heistad, R.M.; Klassen, P.C.; Baker, G.B.; Mousseau, D.D. Alzheimer disease and selected risk factors disrupt a co-regulation of monoamine oxidase-A/B in the hippocampus, but not in the cortex. Front. Neurosci. 2018, 12, 419. [Google Scholar] [CrossRef]
- Park, J.-H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef] [Green Version]
- Borroni, E.; Bohrmann, B.; Grueninger, F.; Prinssen, E.; Nave, S.; Loetscher, H.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Siddiqui, A. Sembragiline: A novel, selective monoamine oxidase type B inhibitor for the treatment of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2017, 362, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Lam, R.W.; Milev, R.; Rotzinger, S.; Andreazza, A.C.; Blier, P.; Brenner, C.; Daskalakis, Z.J.; Dharsee, M.; Downar, J.; Evans, K.R. Discovering biomarkers for antidepressant response: Protocol from the Canadian biomarker integration network in depression (CAN-BIND) and clinical characteristics of the first patient cohort. BMC Psychiatry 2016, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Marchitti, S.A.; Deitrich, R.A.; Vasiliou, V. Neurotoxicity and metabolism of the catecholamine-derived 3, 4-dihydroxyphenylacetaldehyde and 3, 4-dihydroxyphenylglycolaldehyde: The role of aldehyde dehydrogenase. Pharmacol. Rev. 2007, 59, 125–150. [Google Scholar] [CrossRef]
- Cagle, B.S.; Crawford, R.A.; Doorn, J.A. Biogenic aldehyde-mediated mechanisms of toxicity in neurodegenerative disease. Curr. Opin. Toxicol. 2019, 13, 16–21. [Google Scholar] [CrossRef]
- Tábi, T.; Vécsei, L.; Youdim, M.B.; Riederer, P.; Szökő, É. Selegiline: A molecule with innovative potential. J. Neural Transm. 2020, 127, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Riederer, P.; Danielczyk, W.; Grünblatt, E. Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxicology 2004, 25, 271–277. [Google Scholar] [CrossRef]
- Zheng, H.; Fridkin, M.; Youdim, M.B. Site-Activated chelators derived from anti-Parkinson drug rasagiline as a potential safer and more effective approach to the treatment of Alzheimer’s disease. Neurochem. Res. 2010, 35, 2117–2123. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Bar-Am, O.; Amit, T. Iron-Chelating backbone coupled with monoamine oxidase inhibitory moiety as novel pluripotential therapeutic agents for Alzheimer’s disease: A tribute to Moussa Youdim. J. Neural Transm. 2011, 118, 479–492. [Google Scholar] [CrossRef]
- Wang, L.; Esteban, G.; Ojima, M.; Bautista-Aguilera, O.M.; Inokuchi, T.; Moraleda, I.; Iriepa, I.; Samadi, A.; Youdim, M.B.; Romero, A. Donepezil+ propargylamine+ 8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 80, 543–561. [Google Scholar] [CrossRef] [Green Version]
- Wood, P.L. Neurodegeneration and aldehyde load: From concept to therapeutics. J. Psychiatry Neurosci. JPN 2006, 31, 296. [Google Scholar]
- Thanh Nam, D.; Arseneault, M.; Murthy, V.; Ramassamy, C. Potential role of acrolein in neurodegeneration and in Alzheimer’s disease. Curr. Mol. Pharmacol. 2010, 3, 66–78. [Google Scholar] [CrossRef]
- Romano, A.; Serviddio, G.; Calcagnini, S.; Villani, R.; Giudetti, A.M.; Cassano, T.; Gaetani, S. Linking lipid peroxidation and neuropsychiatric disorders: Focus on 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 2017, 111, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Siraki, A.G.; Shangari, N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005, 35, 609–662. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Daim, M.M.; Abo-EL-Sooud, K.; Aleya, L.; Bungău, S.G.; Najda, A.; Saluja, R. Alleviation of drugs and chemicals toxicity: Biomedical value of antioxidants. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Wood, J.A. Thiol metabolism in schizophrenia: Current status. Curr. Psychiatry Rev. 2013, 9, 136–147. [Google Scholar] [CrossRef]
- Gustaw-Rothenberg, K.; Kowalczuk, K.; Stryjecka-Zimmer, M. Lipids’ peroxidation markers in Alzheimer’s disease and vascular dementia. Geriatr. Gerontol. Int. 2010, 10, 161–166. [Google Scholar] [CrossRef]
- Sinem, F.; Dildar, K.; Gokhan, E.; Melda, B.; Orhan, Y.; Filiz, M. The serum protein and lipid oxidation marker levels in Alzheimer’s disease and effects of cholinesterase inhibitors and antipsychotic drugs therapy. Curr. Alzheimer Res. 2010, 7, 463–469. [Google Scholar] [CrossRef]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular mechanisms of acrolein toxicity: Relevance to human disease. Toxicol. Sci. 2015, 143, 242–255. [Google Scholar] [CrossRef]
- Khoramjouy, M.; Naderi, N.; Kobarfard, F.; Heidarli, E.; Faizi, M. An intensified acrolein exposure can affect memory and cognition in rat. Neurotox. Res. 2021, 39, 277–291. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Jin, M.-H.; Pi, R.-B.; Zhang, J.-J.; Ouyang, Y.; Chao, X.-J.; Chen, M.-H.; Liu, P.-Q.; Yu, J.-C.; Ramassamy, C. Acrolein induces Alzheimer’s disease-like pathologies in vitro and in vivo. Toxicol. Lett. 2013, 217, 184–191. [Google Scholar] [CrossRef]
- Li, X.-H.; Xie, J.-Z.; Jiang, X.; Lv, B.-L.; Cheng, X.-S.; Du, L.-L.; Zhang, J.-Y.; Wang, J.-Z.; Zhou, X.-W. Methylglyoxal induces tau hyperphosphorylation via promoting AGEs formation. Neuromol. Med. 2012, 14, 338–348. [Google Scholar] [CrossRef]
- Bradley, M.; Markesbery, W.; Lovell, M. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic. Biol. Med. 2010, 48, 1570–1576. [Google Scholar] [CrossRef] [Green Version]
- Baker, G.; Matveychuk, D.; MacKenzie, E.M.; Holt, A.; Wang, Y.; Kar, S. Attenuation of the effects of oxidative stress by the MAO-inhibiting antidepressant and carbonyl scavenger phenelzine. Chem. Biol. Interact. 2019, 304, 139–147. [Google Scholar] [CrossRef]
- Elhelaly, A.E.; AlBasher, G.; Alfarraj, S.; Almeer, R.; Bahbah, E.I.; Fouda, M.M.A.; Bungau, S.G.; Aleya, L.; Abdel-Daim, M.M. Protective effects of hesperidin and diosmin against acrylamide-induced liver, kidney, and brain oxidative damage in rats. Environ. Sci. Pollut. Res. 2019, 26, 35151–35162. [Google Scholar] [CrossRef]
- Singh, I.N.; Gilmer, L.K.; Miller, D.M.; Cebak, J.E.; Wang, J.A.; Hall, E.D. Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J. Cereb. Blood Flow Metab. 2013, 33, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, A.G.; Alfaqih, M.A.; Al-Shboul, O. The 4-hydroxynonenal mediated oxidative damage of blood proteins and lipids involves secondary lipid peroxidation reactions. Exp. Ther. Med. 2018, 16, 2132–2137. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, E.M.; Song, M.-S.; Dursun, S.M.; Tomlinson, S.; Todd, K.G.; Baker, G.B. Phenelzine: An old drug that may hold clues to the development of new neuroprotective agents. Klin. Psikofarmakol. Bül. Bull. Clin. Psychopharmacol. 2010, 20, 179–186. [Google Scholar] [CrossRef]
- Matveychuk, D.; Nunes, E.; Ullah, N.; Velázquez-Martinez, C.A.; MacKenzie, E.M.; Baker, G.B. Comparison of phenelzine and geometric isomers of its active metabolite, β-phenylethylidenehydrazine, on rat brain levels of amino acids, biogenic amine neurotransmitters and methylamine. J. Neural Transm. 2013, 120, 987–996. [Google Scholar] [CrossRef]
- Paslawski, T.; Knaus, E.; Iqbal, N.; Coutts, R.; Baker, G. β-phenylethylidenehydrazine, a novel inhibitor of GABA transaminase. Drug Dev. Res. 2001, 54, 35–39. [Google Scholar] [CrossRef]
- Leker, R.; Neufeld, M. Anti-epileptic drugs as possible neuroprotectants in cerebral ischemia. Brain Res. Rev. 2003, 42, 187–203. [Google Scholar] [CrossRef]
- Stumm, R.K.; Culmsee, C.; Schäfer, M.K.-H.; Krieglstein, J.; Weihe, E. Adaptive plasticity in tachykinin and tachykinin receptor expression after focal cerebral ischemia is differentially linked to gabaergic and glutamatergic cerebrocortical circuits and cerebrovenular endothelium. J. Neurosci. 2001, 21, 798–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.M.; Tsien, R.W.; Goff, D.C.; Halassa, M.M. The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr. Res. 2015, 167, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerster, B.R.; Pomper, M.G.; Callaghan, B.C.; Petrou, M.; Edden, R.A.; Mohamed, M.A.; Welsh, R.C.; Carlos, R.C.; Barker, P.B.; Feldman, E.L. An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA Neurol. 2013, 70, 1009–1016. [Google Scholar] [CrossRef]
- Kim, Y.S.; Yoon, B.-E. Altered GABAergic signaling in brain disease at various stages of life. Exp. Neurobiol. 2017, 26, 122. [Google Scholar] [CrossRef] [Green Version]
- Potter, L.E.; Paylor, J.W.; Suh, J.S.; Tenorio, G.; Caliaperumal, J.; Colbourne, F.; Baker, G.; Winship, I.; Kerr, B.J. Altered excitatory-inhibitory balance within somatosensory cortex is associated with enhanced plasticity and pain sensitivity in a mouse model of multiple sclerosis. J. Neuroinflamm. 2016, 13, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef] [Green Version]
- Sowa, B.; Rauw, G.; Davood, A.; Fassihi, A.; Knaus, E.E.; Baker, G.B. Design and biological evaluation of phenyl-substituted analogs of β-phenylethylidenehydrazine. Bioorg. Med. Chem. 2005, 13, 4389–4395. [Google Scholar] [CrossRef]
- Sowa, B.; Knaus, E.; Todd, K.; Davood, A.; Baker, G. Biochemical activity of 4-fluorophenylethyl-idenehydrazine and its potential as a neuroprotectant in cerebral ischemia. J. Neurochem. 2003, 85, 10. [Google Scholar]
- Shanahan, P.; O’Sullivan, J.; Tipton, K.F.; Kinsella, G.K.; Ryan, B.J.; Henehan, G.T. Theobromine and related methylxanthines as inhibitors of primary amine oxidase. J. Food Biochem. 2019, 43, e12697. [Google Scholar] [CrossRef] [Green Version]
- Stolen, C.M.; Yegutkin, G.G.; Kurkijärvi, R.; Bono, P.; Alitalo, K.; Jalkanen, S. Origins of serum semicarbazide-sensitive amine oxidase. Circ. Res. 2004, 95, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Maley, J.; Yu, P.H. Potential implications of endogenous aldehydes in β-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 2006, 99, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Pannecoeck, R.; Serruys, D.; Benmeridja, L.; Delanghe, J.R.; Geel, N.V.; Speeckaert, R.; Speeckaert, M.M. Vascular adhesion protein-1: Role in human pathology and application as a biomarker. Crit. Rev. Clin. Lab. Sci. 2015, 52, 284–300. [Google Scholar] [CrossRef] [PubMed]
- Unzeta, M.; Sole, M.; Boada, M.; Hernández, M. Semicarbazide-Sensitive amine oxidase (SSAO) and its possible contribution to vascular damage in Alzheimer’s disease. J. Neural Transm. 2007, 114, 857–862. [Google Scholar] [CrossRef]
- Valente, T.; Gella, A.; Solé, M.; Durany, N.; Unzeta, M. Immunohistochemical study of semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 in the hippocampal vasculature: Pathological synergy of Alzheimer’s disease and diabetes mellitus. J. Neurosci. Res. 2012, 90, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, A.; Wang, E.; Salter-Cid, L.; Huang, L.; Miller, A.; Podar, E.; Gao, H.; Jones, D.; Linnik, M. Benefit of inhibiting SSAO in relapsing experimental autoimmune encephalomyelitis. J. Neural Transm. 2007, 114, 845. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.; Schilter, H.; Liu, G.; Wheeldon, K.; Essilfie, A.T.; Foot, J.; Yow, T.; Jarolimek, W.; Hansbro, P. The inhibitor of semicarbazide-sensitive amine oxidase, PXS-4728A, ameliorates key features of chronic obstructive pulmonary disease in a mouse model. Br. J. Pharmacol. 2016, 173, 3161–3175. [Google Scholar] [CrossRef]
- Horváth, Á.; Menghis, A.; Botz, B.; Borbély, É.; Kemény, Á.; Tékus, V.; Csepregi, J.Z.; Mócsai, A.; Juhász, T.; Zákány, R. Analgesic and anti-inflammatory effects of the novel semicarbazide-sensitive amine-oxidase inhibitor SzV-1287 in chronic arthritis models of the mouse. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Ma, Q.; Manaenko, A.; Khatibi, N.H.; Chen, W.; Zhang, J.H.; Tang, J. Vascular adhesion protein-1 inhibition provides antiinflammatory protection after an intracerebral hemorrhagic stroke in mice. J. Cereb. Blood Flow Metab. 2011, 31, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, A.G.; Al-Shboul, O.; Alfaqih, M.A.; Al-Qudah, M.A.; Al-Dwairi, A.N. Phenelzine reduces the oxidative damage induced by peroxynitrite in plasma lipids and proteins. Arch. Physiol. Biochem. 2018, 124, 418–423. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative stress and cardiovascular risk: Obesity, diabetes, smoking, and pollution: Part 3 of a 3-part series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Hoshaw, B.A.; Malberg, J.E.; Rosenzweig-Lipson, S.; Schechter, L.E.; Lucki, I. Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res. 2008, 1211, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, Y.; Rizavi, H.S.; Pandey, G.N. Antidepressants reverse corticosterone-mediated decrease in brain-derived neurotrophic factor expression: Differential regulation of specific exons by antidepressants and corticosterone. Neuroscience 2006, 139, 1017–1029. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Han, E.S.; Lee, W.B. Antioxidant effect of phenelzine on MPP+-induced cell viability loss in differentiated PC12 cells. Neurochem. Res. 2003, 28, 1833–1841. [Google Scholar] [CrossRef]
- Joseph, T.P.; Nataraj Jagadeesan, L.Y.S.; Lin, S.L.; Sahu, S.; Schachner, M. Adhesion molecule L1 agonist mimetics protect against the pesticide paraquat-induced locomotor deficits and biochemical alterations in zebrafish. Front. Neurosci. 2020, 14, 458. [Google Scholar] [CrossRef]
- Li, R.; Sahu, S.; Schachner, M. Phenelzine, a cell adhesion molecule L1 mimetic small organic compound, promotes functional recovery and axonal regrowth in spinal cord-injured zebrafish. Pharmacol. Biochem. Behav. 2018, 171, 30–38. [Google Scholar] [CrossRef]
- García-Fuster, M.J.; García-Sevilla, J.A. Effects of anti-depressant treatments on FADD and p-FADD protein in rat brain cortex: Enhanced anti-apoptotic p-FADD/FADD ratio after chronic desipramine and fluoxetine administration. Psychopharmacology 2016, 233, 2955–2971. [Google Scholar] [CrossRef]
- Demougeot, C.; Garnier, P.; Mossiat, C.; Bertrand, N.; Giroud, M.; Beley, A.; Marie, C. N-Acetylaspartate, a marker of both cellular dysfunction and neuronal loss: Its relevance to studies of acute brain injury. J. Neurochem. 2001, 77, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Ladostigil: A novel multimodal neuroprotective drug with cholinesterase and brain-selective monoamine oxidase inhibitory activities for Alzheimer’s disease treatment. Curr. Drug Targets 2012, 13, 483–494. [Google Scholar] [CrossRef]
- Birks, J.; Flicker, L. Selegiline for Alzheimer’s disease. Cochrane Database Syst. Rev. 2003, 1, CD000442. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Amit, T.; Bar-Am, O.; Fridkin, M.; Youdim, M.B.; Mandel, S.A. From anti-Parkinson’s drug rasagiline to novel multitarget iron chelators with acetylcholinesterase and monoamine oxidase inhibitory and neuroprotective properties for Alzheimer’s disease. J. Alzheimers Dis. 2012, 30, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gal, S.; Abassi, Z.A.; Youdim, M.B. Limited potentiation of blood pressure in response to oral tyramine by the anti-Parkinson brain selective multifunctional monoamine oxidase-AB inhibitor, M30. Neurotox. Res. 2010, 18, 143–150. [Google Scholar] [CrossRef]
- Pletscher, A. The discovery of antidepressants: A winding path. Experientia 1991, 47, 4–8. [Google Scholar] [CrossRef]
- Gareri, P.; Falconi, U.; De Fazio, P.; De Sarro, G. Conventional and new antidepressant drugs in the elderly. Prog. Neurobiol. 2000, 61, 353–396. [Google Scholar] [CrossRef]
- Makkar, R.; Behl, T.; Bungau, S.; Zengin, G.; Mehta, V.; Kumar, A.; Uddin, M.S.; Ashraf, G.M.; Abdel-Daim, M.M.; Arora, S.; et al. Nutraceuticals in neurological disorders. Int. J. Mol. Sci. 2020, 21, 4424. [Google Scholar] [CrossRef]
- AlBasher, G.; Abdel-Daim, M.M.; Almeer, R.; Ibrahim, K.A.; Hamza, R.Z.; Bungau, S.; Aleya, L. Synergistic antioxidant effects of resveratrol and curcumin against fipronil-triggered oxidative damage in male albino rats. Environ. Sci. Pollut. Res. 2020, 27, 6505–6514. [Google Scholar] [CrossRef]
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: The PRESTO study. Arch. Neurol. 2005, 62, 241–248. [Google Scholar] [CrossRef]
- Shoulson, I.; Fahn, S.; Oakes, D.; Lang, A.; Langston, J.W.; LeWitt, P.; Olanow, C.W.; Penney, J.B.; Tanner, C.; Kieburtz, K.; et al. Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP subjects not requiring levodopa. Ann. Neurol. 1996, 39, 29–36. [Google Scholar]
- Rascol, O.; Brooks, D.J.; Melamed, E.; Oertel, W.; Poewe, W.; Stocchi, F.; Tolosa, E.; Group, L.S. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): A randomised, double-blind, parallel-group trial. Lancet 2005, 365, 947–954. [Google Scholar] [CrossRef]
- Clarke, C.E. A “cure” for Parkinson’s disease: Can neuroprotection be proven with current trial designs? Mov. Disord. 2004, 19, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.A.J.; Leysen, J.E.; Megens, A.A.H.P.; Awouters, F.H.L. Does phenylethylamine act as an endogenous amphetamine in some patients? Int. J. Neuropsychopharmacol. 1999, 2, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-M.; Juorio, A.V.; Qi, J.; Boulton, A.A. L-Deprenyl induces aromatic l-amino acid decarboxylase (AADC) mRNA in the rat substantia nigra and ventral tegmentum. Mol. Chem. Neuropathol. 1998, 35, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, K.; Channon, S.; Ramponi, C.; Stern, G.M.; Lees, A.J.; Youdim, M.B.H. The therapeutic potential of moclobemide, a reversible selective monoamine oxidase A inhibitor in Parkinson’s disease. J. Clin. Psychopharmacol. 1995, 15, 51S–59S. [Google Scholar] [CrossRef]
- Weinstock, M.; Gorodetsky, E.; Poltyrev, T.; Gross, A.; Sagi, Y.; Youdim, M. A novel cholinesterase and brain-selective monoamine oxidase inhibitor for the treatment of dementia comorbid with depression and Parkinson’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 555–561. [Google Scholar] [CrossRef]
- Poltyrev, T.; Gorodetsky, E.; Bejar, C.; Schorer-Apelbaum, D.; Weinstock, M. Effect of chronic treatment with ladostigil (TV-3326) on anxiogenic and depressive-like behaviour and on activity of the hypothalamic-pituitary-adrenal axis in male and female prenatally stressed rats. Psychopharmacology 2005, 181, 118–125. [Google Scholar] [CrossRef]
- Waibel, S.; Reuter, A.; Malessa, S.; Blaugrund, E.; Ludolph, A.C. Rasagiline alone and in combination with riluzole prolongs survival in an ALS mouse model. J. Neurol. 2004, 251, 1080–1084. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Model | Controls | Stimulus/Drugs Employed | Method of Determination | Result/Conclusion | Ref. |
|---|---|---|---|---|---|
| 11 autopsied brains of AD individuals | 5 non-AD individuals | Lazabemide (MAO-B inhibitor) Ro41-1049 (MAO-A inhibitor) | Enzyme radiography | The concentration of MAO enzyme in the parietal cortex of AD was substantially higher as compared to controls. | [46] |
| 3 autopsied brains of AD patients | 3 autopsied brains of controls | L-deprenyl | Cryo-microtomy (autoradiography) | The AD patients had higher MAO-B expression than the controls | [47] |
| Plaque-associated astrocytes of brains of AD patients | Literature data | L-deprenyl, Pargyline, Iproniazid | Enzymatic assay | Increased MAO expression in brains of AD pIC50 value was determined of all drugs | [48] |
| Postmortem brains of AD | Autopsied brains of non-AD individuals | Gene silencing (sense and anti-sense siRNA) | PLA (in situ proximity ligation assay) Immunocytochemistry. | Immunocytochemistry revealed MAO-B staining in the frontal cortex, hippocampus, and entorhinal cortex whose intensity is higher in AD brains than in controls. | [49] |
| 60 autopsied brains of AD patients | 60 autopsied brains of controls | Apo-E4 status | HPLC (high-performance liquid chromatography) and immunodetection (SDS-PAGE) | Increased MAO-A activity in AD brains has been associated with prodromal and co-morbid neuropsychiatric symptoms and with neurodegeneration | [50] |
| APP/PS1 mice | Control mice (wild type) | Selegiline (10 mg/kg per day for 4 weeks) | Morris water maze test | The aberrant GABA level in APP/PS1 mice was significantly decreased to the levels of GABA observed in wild type control mice following selegiline treatment | [51] |
| Adult male Wistar rats with AD | Male Wistar rats as controls | Sembragiline (0.3%) | HPLC + ion-spray tandem mass spectrometry | Administration of sembragiline resulted in substantially decreased levels of ROS and prevented reduced dopaminergic neuron numbers in substantia nigra, when compared with vehicle-treated mice | [52] |
| Mouse model of AD | Control mice | 2-photon MAO probe | Fluorescence TPM imaging | In vivo correlation between MAO and progression of AD-indicating MAO as a potential biomarker of AD. | [44] |
| Drug | Category of the Drug | Mechanism of Action | Clinical Phase | Ref. |
|---|---|---|---|---|
| Ladostigil | Inhibitor of monoamine oxidases A and B | Prevention of age-related glial activation and spatial memory deficits Improvement of cognitive performance Modulation of amyloid precursor protein processing Upregulation of antioxidant activity and mRNA expression of antioxidant enzymes. | Phase 2 | [111] |
| Selegiline | Selective and irreversible inhibitor of monoamine oxidase B | Regulation of cleavage of amyloid precursor protein Activation of phosphokinase C Inhibits amyloid-beta plaque formation antioxidant. | Phase 3 | [42,112] |
| Rasagiline | Irreversible inhibitor of monoamine oxidase B | Modulates amyloid precursor protein and amyloid-beta processing Iron chelator Regulates the cell cycle | Phase 2 | [113] |
| M-30 | Inhibitor of monoamine oxidases A and B Iron chelator | Regulates proteolytic processing of amyloid precursor protein Iron chelator Reduces neuronal death and apoptotic DNA damage | Preclinical and clinical phase | [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors. Molecules 2021, 26, 3724. https://doi.org/10.3390/molecules26123724
Behl T, Kaur D, Sehgal A, Singh S, Sharma N, Zengin G, Andronie-Cioara FL, Toma MM, Bungau S, Bumbu AG. Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors. Molecules. 2021; 26(12):3724. https://doi.org/10.3390/molecules26123724
Chicago/Turabian StyleBehl, Tapan, Dapinder Kaur, Aayush Sehgal, Sukhbir Singh, Neelam Sharma, Gokhan Zengin, Felicia Liana Andronie-Cioara, Mirela Marioara Toma, Simona Bungau, and Adrian Gheorghe Bumbu. 2021. "Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors" Molecules 26, no. 12: 3724. https://doi.org/10.3390/molecules26123724