
Quantitative Proteomics and Differential Protein Abundance Analysis after Depletion of Putative mRNA Receptors in the ER Membrane of Human Cells Identifies Novel Aspects of mRNA Targeting to the ER

 ,
,  , and
, and
Abstract
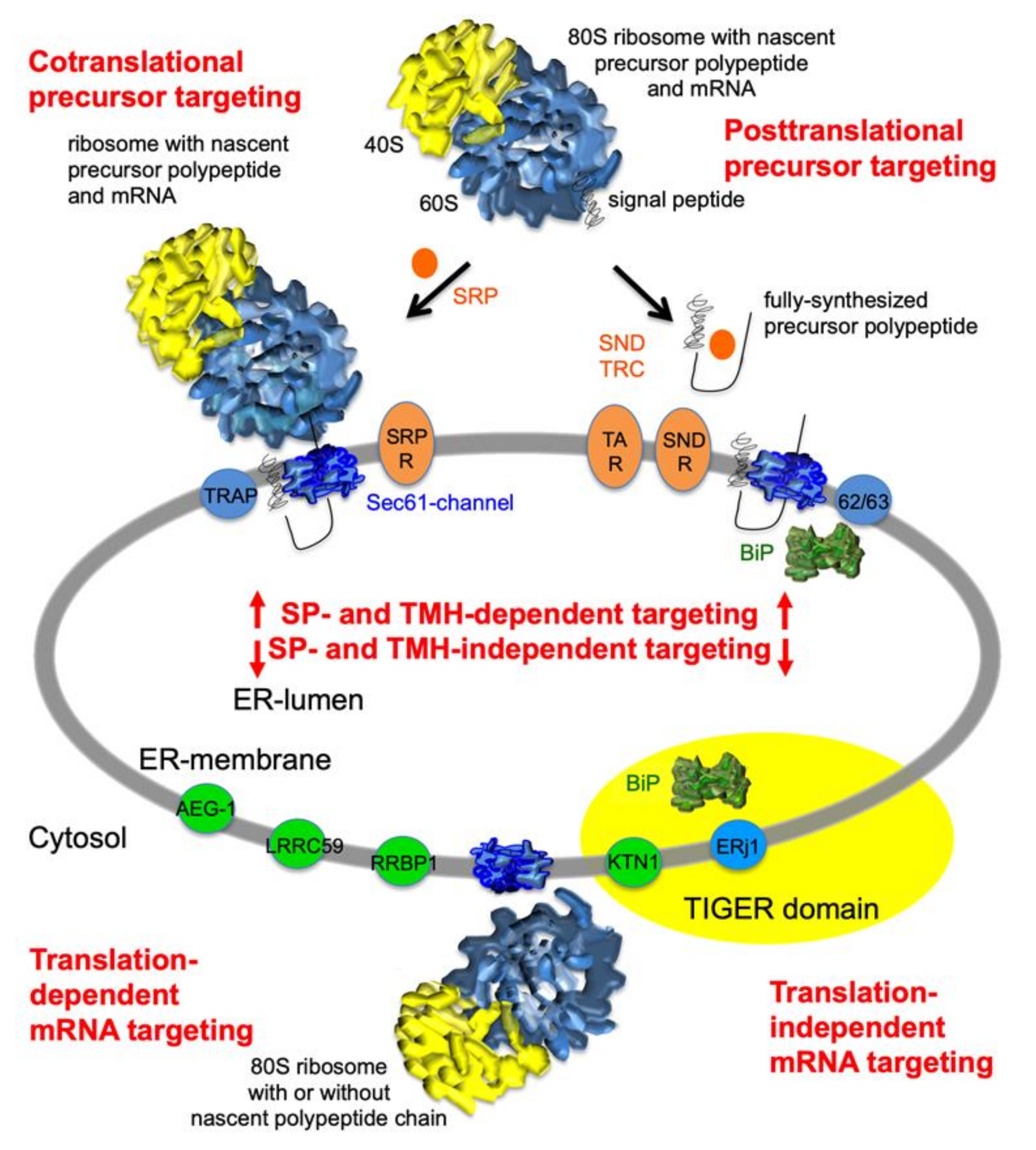
:1. Introduction
2. Results
2.1. The Experimental Approach
2.2. Quantitative Proteomic Analysis of HeLa Cells after Transient and Partial Depletion of RRBP1 by siRNA
2.3. Quantitative Proteomic Analysis of HeLa Cells after Transient and Partial Depletion of KTN1 by siRNA
2.4. Quantitative Proteomic Analysis of HeLa Cells after Transient and Partial Depletion of ERj1 by siRNA
3. Discussion
3.1. Discussion of the Experimental Approach
3.2. Discussion of the Results on Possible Clients for mRNA Targeting to the ER
3.3. Discussion of Compensatory Mechanisms after Depletion of mRNA Receptors on the ER
3.4. Possible Implications for the TIGER Domain
4. Materials and Methods
4.1. Materials
4.2. Cell Manipulation and Analysis
| RRBP1 | siRNA#1, GGAUAUUUACGACACUCAAdTdT; |
| RRBP1 | siRNA#2, GAGAUUGUAGAGAAGCUAAdTdT; |
| KTN1 | siRNA#3, CAGUUGGAGCAAAGACUAAdTdT; |
| KTN1 | iRNA#4, GCCUCUGACUUCAACUCAAdTdT; |
| ERJ1 | siRNA#5, CCUCAAUAUUUCUACGUCAdTdT; |
| ERj1 | siRNA#6, GGUAUGAUGAUAUUCUGAUdTdT. |
4.3. Label-Free Quantitative Proteomic Analysis
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BiP | Immunoglobulin heavy chain binding protein |
| EMC | ER membrane complex |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated (protein) degradation |
| ERj | ER (resident) J-domain protein |
| GET | Guided entry of tail-anchored proteins |
| GPI | Glycosylphosphatidylinositol |
| GRP | Glucose-regulated protein |
| HSP | Heat shock protein |
| JDP | J-domain protein |
| PEX | Peroxisome (protein) |
| RAMP | Ribosome-associated membrane protein |
| RNC | Ribosome-nascent chain |
| SEC | (Protein involved in) secretion |
| SND | SRP-independent |
| SP | Signal peptide |
| SR | SRP receptor |
| SRP | Signal recognition particle |
| SSR | Signal sequence receptor |
| TA | Tail-anchore(d) |
| TMEM | Transmembrane (protein) |
| TMH | Transmembrane helix |
| TRAM | translocating chain-associating membrane (protein) |
| TRAP | Translocon-associated protein |
| TRC | Transmembrane recognition complex |
| UPR | Unfolded protein response |
| UTR | Untranslated region |
References
- Palade, G. Intracellular aspects of protein synthesis. Science 1975, 189, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes: I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 1975, 67, 835–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blobel, G.; Dobberstein, B. Transfer of proteins across membranes: II. Reconstitution of functional rough microsomes from heterologous components. J. Cell Biol. 1975, 67, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Aviram, N.; Schuldiner, M. Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J. Cell Sci. 2017, 130, 4079–4085. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Pfeffer, S.; Lee, P.-H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An update on Sec61 channel function, mechanisms, and related diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Nguyen, D.; Pfeffer, S.; Förster, F.; Helms, V.; Zimmermann, R. Functions and mechanisms of the human ribosome-translocon complex, in Macromolecular Complexes II: Structure and Function, Harris, J.R.; Marles-Wrigth, J., Eds. Subcell. Biochem. 2019, 93, 83–141. [Google Scholar] [CrossRef]
- von Heijne, G. Signal sequences. J. Mol. Biol. 1985, 184, 99–105. [Google Scholar] [CrossRef]
- von Heijne, G. Towards a comparative anatomy of N-terminal topogenic protein sequences. J. Mol. Biol. 1986, 189, 239–242. [Google Scholar] [CrossRef]
- von Heijne, G.; Gavel, Y. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 1988, 174, 671–678. [Google Scholar] [CrossRef]
- Ng, D.T.; Brown, J.D.; Walter, P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell Biol. 1996, 134, 269–278. [Google Scholar] [CrossRef]
- Hegde, R.S.; Bernstein, H. The surprising complexity of signal peptides. Trends Biochem. Sci. 2006, 31, 563–571. [Google Scholar] [CrossRef]
- Görlich, D.; Prehn, S.; Hartmann, E.; Kalies, K.-U.; Rapoport, T.A. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell 1992, 71, 489–503. [Google Scholar] [CrossRef]
- Görlich, D.; Rapoport, T.A. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell 1993, 75, 615–630. [Google Scholar] [CrossRef]
- Hartmann, E.; Sommer, T.; Prehn, S.; Görlich, D.; Jentsch, S.; Rapoport, T.A. Evolutionary conservation of components of the protein translocation complex. Nature 1994, 367, 654–657. [Google Scholar] [CrossRef]
- Simon, S.M.; Blobel, G. A protein-conducting channel in the endoplasmic reticulum. Cell 1991, 65, 371–380. [Google Scholar] [CrossRef]
- Wirth, A.; Jung, M.; Bies, C.; Frien, M.; Tyedmers, J.; Zimmermann, R.; Wagner, R. The Sec61p complex is a dynamic precursor activated channel. Mol. Cell. 2003, 12, 261–268. [Google Scholar] [CrossRef]
- Beckmann, R.; Spahn, C.M.; Eswar, N.; Helmers, J.; Penczek, P.A.; Sali, A.; Frank, J.; Blobel, G. Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell 2001, 107, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, B.; Clemons, W.M.; Collinson, I.; Modis, Y.; Hartmann, E.; Harrison, S.C.; Rapoport, T.A. X-ray structure of a protein-conducting channel. Nature 2004, 427, 36–44. [Google Scholar] [CrossRef]
- Pfeffer, S.; Brandt, F.; Hrabe, T.; Lang, S.; Eibauer, M.; Zimmermann, R.; Förster, F. Structure and 3D arrangement of ER-membrane associated ribosomes. Structure 2012, 20, 1508–1518. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, R.M.; Fernández, I.S.; Scheres, S.H.W.; Hegde, R.S. Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 2014, 157, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Förster, F. Structure of the mammalian oligosaccharyltransferase in the native ER protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Burbaum, L.; Unverdorben, P.; Pech, M.; Chen, Y.; Zimmermann, R.; Beckmann, R.; Förster, F. Structure of the native Sec61 protein-conducting channel. Nat. Commun. 2015, 6, 8403. [Google Scholar] [CrossRef] [Green Version]
- Voorhees R M, Hegde R S Structure of the Sec61 channel opened by a signal peptide. Science 2016, 351, 88–91. [CrossRef] [Green Version]
- Pfeffer, S.; Dudek, J.; Ng, B.; Schaffa, M.; Albert, S.; Plitzko, J.; Baumeister, W.; Zimmermann, R.; Freeze, H.; Engel, B.D.; et al. Dissecting the molecular organization of the translocon-associatecd protein complex. Nat. Commun. 2017, 8, 14516. [Google Scholar] [CrossRef]
- Gemmer, M.; Förster, F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020, 133, jcs231340. [Google Scholar] [CrossRef]
- Shurtleff, M.J.; Itzhak, D.N.; Hussmann, J.A.; Schirle Oakdale, N.T.; Costa, E.A.; Jonikas, M.; Weibezahn, J.; Popova, K.D.; Jan, C.H.; Sinitcyn, P.; et al. The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. eLife 2018, 7, e37018. [Google Scholar] [CrossRef]
- Chitwood, P.J.; Juszkiewicz, S.; Guna, A.; Shao, S.; Hegde, R.S. EMC is required to initiate accurate membrane protein topogenesis. Cell 2018, 175, 1507–1519. [Google Scholar] [CrossRef]
- Pleiner, T.; Tomaleri, G.P.; Januszyk, K.; Inglis, A.J.; Hazu, M.; Voorhees, R.M. Structural basis for membrane insertion by the human ER membrane protein complex. Science 2020, 369, 433–436. [Google Scholar] [CrossRef]
- Bai, L.; You, Q.; Feng, X.; Kovach, A.; Li, H. Structure of the ER membrane complex, a transmembrane insertase. Nature 2020, 584, 475–478. [Google Scholar] [CrossRef]
- O´Donnel, J.P.; Philips, B.P.; Yagita, Y.; Juszkiewicz, S.; Wagner, A.; Malinverni, D.; Keenan, R.J.; Mille, E.A.; Hegde, R.S. The architecture of EMC reveals a path for membrane protein nsertion. eLife 2020, 9, e57887. [Google Scholar] [CrossRef]
- Wang, Q.-C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 is an ER Ca2+ load-activated Ca2+ channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anghel, S.A.; McGilvray, P.T.; Hegde, R.S.; Keenan, R.J. Identification of Oxa1 homologs operating in the eukaryotic endoplasmic reticulum. Cell Rep. 2017, 21, 3708–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGilvray, P.T.; Anghel, S.A.; Sundaram, A.; Zhong, F.; Trnka, M.J.; Fuller, J.R.; Hu, H.; Burlingame, A.L.; Keenan, R.J. An ER translocon for multi-pass mambrane protein biogenesis. eLife 2020, 9, e56889. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; VanValkenburgh, C.; Liang, H.; Fang, H.; Green, N. Signal peptidase and oligosaccharyltransferase interact in a sequential and dependent manner within the endoplasmic reticulum. J. Biol. Chem. 2001, 276, 2411–2416. [Google Scholar] [CrossRef]
- Kalies, K.-U.; Rapoport, T.A.; Hartmann, E. The beta-subunit of the Sec61 complex facilitates cotranslational protein transport and interacts with the signal peptidase during translocation. J. Cell Biol. 1998, 141, 887–894. [Google Scholar] [CrossRef]
- Liaci, A.M.; Steigenberger, B.; Tamara, S.; de Souza, P.T.; Gröllers-Mulderij, M.; Ogrissek, P.; Marrink, S.-J.; Scheltema, R.A.; Förster, F. Structure of the human signal peptidase complex reveals the determinants for signal peptide cleavage. Cell 2021, in press. [Google Scholar] [CrossRef]
- Siegel, V.; Walter, P. Functional dissection of the signal recognition particle. Trends Biochem. Sci. 1988, 13, 314–316. [Google Scholar] [CrossRef]
- Egea, P.F.; Stroud, R.M.; Walter, P. Targeting proteins to membranes: Structure of the signal recognition particle. Curr. Opinion Struct. Biol. 2005, 15, 213–220. [Google Scholar] [CrossRef]
- Halic, M.; Beckmann, R. The signal recognition particle and its interactions during protein targeting. Curr. Opinion Struct. Biol. 2005, 15, 116–125. [Google Scholar] [CrossRef]
- Halic, M.; Blau, M.; Becker, T.; Mielke, T.; Pool, M.R.; Wild, K.; Sinning, I.; Beckmann, R. Following the signal sequence from ribosomal tunnel exit to signal recognition particle. Nature 2006, 444, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.I.; Dobberstein, B. A membrane component essential for vectorial translocation of nascent proteins across the endoplasmic reticulum: Requirements for its extraction and reassociation with the membrane. J. Cell. Biol. 1980, 87, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.I.; Dobberstein, B. Identification and characterization of a membrane component essential for the translocation of nascent proteins across the membrane of the endoplasmic reticulum. J. Cell Biol. 1980, 87, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Meyer, D.I.; Krause, E.; Dobberstein, B. Secretory protein translocation across membranes-the role of the "docking protein". Nature 1982, 297, 647–650. [Google Scholar] [CrossRef]
- Gilmore, R.; Blobel, G.; Walter, P. Protein translocation across the endoplasmic reticulum. I. Detection in the microsomal membrane of a receptor for the signal recognition particle. J. Cell Biol. 1982, 95, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, R.; Walter, P.; Blobel, G. Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor. J. Cell Biol. 1982, 95, 470–477. [Google Scholar] [CrossRef]
- Gilmore, R.; Blobel, G. Transient involvement of signal recognition particle and its receptor in the microsomal membrane prior to protein translocation. Cell 1983, 35, 677–685. [Google Scholar] [CrossRef]
- Tajima, S.; Lauffer, L.; Rath, V.L.; Walter, P. The signal recognition particle receptor is a complex that contains two distinct polypeptide chains. J. Cell Biol. 1986, 103, 1167–1178. [Google Scholar] [CrossRef]
- Aviram, N.; Ast, T.; Costa, E.A.; Arakel, E.; Chuartzman, S.G.; Jan, C.H.; Haßdenteufel, S.; Dudek, J.; Jung, M.; Schorr, S.; et al. The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 2016, 540, 134–138. [Google Scholar] [CrossRef]
- Casson, J.; McKenna, M.; Haßdenteufel, S.; Aviram, N.; Zimmermann, R.; High, S. Multiple pathways facilitate the biogenesis of mammalian tail-anchored proteins. J. Cell Sci. 2017, 130, 3851–3861. [Google Scholar] [CrossRef] [Green Version]
- Haßdenteufel, S.; Sicking, M.; Schorr, S.; Aviram, N.; Fecher-Trost, C.; Schuldiner, M.; Jung, M.; Zimmermann, R.; Lang, S. hSnd2 protein represents an alternative targeting factor to the endoplasmic reticulum in human cells. FEBS Lett. 2017, 591, 3211–3224. [Google Scholar] [CrossRef] [Green Version]
- Haßdenteufel, S.; Johnson, N.; Paton, A.W.; Paton, J.C.; High, S.; Zimmermann, R. Chaperone-mediated Sec61 channel gating during ER import of small precursor proteins overcomes Sec61 inhibitor-reinforced energy barrier. Cell Rep. 2018, 23, 1373–1386. [Google Scholar] [CrossRef]
- Haßdenteufel, S.; Nguyen, D.; Helms, V.; Lang, S.; Zimmermann, R. Components and mechanisms for ER import of small human presecretory proteins. FEBS Lett. 2019, 593, 2506–2524. [Google Scholar] [CrossRef]
- Lakkaraju, A.K.K.; Thankappan, R.; Mary, C.; Garrison, J.L.; Taunton, J.; Strub, K. Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Mol. Biol. Cell 2012, 23, 2712–2722. [Google Scholar] [CrossRef]
- Kutay, U.; Hartmann, E.; Rapoport, T.A. A class of membrane proteins with a C-terminal anchor. Trends Cell Biol. 1993, 3, 72–75. [Google Scholar] [CrossRef]
- Schuldiner, M.; Metz, J.; Schmid, V.; Denic, V.; Rakwalska, M.; Schmitt, H.D.; Schwappach, B.; Weissman, J.S. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 2008, 134, 634–645. [Google Scholar] [CrossRef]
- Mariappan, M.; Li, X.; Stefanovic, S.; Sharma, A.; Mateja, A.; Keenan, R.J.; Hegde, R.S. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature 2010, 466, 1120–1124. [Google Scholar] [CrossRef] [Green Version]
- Borgese, N.; Righi, M. Remote origins of tail-anchored proteins. Traffic 2010, 11, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Borgese, N.; Fasana, E. Targeting pathways of C-tail-anchored proteins. Biochim. Biophys. Acta 2011, 1808, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Vilardi, F.; Lorenz, H.; Dobberstein, B. WRB is the receptor for TRC40/Asna1-mediated insertion of tail-anchored proteins into the ER membrane. J. Cell Sci. 2011, 124, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Sakisaka, T. Molecular machinery for insertion of tail-anchored membrane proteins into the endoplasmic reticulum membrane in mammalian cells. Mol. Cell 2012, 48, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chan, C.; Weir, N.R.; Denic, V. The Get1/2 transmembrane complex is an endoplasmic-reticulum membrane protein insertase. Nature 2014, 512, 441–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgese, N.; Coy-Vergara, J.; Colombo, S.F.; Schwappach, B. The ways of tails: The GET pathway and more. Proteins 2019, 38, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Schrul, B.; Kopito, R.R. Peroxin-dependent targeting of a lipid-droplet-destined membrane protein to ER subdomains. Nat. Cell Biol. 2016, 18, 740. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Sakisaka, T. The peroxisome biogenesis factors posttranslationally target reticulon homology domain-containing proteins to the endoplasmic reticulum. Sci. Rep. 2018, 8, 2322. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.A.; Zhang, H.; Ilan, L.; Liu., A.X.; Kharchuk, I.; Palazzo, A.F. mRNA encoding Sec61beta, a tail-anchored protein, is localized on the endoplasmic reticulum. J. Cell Sci. 2015, 128, 3398–3410. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.G.; Aviram, N.; Laborenz, J.; Bibi, C.; Meyer, M.; Spang, A.; Schuldiner, M.; Herrmann, J.M. An ER surface retrieval pathway safeguaerds the import of mitochondrial membrane proteins in yeast. Science 2018, 361, 1118–1122. [Google Scholar] [CrossRef] [Green Version]
- Savitz, A.J.; Meyer, D.I. Identification of a ribosome receptor in the rough endoplasmic reticulum. Nature 1990, 346, 540–544. [Google Scholar] [CrossRef]
- Tazawa, S.; Unuma, M.; Tondokoro, N.; Asano, Y.; Ohsumi, T.; Ichimura, T.; Sugano, H. Identification of a membrane protein responsible for ribosome binding in rough microsomal membranes. J. Biochem. 1991, 109, 89–98. [Google Scholar]
- Savitz, A.J.; Meyer, D.I. 180-kD ribosome receptor is essential for both ribosome binding and protein translocation. J. Cell Biol. 1993, 120, 853–863. [Google Scholar] [CrossRef]
- Ohsumi, T.; Ichimura, T.; Sugano, H.; Omata, S.; Isobe, T.; Kumwano, R. Ribosome-binding protein p34 is a membrer of the leucine-rich-repeat-protein superfamily. Biochem. J. 1993, 294, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Wanker, E.E.; Sun, Y.; Savitz, A.J.; Meyer, D.I. Functional characterization of the 180-kD ribosome receptor in vivo. J. Cell Biol. 1995, 130, 29–39. [Google Scholar] [CrossRef]
- Seiser, R.M.; Nicchitta, C.V. The fate of membrane-bound ribosomes following the termination of protein synthesis. J. Biol. Chem. 2000, 275, 33820–33827. [Google Scholar] [CrossRef] [Green Version]
- Potter, M.D.; Seiser, R.M.; Nicchitta, C.V. Ribosome exchange revisited: A mechanism for translation-coupled ribosome detachment from the ER membrane. Trends Cell Biol. 2001, 11, 112–115. [Google Scholar] [CrossRef]
- Potter, M.D.; Nicchitta, C.V. Endoplasmic reticulum-bound ribosomes reside in stable association with the translocon following termination of protein synthesis. J. Biol. Chem. 2002, 277, 23314–23320. [Google Scholar] [CrossRef] [Green Version]
- Ong, L.L.; Er, C.P.; Aung, M.T.; Yu, H. Kinectin anchors the translation elongation factor-1 delta to the endoplasmic reticulum. J. Biol. Chem. 2003, 278, 32115–32123. [Google Scholar] [CrossRef] [Green Version]
- Crofts, A.J.; Washida, H.; Okita, T.W.; Ogawa, M.; Kumamaru, T.; Satoh, H. Targeting of proteins to endoplasmic reticulum-derived compartments in plants. The importance of RNA localization. Plant Physiol. 2004, 136, 3414–3419. [Google Scholar] [CrossRef] [Green Version]
- Ong, L.L.; Lin, P.C.; Zhang, X.; Chia, S.M.; Yu, H. Kinectin-dependent assembly of translation elongation factor-1 on endoplasmic reticulum regulates protein synthesis. J. Biol. Chem. 2006, 281, 33621–33634. [Google Scholar] [CrossRef] [Green Version]
- Pyhtila, B.; Zheng, T.; Lager, P.J.; Keene, J.D.; Reedy, M.C.; Nicchitta, C.V. Signal sequence- and translation-independent mRNA localization to the endoplasmic reticulum. RNA 2008, 14, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Kraut-Cohen, J.; Gerst, J.E. Addressing mRNAs to the ER: Cis sequences act up! Trends Biochem. Sci. 2010, 35, 459–469. [Google Scholar] [CrossRef]
- Dejgaard, K.; Theberge, J.-F.; Heath-Engel, H.; Chevet, E.; Tremblay, M.L.; Thomas, D.Y. Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J. Proteome Res. 2010, 9, 1763–1771. [Google Scholar] [CrossRef]
- Ueno, T.; Tanaka, K.; Kaneko, K.; Taga, Y.; Sata, T.; Irie, S.; Shunji Hattori, S.; Ogawa-Goto, K. Enhancement of procollagen biosynthesis by p180 through augmented ribosome association on the endoplasmic reticulum in response to stimulated secretion. J. Biol. Chem. 2010, 285, 29942–29950. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Kaneko, K.; Sata, T.; Hattori, S.; Ogawa-Goto, K. (2011) Regulation of polysome assembly on the endoplasmic reticulum by a coiled-coil protein, p180. Nucleic Acids Res. 2012, 40, 3006–3017. [Google Scholar] [CrossRef] [Green Version]
- Reid, D.W.; Nicchitta, C.V. Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J. Biol. Chem. 2012, 287, 5518–5527. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.A.; Zhang, H.; Palazzo, A.F. p180 promotes the ribosome-independent localization of a subset of mRNA to the endoplasmic reticulum. PLoS Biol. 2012, 10, e1001336. [Google Scholar] [CrossRef]
- Cui, X.A.; Zhang, Y.; Hong, S.J.; Palazzo, A.F. Identification of a region within the placental alkaline phosphatase mRNA that mediates p180-dependent targeting to the endoplasmic reticulum. J. Biol. Chem. 2013, 288, 29633–29641. [Google Scholar] [CrossRef] [Green Version]
- Hermesh, O.; Jansen, R.-P. Take the (RN)A-train: Localization of mRNA to the endoplasmic reticulum. Biochem. Biophys. Acta 2013, 1833, 2519–2525. [Google Scholar] [CrossRef]
- Calvin, H.J.; Williams, C.C.; Weissman, J.S. Principles of ER coranslational translocation revealed by proximity-specific ribosome profiling. Science 2014, 346, 1257521. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan, S.; Hsu, J.C.; Reid, D.W.; Chen, Q.; Thompson, W.J.; Moseley, A.M.; Nicchitta, C.V. Multifunctional roles for the protein translocation machinery in RNA anchoring to the endoplasmic reticulum. J. Biol. Chem. 2014, 289, 25907–25924. [Google Scholar] [CrossRef] [Green Version]
- Voigt, F.; Zhang, H.; Cui, X.A.; Triebold, D.; Liu, A.X.; Eglinger, J.; Lee, E.S.; Chao, J.A.; Palazzo, A.F. Single-molecule quantification of translation-dependent association of mRNAs with the endoplasmic reticulum. Cell Rep. 2017, 21, 3740–3753. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.C.-C.; Reid, D.W.; Hoffman, A.M.; Sarkar, D.; Nicchitta, C.V. Oncoprotein AEG-1 is an endoplasmic reticulum RNA-binding protein whose interactome is enriched in organelle resident protein-encoding mRNAs. RNA 2018, 24, 688–703. [Google Scholar] [CrossRef] [Green Version]
- Béthune, J.; Jansen, R.-P.; Feldbrügge, M.; Zarnack, K. Membrane-associated RNA-binding proteins orchestrate organelle-coupled translation. Trends Cell Biol. 2019, 29, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.M.; Chen, Q.; Zheng, T.; Nicchitta, C.V. Heterogeneous translational landscape of the endoplasmic reticulum revealed by ribosome proximity labeling and transcriptome analysis. J. Biol. Chem. 2019, 294, 18863–18872. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Zontag, O.; Baez, C.; Lim, L.Q.J.; Olender, T.; Schirman, D.; Dahary, D.; Pilpel, Y.; Gerst, J.E. A secretion-enhancing cis regulatory targeting element (SECReTE) involved in mRNA localization and protein synthesis. PLoS Genet. 2019, 15, e1008248. [Google Scholar] [CrossRef] [PubMed]
- Li Tian, L.; Chou, H.-L.; Zhang, L.; Okita, T.W. Targeted Endoplasmic Reticulum Localization of Storage Protein mRNAs Requires the RNA-Binding Protein RBP-L1. Plant Physiol. 2019, 179, 1111–1131. [Google Scholar] [CrossRef] [Green Version]
- Blenski, M.; Kehlenbach, R.H. Targeting of LRRC59 to the endoplasmic reticulum and the inner nuclear membrane. Int. J. Mol. Sci. 2019, 20, 334. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, M.M.; Hoffman, A.M.; Thompson, J.W.; Zheng, T.; Nicchitta, C.V. Quantitative proteomics links the LRRC59 interactome to mRNA translation on the ER membrane. Mol. Cell. Proteom. 2020, 19, 1826–1849. [Google Scholar] [CrossRef]
- Dudek, J.; Volkmer, J.; Bies, C.; Guth, S.; Müller, A.; Lerner, M.; Feick, P.; Schäfer, K.H.; Morgenstern, E.; Hennessy, F.; et al. A novel type of cochaperone mediates transmembrane recruitment of DnaK-like chaperones to ribosomes. EMBO J. 2002, 21, 2958–2967. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J.; Greiner, M.; Müller, A.; Hendershot, L.M.; Kopsch, K.; Nastainczyk, W.; Zimmermann, R. ERj1p plays a basic role in protein biogenesis at the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2005, 12, 1008–1014. [Google Scholar] [CrossRef]
- Blau, M.; Mullapudi, S.; Becker, T.; Dudek, J.; Zimmermann, R.; Penczek, P.A.; Beckmann, R. ERj1p uses a universal ribosomal adaptor site to coordinate the 80S ribosome at the membrane. Nat. Struct. Mol. Biol. 2005, 12, 1015–1016. [Google Scholar] [CrossRef]
- Benedix, J.; Lajoie, P.; Jaiswal, H.; Burgard, C.; Greiner, M.; Zimmermann, R.; Rospert, S.; Snapp, E.L.; Dudek, J. BiP modulates the affinity of its co-chaperone ERj1 to ribosomes. J Biol. Chem. 2010, 285, 36427–36433. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Stutz, R.; Schorr, S.; Lang, S.; Pfeffer, S.; Freeze, H.F.; Förster, F.; Helms, V.; Dudek, J.; Zimmermann, R. Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat. Commun. 2018, 9, 37639. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.-C.; Lerner, M.; Nguyen, D.; Pfeffer, S.; Dudek, J.; Förster, F.; Helms, V.; Lang, S.; Zimmermann, R. TRAM1 protein may support ER protein import by modulating the phospholipid bilayer near the lateral gate of the Sec61 channel. Channels 2020, 14, 28–44. [Google Scholar] [CrossRef] [Green Version]
- Schorr, S.; Nguyen, D.; Haßdenteufel, S.; Nagaraj, N.; Cavalié, A.; Greiner, M.; Weissgerber, P.; Loi, M.; Paton, A.W.; Paton, J.C.; et al. Proteomics identifies signal peptide features determining the substrate specificity in human Sec62/Sec63-dependent ER protein import. FEBS J. 2020, 287, 4612–4640. [Google Scholar] [CrossRef] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Nagaraj, N.; Kulak, N.A.; Cox, J.; Neuhauser, N.; Mayr, K.; Hoerning, O.; Vorm, O.; Mann, M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteom. 2012, 11, M111.013722. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Pääbo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Sys. Biol. 2011, 7, 548. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Coc, J. The Perseus computational platform for comprehensive analysis of (proteo)omics data. Nat. Meth. 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Hyunsoo, K.; Golub, G.H.; Park, H. Missing value estimation for DNA microarray gene expression data: Local least squares imputation. Bioinformatics 2005, 21, 187–198. [Google Scholar] [CrossRef]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. Gorilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Skowronek, M.H.; Rotter, M.; Haas, I.G. Molecular characterization of a novel mammalian DnaJ-like Sec63p homolog. Biol. Chem. 1999, 380, 1133–1138. [Google Scholar] [CrossRef]
- Mayer, H.-A.; Grau, H.; Kraft, R.; Prehn, S.; Kalies, K.-U.; Hartmann, E. Mammalian Sec61 is associated with Sec62 and Sec63. J. Biol. Chem. 2000, 275, 14550–14557. [Google Scholar] [CrossRef] [Green Version]
- Tyedmers, J.; Lerner, M.; Bies, C.; Dudek, J.; Skowronek, M.H.; Haas, I.G.; Heim, N.; Nastainczyk, W.; Volkmer, J.; Zimmermann, R. Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. USA 2000, 97, 7214–7219. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.; Diaz de Escauriaza, M.; Lajoie, P.; Theis, M.; Jung, M.; Müller, A.; Burgard, C.; Greiner, M.; Snapp, E.L.; Dudek, J.; et al. Evolutionary gain of function of the ER membrane protein Sec62 from yeast to humans. Mol. Biol. Cell 2010, 21, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.; Benedix, J.; Fedeles, S.V.; Schorr, S.; Schirra, C.; Schäuble, N.; Jalal, C.; Greiner, M.; Haßdenteufel, S.; Tatzelt, J.; et al. Different effects of Sec61-, Sec62 and Sec63-depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 2012, 125, 1958–1969. [Google Scholar] [CrossRef] [Green Version]
- Schäuble, N.; Lang, S.; Jung, M.; Cappel, S.; Schorr, S.; Ulucan, Ö.; Linxweiler, J.; Dudek., J.; Blum, R.; Helms, V.; et al. BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 2012, 31, 3282–3296. [Google Scholar] [CrossRef] [Green Version]
- Kroczynska, B.; Evangelista, C.M.; Samant, S.S.; Elguindi, E.C.; Blond, S.Y. The SANT2 domain of murine tumor cell DnaJ-like protein 1 human homologue interacts with 1-antichymotrypsin and kinetically interferes with its serpin inhibitory activity. J. Biol. Chem. 2004, 279, 11432–11443. [Google Scholar] [CrossRef] [Green Version]
- Zupicich, J.; Brenner, S.E.; Skarnes, W.C. Computational prediction of membrane-tethered transcription factors. Genome Biol. 2001, 2, 501–506. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012, 3, 3157–3168. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Voss, M.; Haug-Kröper, M.; Schröder, B.; Schepers, U.; Bräse, S.; Haass, C.; Lichtenthaler, S.F.; Fluhrer, R. Secretome analysis identifies novel signal Peptide peptidase-like 3 (Sppl3) substrates and reveals a role of Sppl3 in multiple Golgi glycosylation pathways. Mol. Cell Proteom. 2015, 14, 1584–1598. [Google Scholar] [CrossRef] [Green Version]
- Peikert, C.D.; Mani, J.; Morgenstern, M.; Käser, S.; Knapp, B.; Wenger, C.; Harsman, A.; Oeljeklaus, S.; Schneider, A.; Warscheid, B. Charting organellar importomes by quantitative mass spectrometry. Nat Commun. 2017, 8, 15272. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Mayr, C. A membraneless organelle associated with the endoplasmicr reticulum enables 3´UTR-mediated protein-protein interactions. Cell 2018, 175, 1492–1506. [Google Scholar] [CrossRef] [Green Version]
- Berkovits, B.D.; Mayr, C. Alternative 3´UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, N.V.; Dirndorfer, D.; Lang, S.; Resenberger, U.K.; Restelli, L.M.; Hemion, C.; Miesbauer, M.; Frank, S.; Neutzner, A.; Zimmermann, R.; et al. Structural features within the nascent chain regulate alternative targeting of secretory proteins to mitochondria. EMBO J. 2013, 32, 1036–1051. [Google Scholar] [CrossRef] [Green Version]
- Bakheet, T.; Williams, B.R.G.; Khabar, K.S.A. ARED 3.0: The large and diverse AU-rich transcriptome. Nucl. Acids Res. 2006, 34, D111–D114. [Google Scholar] [CrossRef]
- Fallmann, J.; Sedlyarov, V.; Tanzer, A.; Kovarik, P.; Hofacker, I.L. AREsite2: An enhanced database for the comprehensive investigation of AU/GU/U-rich elements. Nucl. Acids Res. 2015, 44, D90–D95. [Google Scholar] [CrossRef] [Green Version]
- Moeller, I.; Jung, M.; Beatrix, B.; Levy, R.; Kreibich, G.; Zimmermann, R.; Wiedmann, M.; Lauring, B. A general mechanism for regulation of access to the translocon: Competition for a membrane attachment site on ribosomes. Proc. Natl. Acad. Sci. USA 1998, 95, 13425–13430. [Google Scholar] [CrossRef] [Green Version]
- Mudunuri, U.; Che, A.; Yi, A.C.; Stephens, R.M. bioDBnet: The biological database network. Bioinformatics 2009, 25, 555–556. [Google Scholar] [CrossRef] [Green Version]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component/Subunit | AREs | Abundance | Location | Linked Diseases |
|---|---|---|---|---|
| # p34 (LRC59, LRRC59) | 1 | 2480 | ERM | |
| # p180 (RRBP1) | 0 | 135 | ERM | Hepatocellular Carcinoma, Colorectal Cancer |
| Kinectin 1 (KTN1) | 3 | 263 | ERM | |
| AEG-1 (LYRIC, MTDH) | 11 | 575 | ERM | |
| # SRP | C | |||
| - SRP68 | 1 | 197 | ||
| - SRP54 | 0 | 228 | Neutropenia, Pancreas Insufficiency | |
| - SRP19 | 8 | 33 | ||
| - SRP14 | 0 | 4295 | ||
| - SRP9 | 12 | 3436 | ||
| -SRP72 | 5 | 355 | Aplasia, Myelodysplasia | |
| - 7SL RNA | ||||
| SRP receptor | ERM | |||
| - SRα | 1 | 249 | ||
| - SRβ | 1 | 173 | ||
| hSnd1 | unknown | |||
| hSnd receptor | ERM | |||
| - hSnd2 (TMEM208) | 0 | 81 | ||
| - hSnd3 | 1 | 49 | ||
| # Bag6 complex | C | |||
| - TRC35 (Get4) | 2 | 171 | ||
| - Ubl4A | NA | 177 | ||
| - Bag6 (Bat3) | 1 | 133 | C C ERM | |
| SGTA | 2 | 549 | ||
| TRC40 (Asna1, Get3) | 0 | 381 | ||
| TA receptor | ||||
| - CAML (CAMLG, Get2) | 3 | 5 | ||
| - WRB (CHD5, Get1) | 4 | 4 | Congenital Heart Disease | |
| PEX1 9PEX3 | 2 3 | 80 103 | C ERM | Zellweger Syndrome Zellweger Syndrome |
| # Sec61 complex | ERM | |||
| - Sec61α1 | 1 | 139 | CVID, TKD, Neutropenia | |
| - Sec61β | 1 | 456 | PCLD, Colorectal cancer | |
| - Sec61γ | 0 | 400 | GBM, Hepatocellular carcinoma | |
| Sec62/Sec63 complex # Sec62 (TLOC1) Sec63 | 14 13 | 26 168 | ERM | Breast-, Prostate-, Cervix-, Lung-Cancer et al. PLD, Colorectal cancer et al. |
| # ERj1 (DNAJC1) | 0 | 8 | ERM | |
| # TRAM1 | 6 | 26 | ERM | |
| TRAM2 | 3 | 40 | ERM | |
| # TRAP complex | ERM | |||
| - TRAPα (SSR1) | 21 | 568 | ||
| - TRAPβ (SSR2) | 0 | |||
| - TRAPγ (SSR3) | 7 | 1701 | CDG, Hepatocellular Carcinoma | |
| - TRAPδ (SSR4) | NA | 3212 | CDG |
| Proteins | SEC61A1 | RRBP1 | KTN1 | ERj1 |
|---|---|---|---|---|
| Quantified proteins | 7212 | 4813 | 4947 | 4947 |
| Statistically analyzed proteins | 5129 | 4813 | 4947 | 4947 |
| Representing the secretory pathway (%) | 26 | 26 | 27 | 27 |
| Proteins with SP (%) | 6 | 6 | 6 | 6 |
| N-Glycoproteins (%) | 8 | 8 | 8 | 8 |
| Membrane proteins (%) | 12 | 12 | 13 | 13 |
| Positively affected proteins | 342 | 157 | 25 | 80 |
| Negatively affected proteins | 482 | 141 | 45 | 92 |
| Representing the secretory pathway (%) | 61 | 37 | 41 | 30 |
| Negatively affected proteins with SP (%) | 41 | 18 | 7 | 8 |
| Negatively affected N-glycoproteins (%) | 45 | 17 | 18 | 13 |
| Negatively affected membrane proteins (%) | 36 | 18 | 22 | 11 |
| Negatively affected proteins with SP | 197 | 21 | 3 | 7 |
| Including N-glycoproteins | 158 | 16 | 3 | 7 |
| Corresponding to % | 80 | 76 | 100 | 100 |
| Including membrane proteins | 77 | 6 | 1 | 2 |
| Corresponding to % | 39 | 29 | 33 | 29 |
| Negatively affected proteins with TMH | 98 | 18 | 8 | 8 |
| Including N-glycoproteins | 56 | 7 | 4 | 3 |
| Corresponding to % | 57 | 39 | 50 | 38 |
| Proteins | BiP | SEC62 | SEC63 | TRAM1 | TRAP |
|---|---|---|---|---|---|
| Quantified proteins | 5543 | 6686 | 6655 | 7502 | 7670 |
| Statistically analyzed proteins | 5543 | 4819 | 6655 | 5961 | 5911 |
| Representing the secretory pathway (%) | 28 | 28 | 28 | 28 | 27 |
| Proteins with SP (%) | 7 | 7 | 7 | 7 | 7 |
| N-Glycoproteins (%) | 9 | 9 | 9 | 9 | 8 |
| Membrane proteins (%) | 14 | 14 | 14 | 14 | 13 |
| Positively affected proteins | 406 | 196 | 13 | 118 | 77 |
| Negatively affected proteins | 340 | 155 | 21 | 86 | 180 |
| Representing the secretory pathway (%) | 28 | 25 | 50 | 48 | 40 |
| Negatively affected proteins with SP (%) | 10 | 12 | 14 | 16 | 22 |
| Negatively affected N-glycoproteins (%) | 11 | 13 | 24 | 21 | 23 |
| Negatively affected membrane proteins (%) | 13 | 8 | 38 | 24 | 26 |
| Negatively affected proteins with SP | 33 | 18 | 3 | 13 | 38 |
| Including N-glycoproteins | 25 | 12 | 2 | 7 | 28 |
| Corresponding to % | 76 | 67 | 67 | 54 | 74 |
| Including membrane proteins | 15 | 2 | 2 | 4 | 19 |
| Corresponding to % | 45 | 11 | 67 | 31 | 50 |
| Negatively affected proteins with TMH | 22 | 6 | 6 | 17 | 22 |
| Including N-glycoproteins | 9 | 5 | 3 | 9 | 11 |
| Corresponding to % | 41 | 83 | 50 | 53 | 50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhadra, P.; Schorr, S.; Lerner, M.; Nguyen, D.; Dudek, J.; Förster, F.; Helms, V.; Lang, S.; Zimmermann, R. Quantitative Proteomics and Differential Protein Abundance Analysis after Depletion of Putative mRNA Receptors in the ER Membrane of Human Cells Identifies Novel Aspects of mRNA Targeting to the ER. Molecules 2021, 26, 3591. https://doi.org/10.3390/molecules26123591
Bhadra P, Schorr S, Lerner M, Nguyen D, Dudek J, Förster F, Helms V, Lang S, Zimmermann R. Quantitative Proteomics and Differential Protein Abundance Analysis after Depletion of Putative mRNA Receptors in the ER Membrane of Human Cells Identifies Novel Aspects of mRNA Targeting to the ER. Molecules. 2021; 26(12):3591. https://doi.org/10.3390/molecules26123591
Chicago/Turabian StyleBhadra, Pratiti, Stefan Schorr, Monika Lerner, Duy Nguyen, Johanna Dudek, Friedrich Förster, Volkhard Helms, Sven Lang, and Richard Zimmermann. 2021. "Quantitative Proteomics and Differential Protein Abundance Analysis after Depletion of Putative mRNA Receptors in the ER Membrane of Human Cells Identifies Novel Aspects of mRNA Targeting to the ER" Molecules 26, no. 12: 3591. https://doi.org/10.3390/molecules26123591