
Triazolyl Conjugated (Oligo)Phenothiazines Building Blocks for Hybrid Materials—Synthesis and Electronic Properties


Abstract
:1. Introduction
2. Results and Discussion
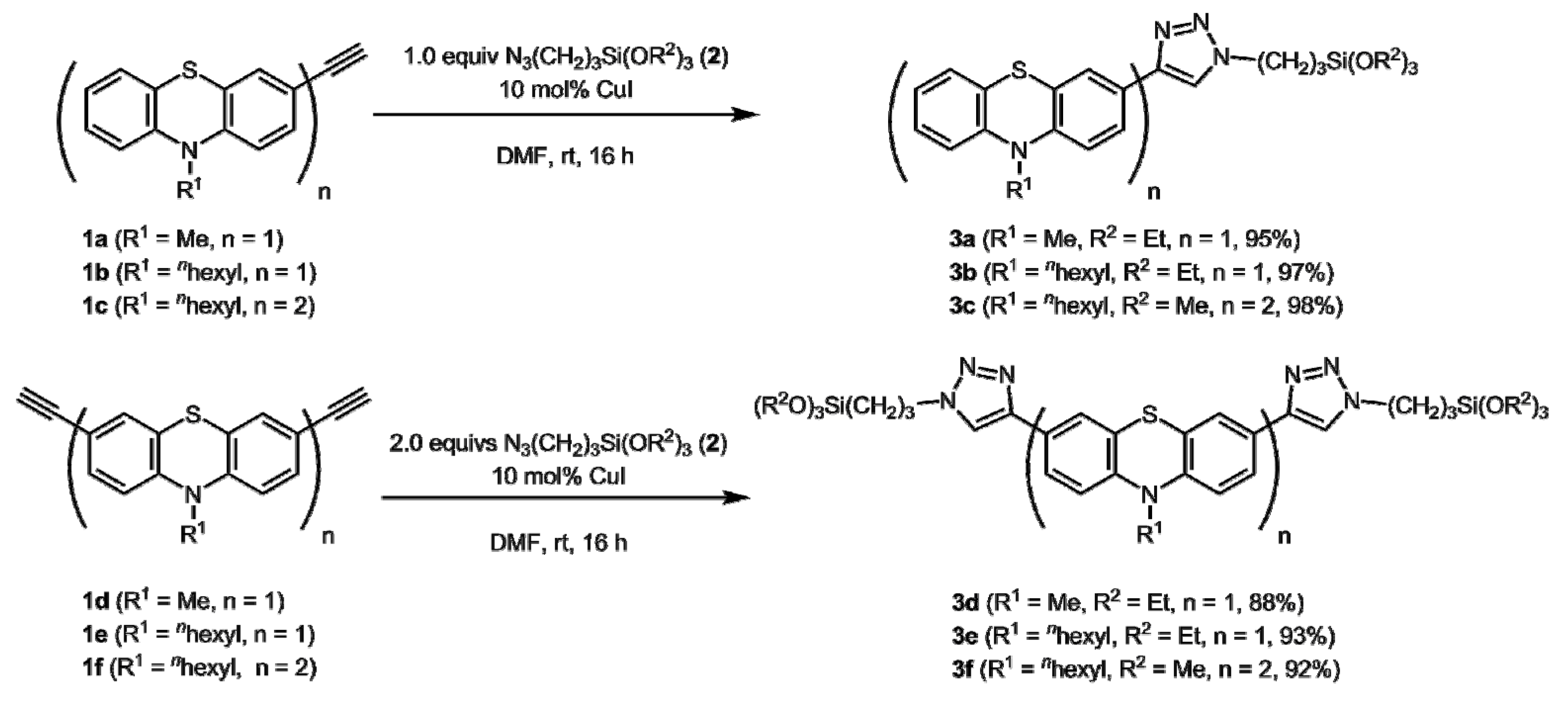
2.1. Synthesis
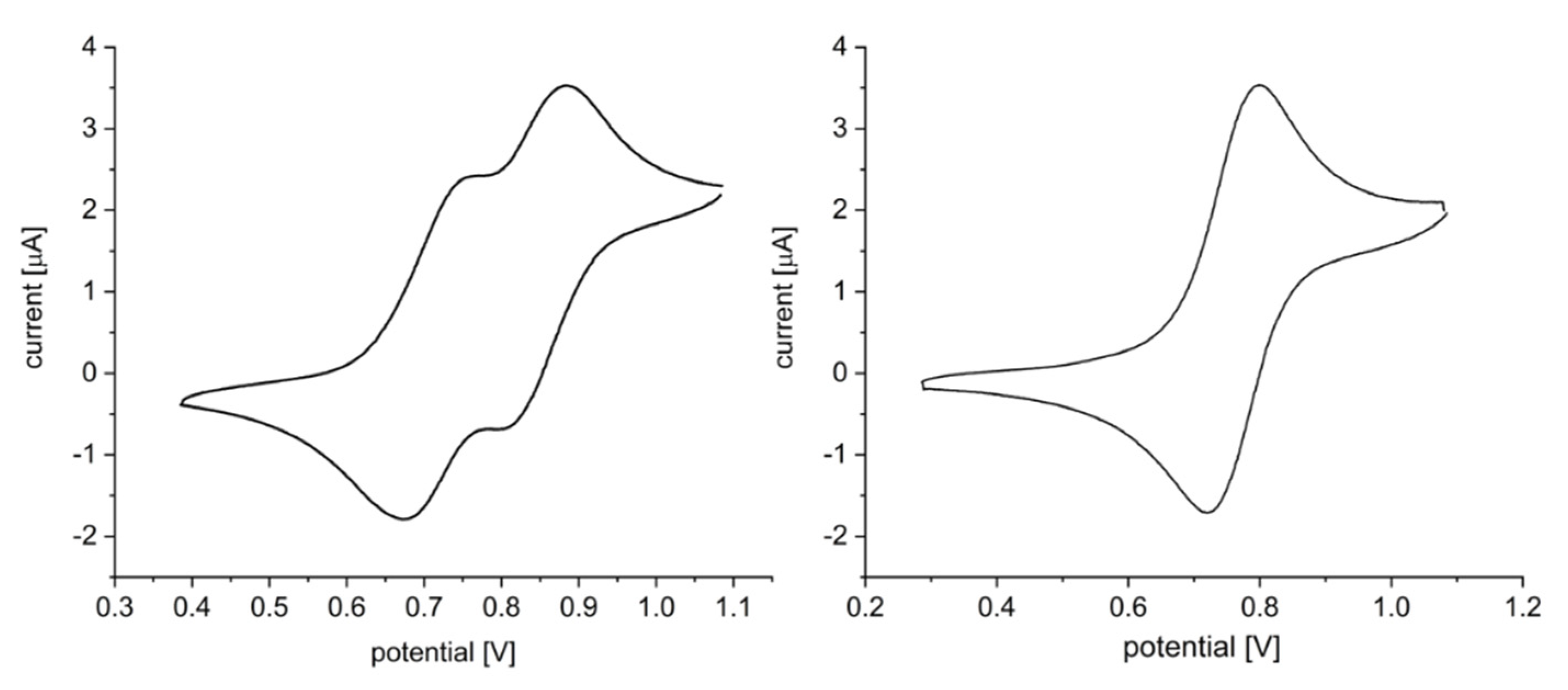
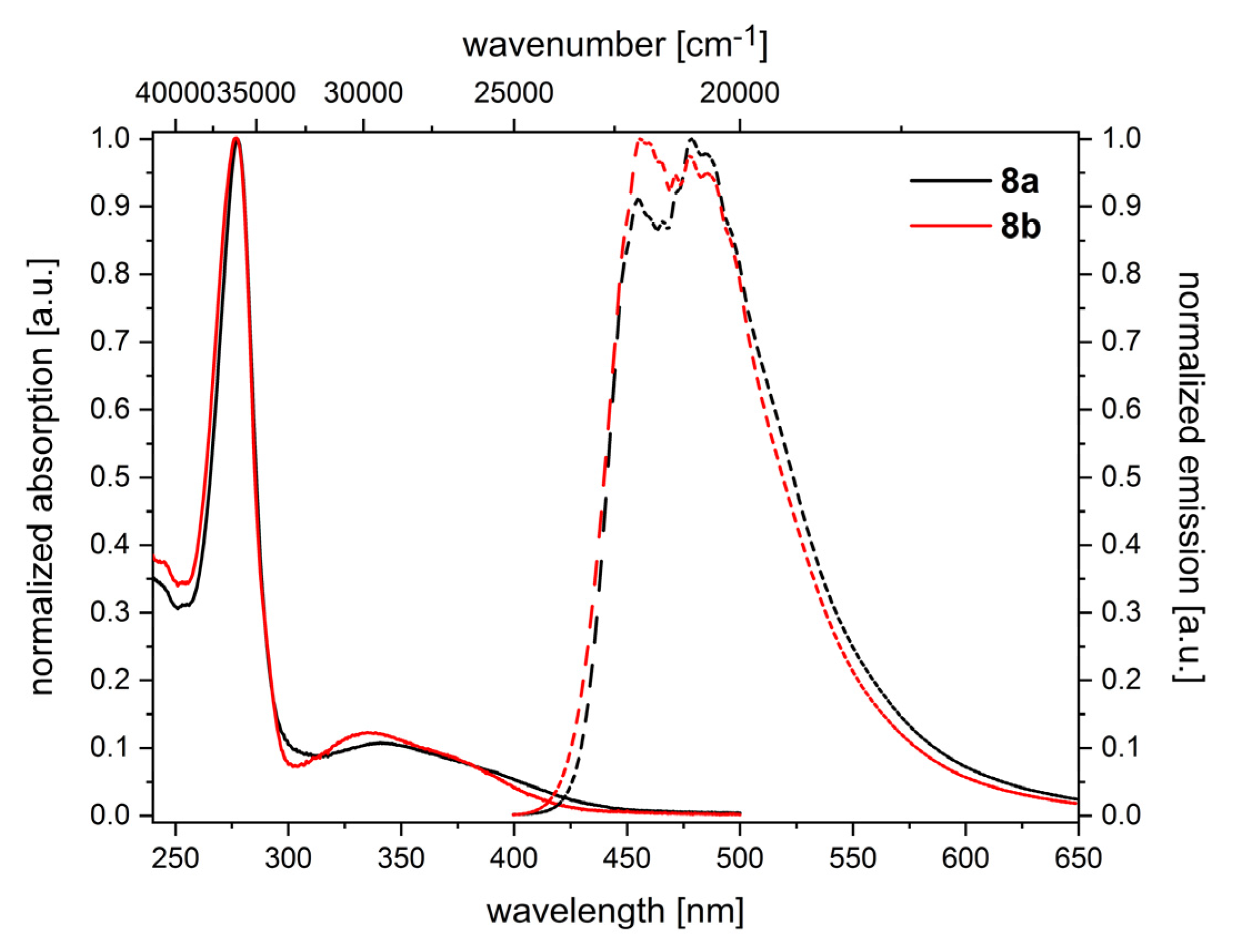
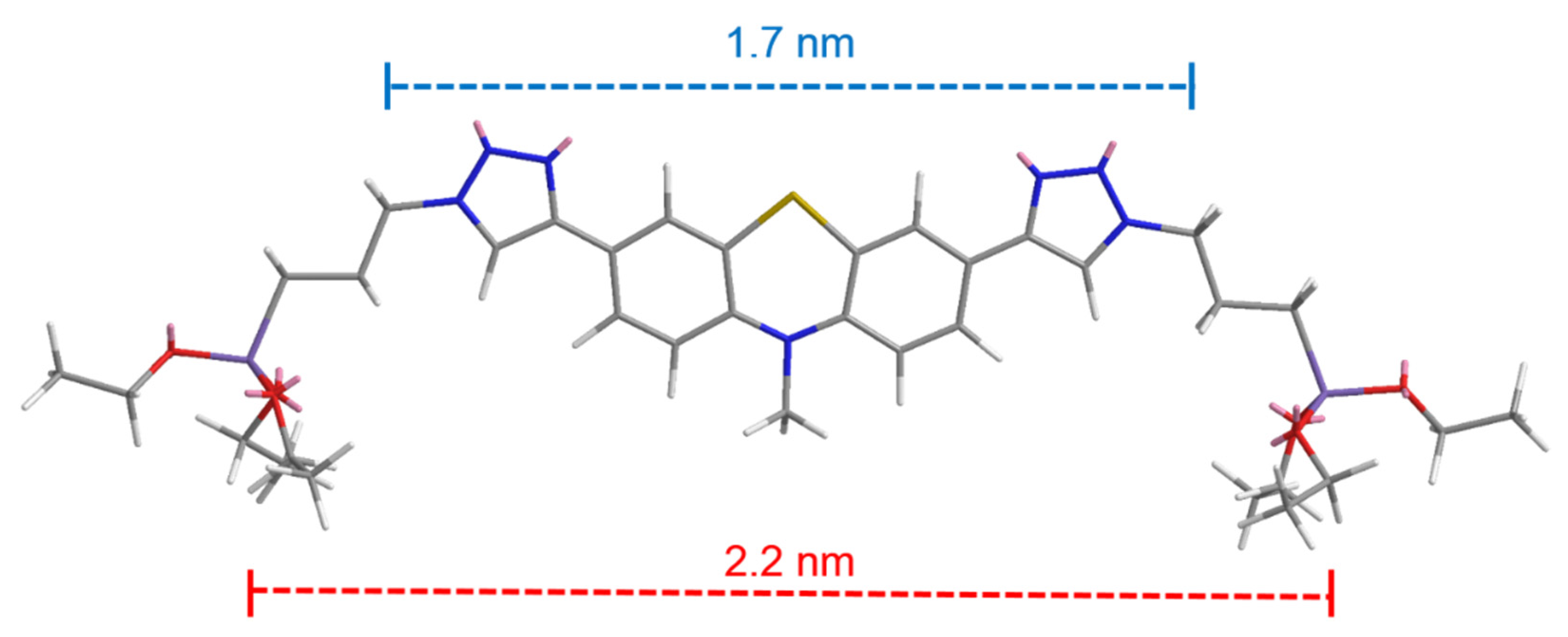
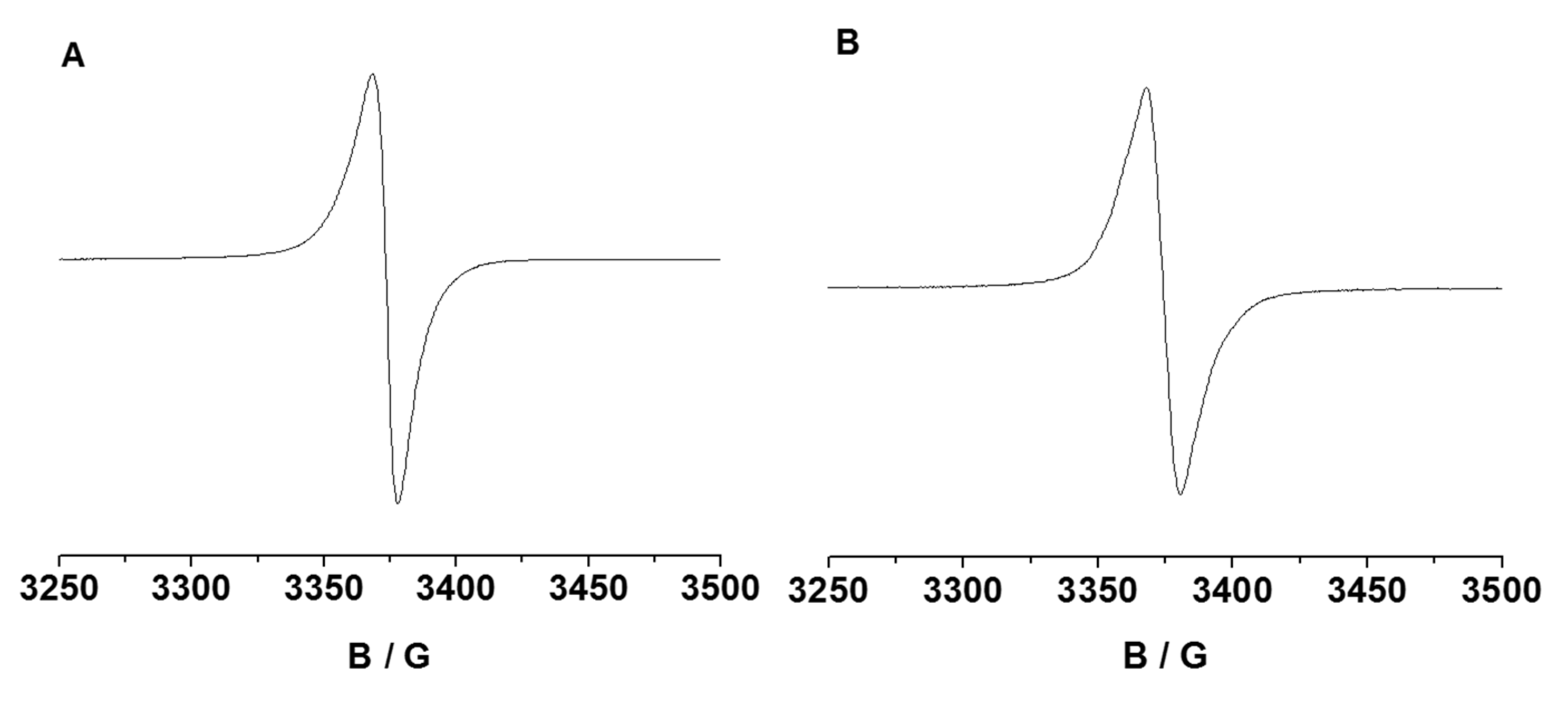
2.2. Electronic Properties of the Triazolyl Conjugated (Oligo)Phenothiazines
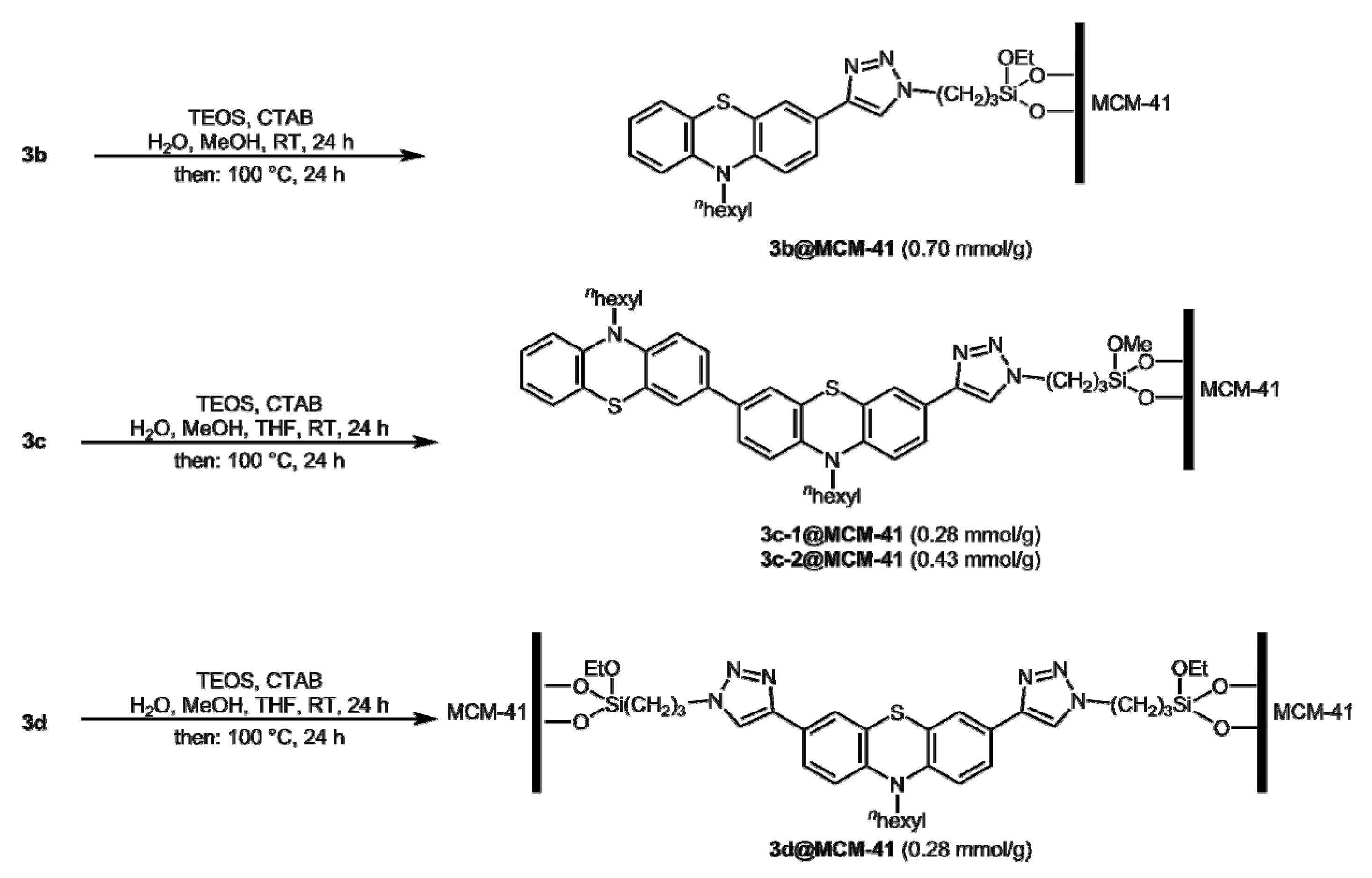
2.3. Silica Hybrid Materials from Selected Triazolyl Conjugated (Oligo)Phenothiazines 3b-d
2.4. Structural Characterization of Silica Hybrid Materials 3b–d@MCM-41
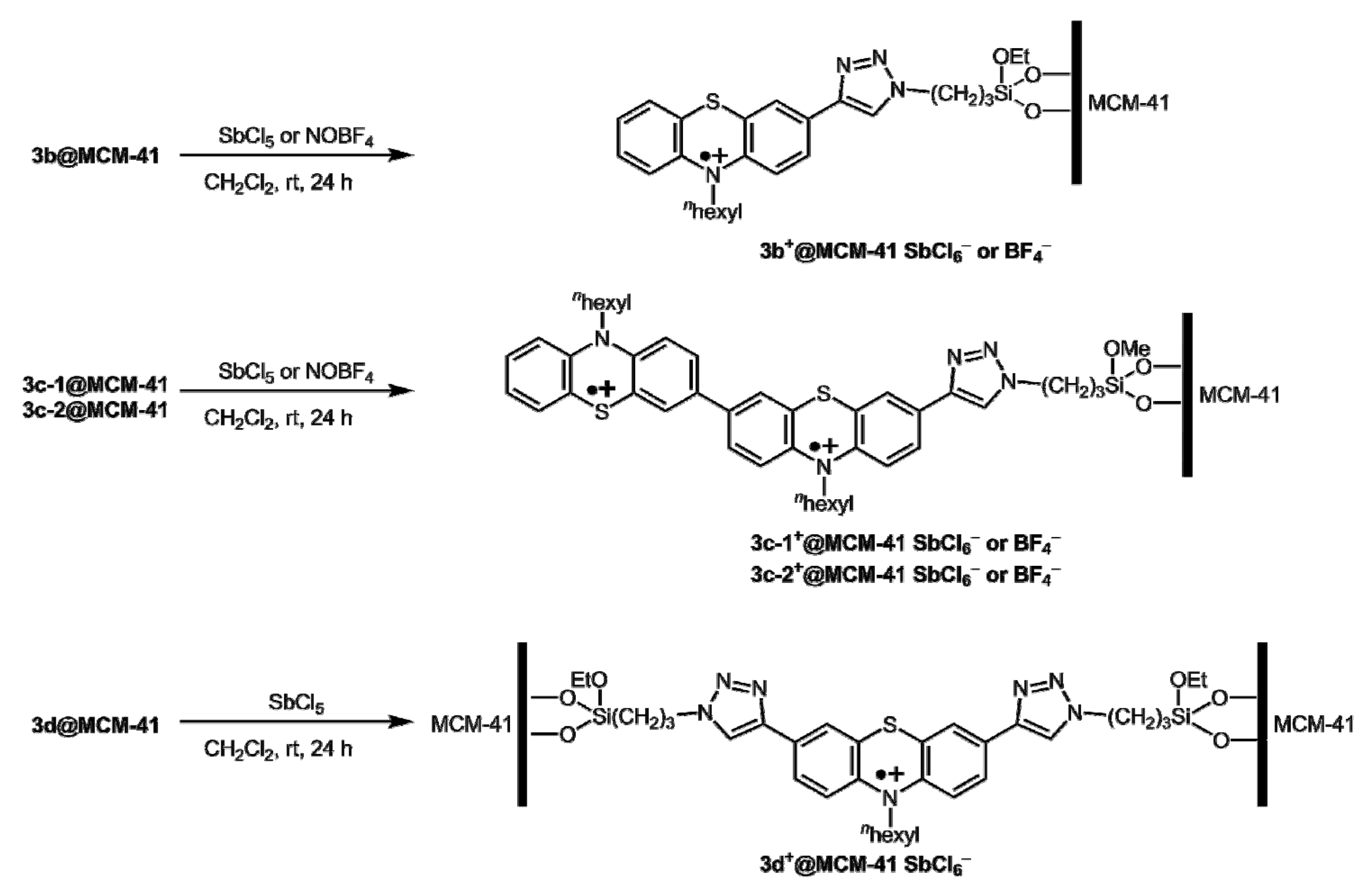
2.5. Optical Properties and Chemical Oxidation of Hybrid Materials 3b–d@MCM-41
3. Materials and Methods
3.1. General Procedure (GP1) for CuAAC Synthesis of 1-(3-(Triethoxysilyl)propyl)-1,2,3-Triazol-4-yl Substituted (Oligo)phenothiazine Precursors 3
3.1.1. 10-Methyl-3-(1-(3-(triethoxysilyl)propyl)-1H-1,2,3-triazole-4-yl)-10H-phenothiazine (3a)
3.1.2. 10-Hexyl-3-(1-(3-(triethoxysilyl)propyl)-1H-1,2,3-triazole-4-yl)-10H-phenothiazine (3b)
3.1.3. 10,10’-Dihexyl-7-(1-(3-(trimethoxysilyl)propyl)-1H-1,2,3-triazole-4-yl)-10H,10’H-3,3’-biphenothiazine (3c)
3.1.4. 10-Methyl-3,7-bis(1-(3-(triethoxysilyl)propyl)-1H-1,2,3-triazole-4-yl)-10H-phenothiazine (3d)
3.1.5. 10-Hexyl-3,7-bis(1-(3-(triethoxysilyl)propyl)-1H-1,2,3-triazole-4-yl)-10H-phenothiazine (3e)
3.1.6. 10,10’Dihexyl-7,7’-bis(1-(3-(trimethoxysilyl)propyl)-1H-1,2,3-triazole-4-yl)-10H,10’H-3,3’-biphenothiazine (3f)
3.2. 1,4-Di(10H-phenothiazin-10-yl)benzene
3.3. 1,4-Bis(3,7-dibromo-10H-phenothiazin-10-yl)benzene (4a)
3.4. 1,4-Bis((3,7-dibromo-10H-phenothiazin-10-yl)methyl)benzene (4b)
3.5. General Procedure (GP2) of Synthesis of Tetrakis(ethynyl)diphenothiazines 7
3.5.1. 1,4-Bis(3,7-diethynyl-10H-phenothiazin-10-yl)benzene (7a)
3.5.2. 1,4-Bis((3,7-diethynyl-10H-phenothiazin-10-yl)methyl)benzene (7b)
3.6. General Procedure (GP3) of the CuAAC Synthesis of Tetrakis[(triethoxysilylpropyl)triazole] Substituted Diphenothiazines 8
3.6.1. 1,4-Bis(3,7-bis(1-(3-(triethoxysilyl)propyl)-1H-1,2,3-triazol-4-yl)-10H-phenothiazin-10-yl)benzol (8a)
3.6.2. 1,4-Bis((3,7-bis(1-(3-(triethoxysilyl)propyl)-1H-1,2,3-triazol-4-yl)-10H-phenothiazin-10-yl)methyl)benzene (8b)
3.7. Synthesis of Hybrid Materials 3b–d@MCM-41
3.7.1. Synthesis of Hybrid Material 3b@MCM-41
3.7.2. Synthesis of Hybrid Material 3c-1@MCM-41
3.7.3. Synthesis of Hybrid Material 3c-2@MCM-41
3.7.4. Synthesis of Hybrid Material 3d@MCM-41
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Breugst, M.; Reißig, H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Huisgen, R.; Szeimies, G.; Mobius, L. 1.3-Dipolare Cycloadditionen, XXXII: Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen. Chem. Ber. 1967, 100, 2494–2507. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Meldal, M.; Tornoe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Bock, V.D.; Hiemstra, H.; van Maarseveen, J.H. CuI-Catalyzed Alkyne–Azide “Click” Cycloadditions from a Mechanistic and Synthetic Perspective. Eur. J. Org. Chem. 2006, 51–68. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Müller, T.J.J. Multicomponent Syntheses Based Upon Cu-Catalyzed Alkyne-Azide Cycloaddition (CuAAC). Adv. Synth. Catal. 2015, 357, 617–666. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.F.; Zarafshani, Z. Efficient construction of therapeutics, bioconjugates, biomaterials and bioactive surfaces using azide-alkyne “click” chemistry. Adv. Drug Deliv. Rev. 2008, 60, 958–970. [Google Scholar] [CrossRef]
- Amblard, F.; Cho, J.H.; Schinazi, R.F. Cu(I)-Catalyzed Huisgen Azide-Alkyne 1,3-Dipolar Cycloaddition Reaction in Nucleoside, Nucleotide, and Oligonucleotide Chemistry. Chem. Rev. 2009, 109, 4207–4220. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.-F. 1,3-Dipolar Cycloadditions of Azides and Alkynes: A Universal Ligation Tool in Polymer and Materials Science. Angew. Chem. Int. Ed. 2007, 46, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ Chemistry in Polymer and Materials Science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Meldal, M. Polymer “Clicking” by CuAAC Reactions. Macromol. Rapid Commun. 2008, 29, 1016–1051. [Google Scholar] [CrossRef]
- Juríček, M.; Kouwer, P.H.J.; Rowan, A.E. Triazole: A unique building block for the construction of functional materials. Chem. Commun. 2011, 47, 8740–8749. [Google Scholar] [CrossRef] [Green Version]
- Espeel, P.; Du Prez, F.E. “Click”-Inspired Chemistry in Macromolecular Science: Matching Recent Progress and User Expectations. Macromolecules 2015, 48, 2–14. [Google Scholar] [CrossRef]
- Delaittre, G.; Guimard, N.K.; Barner-Kowollik, C. Cycloadditions in Modern Polymer Chemistry. Acc. Chem. Res. 2015, 48, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Kautny, P.; Bader, D.; Stöger, B.; Reider, G.A.; Fröhlich, J.; Lumpi, D. Structure-Property Relationships in Click-Derived Donor-Triazole-Acceptor Materials. Chem. Eur. J. 2016, 22, 18887–18898. [Google Scholar] [CrossRef]
- Brunel, D.; Dumur, F. Recent advances in organic dyes and fluorophores comprising a 1,2,3-triazole moiety. New J. Chem. 2020, 44, 3546–3561. [Google Scholar] [CrossRef]
- Malvi, B.; Sarkar, B.R.; Pati, D.; Mathew, R.; Ajithkumarb, T.G.; Sen Gupta, S. “Clickable” SBA-15 mesoporous materials: Synthesis, characterization and their reaction with alkynes. J. Mater. Chem. 2009, 19, 1409–1416. [Google Scholar] [CrossRef]
- Surfaces in Mesoporous Materials for Biosensing Applications Using “Click” Chemistry. Langmuir 2011, 27, 328–334. [CrossRef]
- Börgardts, M.; Verlinden, K.; Neidhardt, M.; Wöhrle, T.; Herbst, A.; Laschat, S.; Janiak, C.; Müller, T.J.J. Synthesis and optical properties of covalently bound Nile Red in mesoporous silica hybrids. RSC Adv. 2016, 6, 6209–6222. [Google Scholar] [CrossRef] [Green Version]
- Börgardts, M.; Müller, T.J.J. Organic fluorophore functionalized mesoporous silica hybrid materials for energy down converting phosphors and application in monolith coated light emitting diodes. Beilstein J. Org. Chem. 2017, 13, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Franz, A.W.; Hartmann, M.; Seifert, A.; Müller, T.J.J.; Thiel, W.R. Novel Organic/Inorganic Hybrid Materials by Covalent Anchoring of Phenothiazines on MCM-41. Chem. Mater. 2008, 20, 4986–4992. [Google Scholar] [CrossRef]
- Franz, A.W.; Zhou, Z.; Turdean, R.; Wagener, A.; Sarkar, B.; Hartmann, M.; Ernst, S.; Thiel, W.R.; Müller, T.J.J. Carbamate-linked (oligo)phenothiazines in mesoporous silica by post-synthetic grafting—A novel type of redox active hybrid materials. Eur. J. Org. Chem. 2009, 3895–3905. [Google Scholar] [CrossRef]
- Zhou, Z.; Franz, A.W.; Bay, S.; Sarkar, B.; Seifert, A.; Yang, P.; Wagener, A.; Ernst, S.; Pagels, M.; Müller, T.J.J.; et al. Redox Active Mesoporous Hybrid Materials by in situ Syntheses with Urea-linked Triethoxysilylated Phenothiazines. Chem. Asian J. 2010, 5, 2001–2015. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.J.J. First Synthesis and Electronic Properties of Ring-Alkynylated Phenothiazines. Tetrahedron Lett. 1999, 40, 6563–6566. [Google Scholar] [CrossRef]
- Krämer, C.S.; Müller, T.J.J. Synthesis and Electronic Properties of Alkynylated Phenothiazines. Eur. J. Org. Chem. 2003, 3534–3548. [Google Scholar] [CrossRef]
- Bucci, N.; Müller, T.J.J. Synthesis and Electronic Properties of (Oligo)Phenothiazine-Ethynyl-Hydro-C60 Dyads. Tetrahedron Lett. 2006, 47, 8329–8332. [Google Scholar] [CrossRef]
- Urselmann, D.; Deilhof, K.; Mayer, B.; Müller, T.J.J. Thiophene-Forming One-pot Synthesis of Three Thienyl-bridged Oligophenothiazines and Their Electronic Properties. Beilstein J. Org. Chem. 2016, 12, 2055–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Munoz, M.; Lopez-Jaramillo, J.; Hernandez-Mateo, F.; Santoyo-Gonzaleza, F. Synthesis of Glyco-Silicas by Cu(I)-Catalyzed “Click-Chemistry” and their Applications in Affinity Chromatography. Adv. Synth. Catal. 2006, 348, 2410–2420. [Google Scholar] [CrossRef]
- Sailer, M.; Franz, A.W.; Müller, T.J.J. Synthesis and Electronic Properties of Monodisperse Oligophenothiazines. Chem. Eur. J. 2008, 14, 2602–2614. [Google Scholar] [CrossRef]
- Yang, L.; Feng, J.-K.; Ren, A.-M. Theoretical Study on Electronic Structure and Optical Properties of Phenothiazine-Containing Conjugated Oligomers and Polymers. J. Org. Chem. 2005, 70, 5987–5996. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Jia, M.; Seifert, A.; Berger, M.; Giegengack, H.; Schulze, S.; Thiel, W.R. Hybrid Mesoporous Materials with a Uniform Ligand Distribution: Synthesis, Characterization, and Application in Epoxidation Catalysis. Chem. Mater. 2004, 16, 877–882. [Google Scholar] [CrossRef]
- Marler, B.; Oberhagemann, U.; Vortmann, S.; Gies, H. Influence of the sorbate type on the XRD peak intensities of loaded MCM-41. Microporous Mater. 1996, 6, 375–383. [Google Scholar] [CrossRef]
- Soler-Illia, G.J.d.A.; Sanchez, C.; Lebeau, B.; Patarin, J. Chemical Strategies to Design Textured Materials: from Microporous and Mesoporous Oxides to Nanonetworks and Hierarchical Structures. Chem. Rev. 2002, 102, 4093–4138. [Google Scholar] [CrossRef] [PubMed]
- Tanev, P.T.; Pinnavaia, T.J. A Neutral Templating Route to Mesoporous Molecular Sieves. Science 1995, 267, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.S.; Lu, G.Q.; Whittaker, A.K.; Millar, G.J.; Zhu, H.Y. Comprehensive Study of Surface Chemistry of MCM-41 Using 29Si CP/MAS NMR, FTIR, Pyridine-TPD, and TGA. J. Phys. Chem. B 1997, 101, 6525–6531. [Google Scholar] [CrossRef]
- Nabokoff, P.; Gastaldi, S.; Besson, E. Probing the efficiency of thermal and photochemical bond homolysis in functionalized nanostructured SBA-15 silicas. Microporous Mesoporous Mat. 2021, 311, 110674. [Google Scholar] [CrossRef]
- Franz, A.W.; Popa, L.N.; Rominger, F.; Müller, T.J.J. Synthesis and electronic communication in diphenothiazine dumbbells bridged by heterocycles. Org. Biomol. Chem. 2009, 7, 469–475. [Google Scholar] [CrossRef]
- Franz, A.W.; Rominger, F.; Müller, T.J.J. Synthesis and Electronic Properties of Sterically Demanding N-Arylphenothiazines and Unexpected Buchwald-Hartwig Aminations. J. Org. Chem. 2008, 73, 1795–1802. [Google Scholar] [CrossRef]
- Braun, D.; Langendorf, R. Synthesis and Characterization of Low Molecular Weight Organic Glasses. J. Prakt. Chem. 1999, 341, 128–137. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Oxidation Potentials 1 | Absorption 2 | Emission 3 | Stokes Shift 4 | |
|---|---|---|---|---|---|
| E1/20/+1 | E1/2+1/+2 | λmax,abs | λmax,abs | ||
| [mV] | [mV] | [nm] (ε [m−1cm−1]) | [nm] | [cm−1] | |
| 3a | 750 | - | 266 (49200), 319 (9950) | 446, 469sh | 8900 |
| 3b | 720 | - | 268 (55400), 318 (14250) | 450 | 9200 |
| 3c | 650 | 790 | 278 (43200), 328 (14600), 367 (12300) | 470, 505sh | 6000 |
| 3d | 730 | - | 276 (37800), 334 (7200) | 459 | 8100 |
| 3e | 700 | - | 276 (54500), 335 (10100) | 460, 484sh | 8100 |
| 3f | 650 | 820 | 278 (50700), 336 (16100) | 469, 506sh | 5700 |
| 8a5 | 700 | 880 | 279 (183000), 343 (20000) | 455, 479 | 7200 |
| 8b5 | 800 | - | 274 (83000), 335 (10000) | 456, 485 | 7900 |
| Sample | Content of Phenothiazine 1 | d1002 | a03 | SBET4 | Vp5 | Dp 6 | wt 7 | |
|---|---|---|---|---|---|---|---|---|
| [mmol g–1] | [wt%] | [nm] | [nm] | [m2g–1] | [cm3g–1] | [nm] | [nm] | |
| MCM-41 | − | − | 4.30 | 4.96 | 1123 | 0.98 | 2.83 | 2.13 |
| 3b@MCM-41 | 0.70 | 30.4 | 4.20 | 4.85 | 536 | 0.36 | 2.36 | 2.49 |
| 3c-1@MCM-41 | 0.28 | 18.3 | 4.44 | 5.13 | 1086 | 0.96 | 2.82 | 2.31 |
| 3c-2@MCM-41 | 0.43 | 28.1 | 4.50 | 5.20 | 543 | 0.48 | 2.77 | 2.43 |
| 3d@MCM-41 | 0.28 | 14.9 | 4.48 | 5.17 | 539 | 0.41 | 2.41 | 2.76 |
| Compound | Absorption | Emission | Stokes Shift 3 |
|---|---|---|---|
| λmax,abs | λmax,abs | Δ | |
| [nm] | [nm] | [cm−1] | |
| 3b | 268, 318 | 450 | 9200 |
| 3b@MCM-41 | 260, 320 | 451 | 9100 |
| 3c | 278, 328, 367 | 470, 505sh | 6000 |
| 3c-1@MCM-41 | 276, 330, 370 | 484, 509sh | 6400 |
| 3c-2@MCM-41 | 275, 327, 376 | 500 | 6700 |
| 3d | 276, 334 | 459 | 8100 |
| 3d@MCM-41 | 270, 327, 360 | 390, 445 | 5300 |
| Entry | Alkyne 1 | Azide 2 | CuI | DMF | Product 3 Yield |
|---|---|---|---|---|---|
| [mg] ([μmol]) | [mg] ([μmol]) | [mg] ([μmol]) | [mL] | [mg] (%) | |
| 1 | 100 (422) of 1a | 105 (422) of 2a | 8.0 (42) | 3 mL | 193 (95) of 3a |
| 2 | 129 (422) of 1b | 105 (422) of 2a | 8.0 (42) | 3 mL | 228 (97) of 3b |
| 3 | 735 (1250) of 1c | 256 (1250) of 2b | 24.0 (125) | 7 mL | 970 (98) of 3c |
| 4 | 55 (211) of 1d | 105 (422) of 2a | 4.0 (42) | 3 mL | 140 (88) of 3d |
| 5 | 270 (815) of 1e | 402 (1630) of 2a | 16.0 (82) | 3 mL | 625 (93) of 3e |
| 6 | 59 (96) of 1f | 41 (200) of 2b | 1.9 (10) | 3 mL | 90 (92) of 3f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khelwati, H.; Franz, A.W.; Zhou, Z.; Thiel, W.R.; Müller, T.J.J. Triazolyl Conjugated (Oligo)Phenothiazines Building Blocks for Hybrid Materials—Synthesis and Electronic Properties. Molecules 2021, 26, 2950. https://doi.org/10.3390/molecules26102950
Khelwati H, Franz AW, Zhou Z, Thiel WR, Müller TJJ. Triazolyl Conjugated (Oligo)Phenothiazines Building Blocks for Hybrid Materials—Synthesis and Electronic Properties. Molecules. 2021; 26(10):2950. https://doi.org/10.3390/molecules26102950
Chicago/Turabian StyleKhelwati, Hilla, Adam W. Franz, Zhou Zhou, Werner R. Thiel, and Thomas J. J. Müller. 2021. "Triazolyl Conjugated (Oligo)Phenothiazines Building Blocks for Hybrid Materials—Synthesis and Electronic Properties" Molecules 26, no. 10: 2950. https://doi.org/10.3390/molecules26102950