
The Glycolytic Pathway as a Target for Novel Onco-Immunology Therapies in Pancreatic Cancer

 , ,
, ,  and
and
Abstract
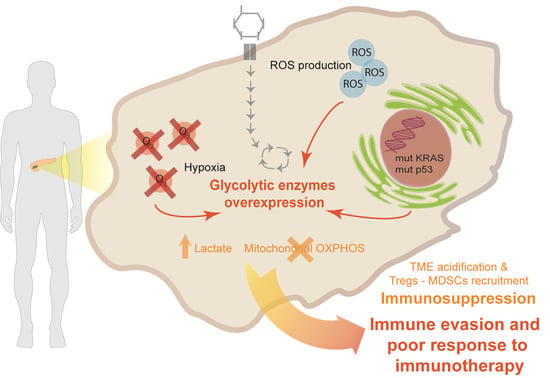
:
1. Introduction
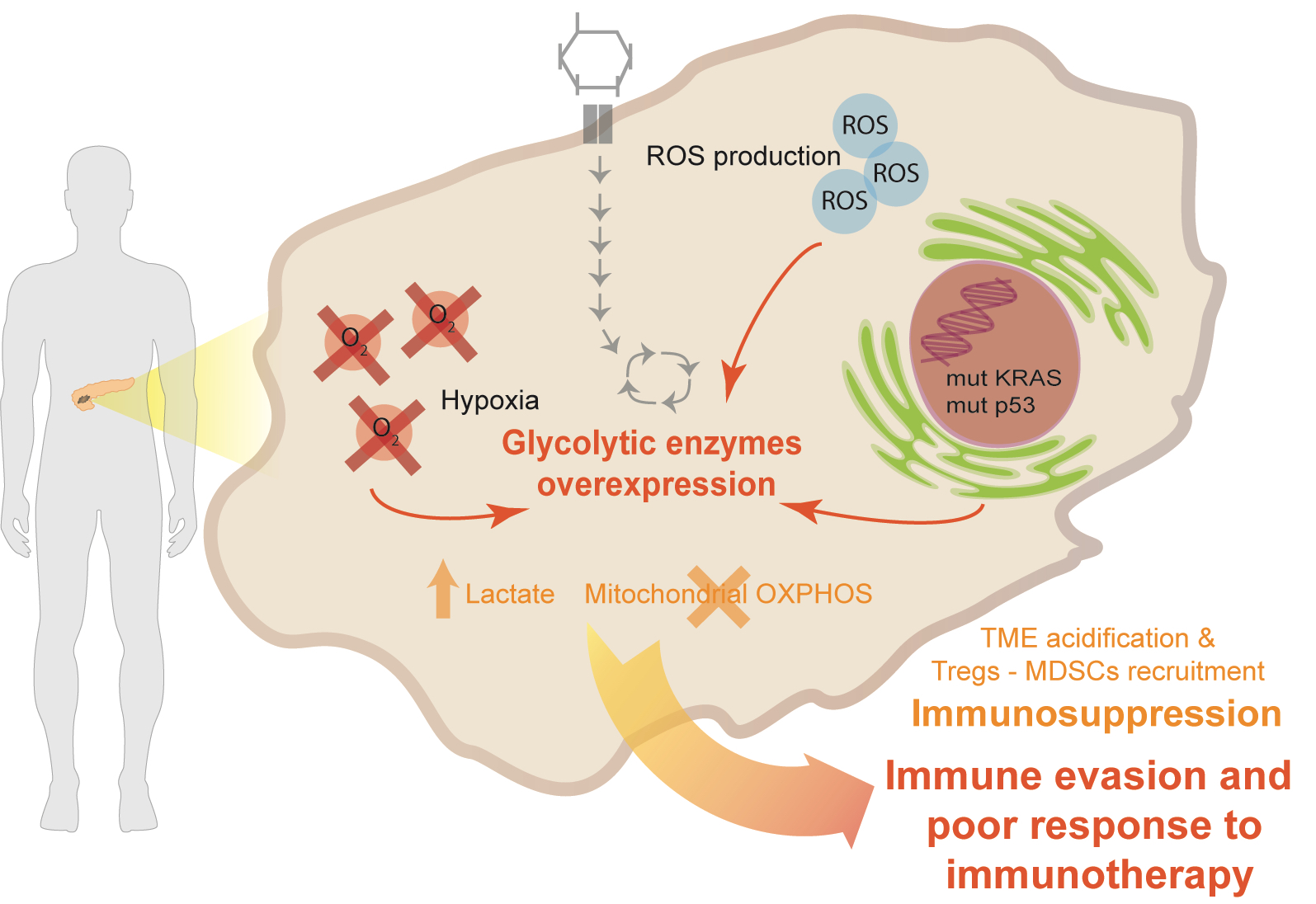
2. KRAS Aberrant Signaling and TP53 Mutation in PDA
TP53 Induced Glycolysis Regulatory Phosphatase (TIGAR)
3. Glucose Transporter 1 (GLUT1)
4. Hexokinase 2 (HK2)
5. Aldolase A (ALDOA)
6. Triose Phosphate Isomerase (TPI) and Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
7. Forkhead Box Protein M1 (FOXM1)
8. Phosphoglycerate Kinase 1 (PGK1)
9. Lactate Dehydrogenase A (LDHA)
10. Alpha Enolase (ENO1)
11. Pyruvate Kinase M2 (PKM2)
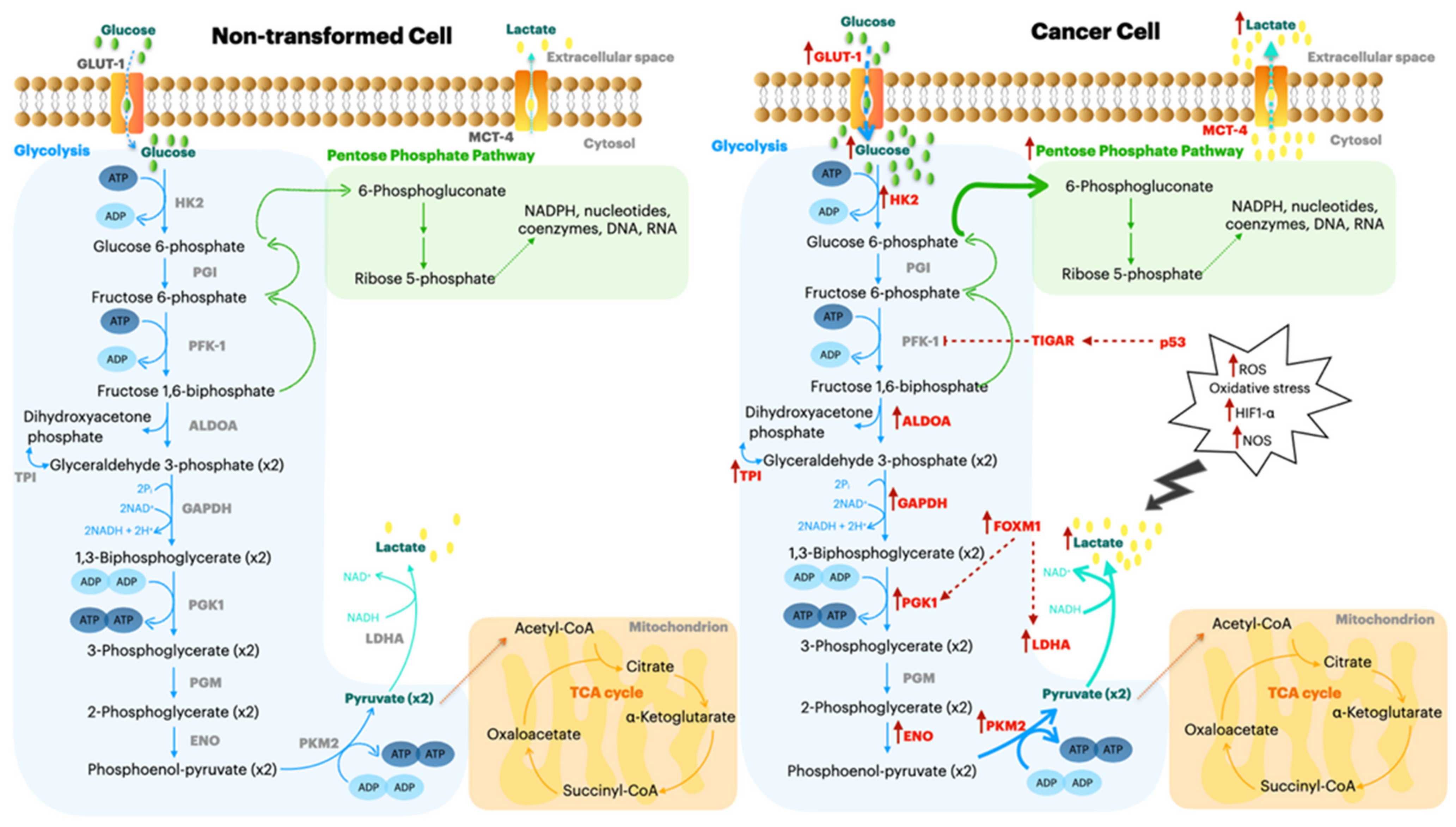
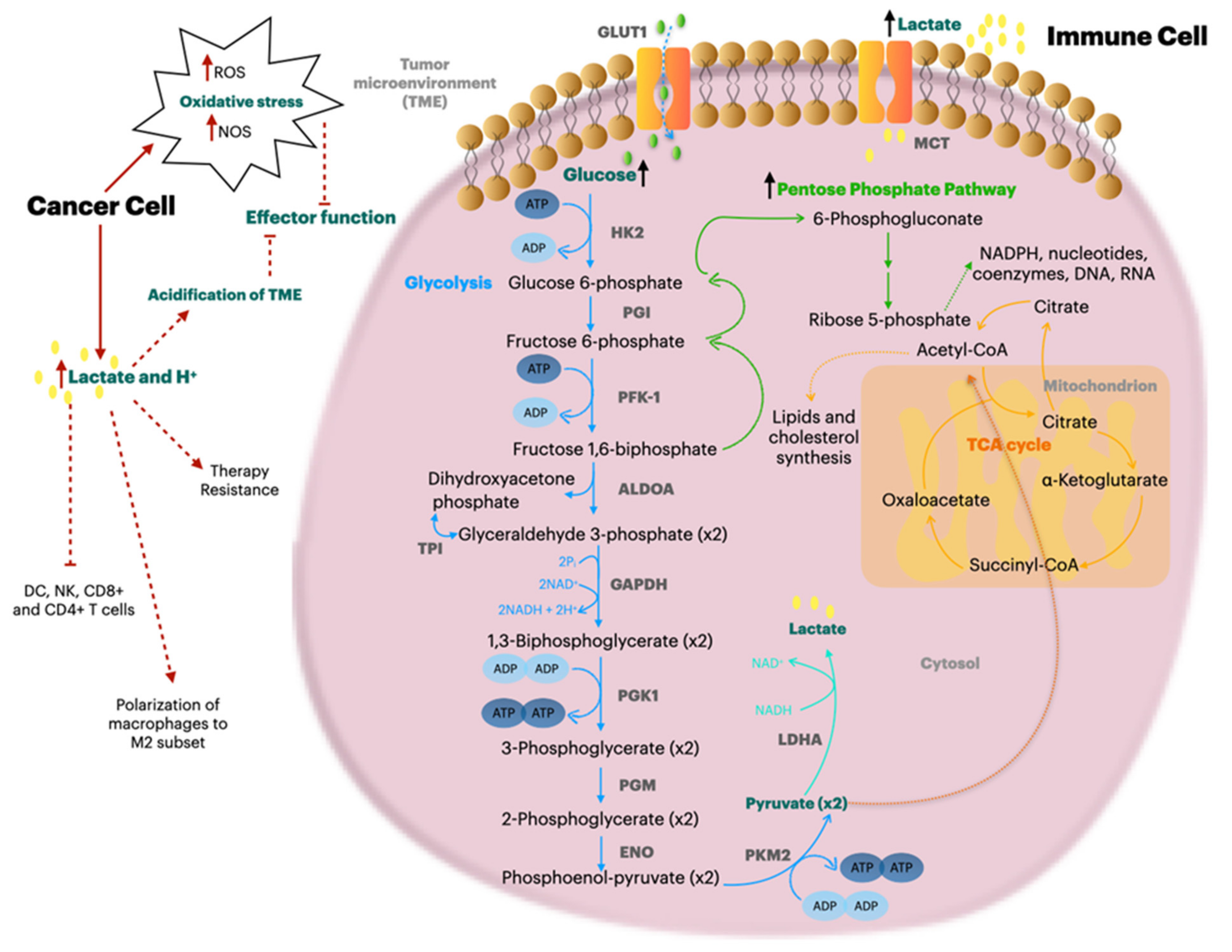
12. Metabolic Reprogramming to Increase the Immune Response in Tumors
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical Implications of Genomic Alterations in the Tumour and Circulation of Pancreatic Cancer Patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy Metabolism in Tumor Cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezene, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened Glycolysis under Hypoxia Supports Tumor Symbiosis and Hexosamine Biosynthesis in Pancreatic Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [Green Version]
- Blum, R.; Kloog, Y. Metabolism Addiction in Pancreatic Cancer. Cell Death Dis. 2014, 5, e1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Liu, Z.; Li, M.; Chen, C.; Wang, X. Increased Glycolysis Correlates with Elevated Immune Activity in Tumor Immune Microenvironment. EBioMedicine 2019, 42, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaino, B.; Cappello, P.; Capello, M.; Fredolini, C.; Ponzetto, A.; Novarino, A.; Ciuffreda, L.; Bertetto, O.; De Angelis, C.; Gaia, E.; et al. Autoantibody Signature in Human Ductal Pancreatic Adenocarcinoma. J. Proteome Res. 2007, 6, 4025–4031. [Google Scholar] [CrossRef]
- Mandili, G.; Curcio, C.; Bulfamante, S.; Follia, L.; Ferrero, G.; Mazza, E.; Principe, M.; Cordero, F.; Satolli, M.A.; Spadi, R.; et al. In Pancreatic Cancer, Chemotherapy Increases Antitumor Responses to Tumor-Associated Antigens and Potentiates DNA Vaccination. J. Immunother. Cancer 2020, 8, e001071. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of Oncogenic KRAS in the Diagnosis, Prognosis and Treatment of Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, N.V.; Dutta, P.; Yabuuchi, S.; de Wilde, R.F.; Martinez, G.V.; Le, A.; Kamphorst, J.J.; Rabinowitz, J.D.; Jain, S.K.; Hidalgo, M.; et al. Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of P53 Function. Cancer Res. 2015, 75, 3355–3364. [Google Scholar] [CrossRef] [Green Version]
- Follia, L.; Ferrero, G.; Mandili, G.; Beccuti, M.; Giordano, D.; Spadi, R.; Satolli, M.A.; Evangelista, A.; Katayama, H.; Hong, W.; et al. Integrative Analysis of Novel Metabolic Subtypes in Pancreatic Cancer Fosters New Prognostic Biomarkers. Front. Oncol. 2019, 9, 115. [Google Scholar] [CrossRef]
- Mosolits, S.; Ullenhag, G.; Mellstedt, H. Therapeutic Vaccination in Patients with Gastrointestinal Malignancies. A Review of Immunological and Clinical Results. Ann. Oncol. 2005, 16, 847–862. [Google Scholar] [CrossRef]
- Zhou, J.; Hui, X.; Mao, Y.; Fan, L. Identification of Novel Genes Associated with a Poor Prognosis in Pancreatic Ductal Adenocarcinoma via a Bioinformatics Analysis. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Wang, K.; Geng, L.; Sun, J.; Xu, W.; Liu, D.; Gong, S.; Zhu, Y. Identification of Candidate Diagnostic and Prognostic Biomarkers for Pancreatic Carcinoma. EBioMedicine 2019, 40, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basturk, O.; Singh, R.; Kaygusuz, E.; Balci, S.; Dursun, N.; Culhaci, N.; Adsay, N.V. GLUT-1 Expression in Pancreatic Neoplasia: Implications in Pathogenesis, Diagnosis, and Prognosis. Pancreas 2011, 40, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, N.; Capretti, G.; Barbagallo, M.; Rigamonti, A.; Takis, P.G.; Castino, G.F.; Vignali, D.; Maggi, G.; Gavazzi, F.; Ridolfi, C.; et al. Metabolome of Pancreatic Juice Delineates Distinct Clinical Profiles of Pancreatic Cancer and Reveals a Link between Glucose Metabolism and PD-1+ Cells. Cancer Immunol. Res. 2020, 8, 493–505. [Google Scholar] [CrossRef]
- Wan, Y.; Zhang, Y.; Wang, G.; Mwangi, P.M.; Cai, H.; Li, R. Recombinant KRAS G12D Protein Vaccines Elicit Significant Anti-Tumor Effects in Mouse CT26 Tumor Models. Front. Oncol. 2020, 10, 1326. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Shi, S.; Liang, C.; Meng, Q.; Xiang, J.; Liang, D.; et al. A New Facet of NDRG1 in Pancreatic Ductal Adenocarcinoma: Suppression of Glycolytic Metabolism. Int. J. Oncol. 2017, 50, 1792–1800. [Google Scholar] [CrossRef]
- Ito, H.; Duxbury, M.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Glucose Transporter-1 Gene Expression Is Associated with Pancreatic Cancer Invasiveness and MMP-2 Activity. Surgery 2004, 136, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Maemura, K.; Mataki, Y.; Sakoda, M.; Iino, S.; Kawasaki, Y.; Arigami, T.; Mori, S.; Kijima, Y.; Ueno, S.; et al. Significance of Glucose Transporter Type 1 (GLUT-1) Expression in the Therapeutic Strategy for Pancreatic Ductal Adenocarcinoma. Ann. Surg. Oncol. 2018, 25, 1432–1439. [Google Scholar] [CrossRef]
- Wang, X.-X.; Yin, G.-Q.; Zhang, Z.-H.; Rong, Z.-H.; Wang, Z.-Y.; Du, D.-D.; Wang, Y.-D.; Gao, R.-X.; Xian, G.-Z. TWIST1 Transcriptionally Regulates Glycolytic Genes to Promote the Warburg Metabolism in Pancreatic Cancer. Exp. Cell Res. 2020, 386, 111713. [Google Scholar] [CrossRef]
- Ren, W.; Qiao, Z.; Wang, H.; Zhu, L.; Zhang, L. Flavonoids: Promising Anticancer Agents. Med. Res. Rev. 2003, 23, 519–534. [Google Scholar] [CrossRef]
- Melstrom, L.; Salabat, M.; Ding, X.-Z.; Milam, B.; Strouch, M.; Pelling, J.; Bentrem, D. Apigenin Inhibits the GLUT-1 Glucose Transporter and the Phosphoinositide 3-Kinase/Akt Pathway in Human Pancreatic Cancer Cells. Pancreas 2008, 37, 426–431. [Google Scholar] [CrossRef]
- Nagarajan, A.; Dogra, S.K.; Sun, L.; Gandotra, N.; Ho, T.; Cai, G.; Cline, G.; Kumar, P.; Cowles, R.A.; Wajapeyee, N. Paraoxonase 2 Facilitates Pancreatic Cancer Growth and Metastasis by Stimulating GLUT1-Mediated Glucose Transport. Mol. Cell 2017, 67, 685–701.e6. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, F.; Vignali, D.; Manini, B.; Rigamonti, A.; Monti, P. Manipulation of Glucose Availability to Boost Cancer Immunotherapies. Cancers 2020, 12. [Google Scholar] [CrossRef]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 Promotes Tumor Growth and Metastasis by Regulating Lactate Production in Pancreatic Cancer. Oncotarget 2016, 8, 56081–56094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Fan, Z.; Cheng, H.; Huang, Q.; Yang, C.; Jin, K.; Luo, G.; Yu, X.; Liu, C. Hexokinase 2 Dimerization and Interaction with Voltage-dependent Anion Channel Promoted Resistance to Cell Apoptosis Induced by Gemcitabine in Pancreatic Cancer. Cancer Med. 2019, 8, 5903–5915. [Google Scholar] [CrossRef]
- Chaika, N.V.; Yu, F.; Purohit, V.; Mehla, K.; Lazenby, A.J.; DiMaio, D.; Anderson, J.M.; Yeh, J.J.; Johnson, K.R.; Hollingsworth, M.A.; et al. Differential Expression of Metabolic Genes in Tumor and Stromal Components of Primary and Metastatic Loci in Pancreatic Adenocarcinoma. PLoS ONE 2012, 7, e32996. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, H.; Nagano, H.; Konno, M.; Eguchi, H.; Koseki, J.; Kawamoto, K.; Nishida, N.; Colvin, H.; Tomokuni, A.; Tomimaru, Y.; et al. The Combination of the Expression of Hexokinase 2 and Pyruvate Kinase M2 Is a Prognostic Marker in Patients with Pancreatic Cancer. Mol. Clin. Oncol. 2015, 3, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyshchik, A.; Higashi, T.; Hara, T.; Nakamoto, Y.; Fujimoto, K.; Doi, R.; Imamura, M.; Saga, T.; Togashi, K. Expression of Glucose Transporter-1, Hexokinase-II, Proliferating Cell Nuclear Antigen and Survival of Patients with Pancreatic Cancer. Cancer Investig. 2007, 25, 154–162. [Google Scholar] [CrossRef]
- Luo, T.; Fredericksen, B.L.; Hasumi, K.; Endo, A.; Garcia, J.V. Human Immunodeficiency Virus Type 1 Nef-Induced CD4 Cell Surface Downregulation Is Inhibited by Ikarugamycin. J. Virol. 2001, 75, 2488–2492. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.-H.; Dong, F.-Y.; Da, L.-T.; Yang, X.-M.; Wang, X.-X.; Weng, J.-Y.; Feng, L.; Zhu, L.-L.; Zhang, Y.-L.; Zhang, Z.-G.; et al. Ikarugamycin Inhibits Pancreatic Cancer Cell Glycolysis by Targeting Hexokinase 2. FASEB J. 2020, 34. [Google Scholar] [CrossRef]
- Ji, S.; Zhang, B.; Liu, J.; Qin, Y.; Liang, C.; Shi, S.; Jin, K.; Liang, D.; Xu, W.; Xu, H.; et al. ALDOA Functions as an Oncogene in the Highly Metastatic Pancreatic Cancer. Cancer Lett. 2016, 374, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, G.; de Jong, P.R.; James, B.P.; Koh, M.Y.; Lemos, R.; Kingston, J.; Aleshin, A.; Bankston, L.A.; Miller, C.P.; Cho, E.J.; et al. Definition of a Novel Feed-Forward Mechanism for Glycolysis-HIF1α Signaling in Hypoxic Tumors Highlights Aldolase A as a Therapeutic Target. Cancer Res. 2016, 76, 4259–4269. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Nishioka, M.; Imura, S.; Batmunkh, E.; Uto, Y.; Nagasawa, H.; Hori, H.; Shimada, M. The Novel Hypoxic Cytotoxin, TX-2098 Has Antitumor Effect in Pancreatic Cancer; Possible Mechanism through Inhibiting VEGF and Hypoxia Inducible Factor-1α Targeted Gene Expression. Exp. Cell Res. 2012, 318, 1554–1563. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, G.; Paccielli Freire, P.; Santiloni Cury, S.; de Moraes, D.; Santos Oliveira, J.; Dal-Pai-Silva, M.; do Reis, P.P.; Francisco Carvalho, R. An Integrated Meta-Analysis of Secretome and Proteome Identify Potential Biomarkers of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Peng, Y.; Zhu, Y.; Xu, D.; Zhu, F.; Xu, W.; Chen, Q.; Zhu, X.; Liu, T.; Hou, C.; et al. Glycolysis Promotes the Progression of Pancreatic Cancer and Reduces Cancer Cell Sensitivity to Gemcitabine. Biomed. Pharmacother. 2020, 121, 109521. [Google Scholar] [CrossRef]
- Schek, N.; Hall, B.L.; Finn, O.J. Increased Glyceraldehyde-3-Phosphate Dehydrogenase Gene Expression in Human Pancreatic Adenocarcinoma. Cancer Res. 1988, 48, 6354–6359. [Google Scholar] [PubMed]
- Mikuriya, K.; Kuramitsu, Y.; Ryozawa, S.; Fujimoto, M.; Mori, S.; Oka, M.; Hamano, K.; Okita, K.; Sakaida, I.; Nakamura, K. Expression of Glycolytic Enzymes Is Increased in Pancreatic Cancerous Tissues as Evidenced by Proteomic Profiling by Two-Dimensional Electrophoresis and Liquid Chromatography-Mass Spectrometry/Mass Spectrometry. Int. J. Oncol. 2007, 30, 849–855. [Google Scholar] [CrossRef] [Green Version]
- Tarbé, N.; Evtimova, V.; Burtscher, H.; Jarsch, M.; Alves, F.; Weidle, U.H. Transcriptional Profiling of Cell Lines Derived from an Orthotopic Pancreatic Tumor Model Reveals Metastasis-Associated Genes. Anticancer Res. 2001, 21, 3221–3228. [Google Scholar] [PubMed]
- Sirover, M.A. New Insights into an Old Protein: The Functional Diversity of Mammalian Glyceraldehyde-3-Phosphate Dehydrogenase. Biochim. Biophys. Acta BBA-Protein Struct. Mol. Enzymol. 1999, 1432, 159–184. [Google Scholar] [CrossRef]
- Li, X.; Li, J.; Zhang, B.; Gu, Y.; Li, Q.; Gu, G.; Xiong, J.; Li, Y.; Yang, X.; Qian, Z. Comparative Peptidome Profiling Reveals Critical Roles for Peptides in the Pathology of Pancreatic Cancer. Int. J. Biochem. Cell Biol. 2020, 120, 105687. [Google Scholar] [CrossRef]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 Inhibition Induces ROS/Akt/MTOR Axis: Role of GAPDH Nuclear Translocation in Genipin/Everolimus Anticancer Synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef]
- Dando, I.; Donadelli, M.; Costanzo, C.; Dalla Pozza, E.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids Inhibit Energetic Metabolism and Induce AMPK-Dependent Autophagy in Pancreatic Cancer Cells. Cell Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Dai, Z.; Wardell, S.E.; Baccile, J.A.; Liu, X.; Gao, X.; Baldi, R.; Mehrmohamadi, M.; Johnson, M.O.; Madhukar, N.S.; et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 2017, 26, 648–659.e8. [Google Scholar] [CrossRef] [Green Version]
- Mandili, G.; Follia, L.; Ferrero, G.; Katayama, H.; Hong, W.; Momin, A.A.; Capello, M.; Giordano, D.; Spadi, R.; Satolli, M.A.; et al. Immune-Complexome Analysis Identifies Immunoglobulin-Bound Biomarkers That Predict the Response to Chemotherapy of Pancreatic Cancer Patients. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilarsky, C.; Wenzig, M.; Specht, T.; Saeger, H.D.; Grützmann, R. Identification and Validation of Commonly Overexpressed Genes in Solid Tumors by Comparison of Microarray Data. Neoplasia N. Y. 2004, 6, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.-T.; Wang, H.; Liang, L.-J.; Peng, B.-G.; Wu, Z.-F.; Chen, L.-Z.; Xue, L.; Li, Z.; Li, W. Overexpression of FOXM1 Is Associated with Poor Prognosis and Clinicopathologic Stage of Pancreatic Ductal Adenocarcinoma. Pancreas 2012, 41, 629–635. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, X.; Jiang, L.; Zhang, L.; Xiang, M.; Ren, H. FoxM1 Induced Paclitaxel Resistance via Activation of the FoxM1/PHB1/RAF-MEK-ERK Pathway and Enhancement of the ABCA2 Transporter. Mol. Ther.-Oncolytics 2019, 14, 196–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Yang, L.; Wang, J.; Xiao, Y.; Ruan, Q. Prognostic Value of FOXM1 in Patients with Malignant Solid Tumor: A Meta-Analysis and System Review. Dis. Markers 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 Promotes the Warburg Effect and Pancreatic Cancer Progression via Transactivation of LDHA Expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Shi, J.; Li, Q.; Li, Z.; Lou, C.; Zhao, Q.; Zhu, Y.; Zhan, F.; Lian, J.; Wang, B.; et al. STAT1-Mediated Inhibition of FOXM1 Enhances Gemcitabine Sensitivity in Pancreatic Cancer. Clin. Sci. 2019, 133, 645–663. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ahmad, A.; Banerjee, S.; Azmi, A.; Kong, D.; Li, Y.; Sarkar, F.H. FoxM1 Is a Novel Target of a Natural Agent in Pancreatic Cancer. Pharm. Res. 2010, 27, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c Promotes Pancreatic Cancer Epithelial-to-Mesenchymal Transition and Metastasis via Upregulation of Expression of the Urokinase Plasminogen Activator System. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Du, J.; Xie, K. FOXM1 and Its Oncogenic Signaling in Pancreatic Cancer Pathogenesis. Biochim. Biophys. Acta 2014, 1845, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Qiu, Z.; Wang, L.; Peng, Z.; Jia, Z.; Logsdon, C.D.; Le, X.; Wei, D.; Huang, S.; Xie, K. A Novel FoxM1-Caveolin Signaling Pathway Promotes Pancreatic Cancer Invasion and Metastasis. Cancer Res. 2012, 72, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Y.; Wu, H.-Y.; Mao, X.-F.; Jiang, L.-X.; Wang, Y.-X. USP5 Promotes Tumorigenesis and Progression of Pancreatic Cancer by Stabilizing FoxM1 Protein. Biochem. Biophys. Res. Commun. 2017, 492, 48–54. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Bhat, U.G.; Hughes, D.E.; Wang, I.-C.; Costa, R.H.; Gartel, A.L. Identification of a Chemical Inhibitor of the Oncogenic Transcription Factor Forkhead Box M1. Cancer Res. 2006, 66, 9731–9735. [Google Scholar] [CrossRef] [Green Version]
- Bhat, U.G.; Halasi, M.; Gartel, A.L. FoxM1 Is a General Target for Proteasome Inhibitors. PLoS ONE 2009, 4, e6593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wente, M.N.; Eibl, G.; Reber, H.A.; Friess, H.; Büchler, M.W.; Hines, O.J. The Proteasome Inhibitor MG132 Induces Apoptosis in Human Pancreatic Cancer Cells. Oncol. Rep. 2005, 14, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Ma, J.; Li, R.; Yang, J.; Yin, X. The Inhibitory Effect of 5,7-DMF on Pancreatic Sphere-Forming Cell Function Mediated by FoxM1 Gene Expression. J. Cell. Biochem. 2018, 119, 1855–1865. [Google Scholar] [CrossRef]
- Ren, X.; Shah, T.A.; Ustiyan, V.; Zhang, Y.; Shinn, J.; Chen, G.; Whitsett, J.A.; Kalin, T.V.; Kalinichenko, V.V. FOXM1 Promotes Allergen-Induced Goblet Cell Metaplasia and Pulmonary Inflammation. Mol. Cell. Biol. 2013, 33, 371–386. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Chen, H.; Xie, R.; Wang, H.; Li, S.; Xu, Q.; Xu, N.; Cheng, Q.; Qian, Y.; Huang, R.; et al. Epigenetically Modulated FOXM1 Suppresses Dendritic Cell Maturation in Pancreatic Cancer and Colon Cancer. Mol. Oncol. 2019, 13, 873–893. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.-L.; Liang, Y.; Chien, K.-Y.; Yu, J.-S. Overexpression and Elevated Serum Levels of Phosphoglycerate Kinase 1 in Pancreatic Ductal Adenocarcinoma. Proteomics 2006, 6, 2259–2272. [Google Scholar] [CrossRef]
- He, Y.; Luo, Y.; Zhang, D.; Wang, X.; Zhang, P.; Li, H.; Ejaz, S.; Liang, S. PGK1-Mediated Cancer Progression and Drug Resistance. Am. J. Cancer Res. 2019, 9, 2280–2302. [Google Scholar] [PubMed]
- Zhou, Y.; Cui, J.; Du, H. Autoantibody-Targeted TAAs in Pancreatic Cancer: A Comprehensive Analysis. Pancreatology 2019, 19, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Shi, S.; Qin, Y.; Meng, Q.; Hua, J.; Hu, Q.; Ji, S.; Zhang, B.; Xu, J.; Yu, X.-J. Localisation of PGK1 Determines Metabolic Phenotype to Balance Metastasis and Proliferation in Patients with SMAD4-Negative Pancreatic Cancer. Gut 2020, 69, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kim, H.-Y.; Vuong, H.; Patwa, T.; Pal, M.; Brand, R.E.; Simeone, D.M.; Lubman, D.M. The Identification of Auto-Antibodies in Pancreatic Cancer Patient Sera Using a Naturally Fractionated Panc-1 Cell Line. Cancer Biomark. Sect. Dis. Markers 2010, 7, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, G.H.; Damink, S.W.M.O.; Malago, M.; Dhar, D.K.; Pereira, S.P. Pyruvate Kinase M2 and Lactate Dehydrogenase A Are Overexpressed in Pancreatic Cancer and Correlate with Poor Outcome. PLoS ONE 2016, 11, e0151635. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Wu, W.; Ni, X.; Kuang, T.; Jin, D.; Wang, D.; Lou, W. Lactate Dehydrogenase A Is Overexpressed in Pancreatic Cancer and Promotes the Growth of Pancreatic Cancer Cells. Tumour Biol. J. Int. Soc. Oncodevelopmental. Biol. Med. 2013, 34, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Cui, J.; Du, J.; Wei, D.; Jia, Z.; Zhang, J.; Zhu, Z.; Gao, Y.; Xie, K. A Novel KLF4/LDHA Signaling Pathway Regulates Aerobic Glycolysis in and Progression of Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4370–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.-L.; Zhang, Y.-J.; Jiang, H.; Li, X.; Zhu, H.; Zheng, K.-L. The C-Myc–LDHA Axis Positively Regulates Aerobic Glycolysis and Promotes Tumor Progression in Pancreatic Cancer. Med. Oncol. 2015, 32, 187. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.-G.; Han, Z.-T.; He, S.-H.; Wu, X.; Chen, T.-R.; Shao, C.-H.; Chen, D.-L.; Su, N.; Chen, Y.-M.; Wang, T.; et al. HIF1/2α Mediates Hypoxia-Induced LDHA Expression in Human Pancreatic Cancer Cells. Oncotarget 2017, 8, 24840–24852. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.-Y.; Zhang, L.; Yee, J.K.; Go, V.-L.W.; Lee, W.-N. Metabolic Consequences of LDHA Inhibition by Epigallocatechin Gallate and Oxamate in MIA PaCa-2 Pancreatic Cancer Cells. Metabolomics Off. J. Metab. Soc. 2015, 11, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.P.; Rachagani, S.; Purohit, V.; Pandey, P.; Joshi, S.; Moore, E.D.; Johansson, S.L.; Singh, P.K.; Ganti, A.K.; Batra, S.K. Graviola: A Novel Promising Natural-Derived Drug That Inhibits Tumorigenicity and Metastasis of Pancreatic Cancer Cells in Vitro and in Vivo through Altering Cell Metabolism. Cancer Lett. 2012, 323, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, G.H.; Vassileva, V.; Acedo, P.; Olde Damink, S.W.M.; Malago, M.; Dhar, D.K.; Pereira, S.P. Targeting Pyruvate Kinase M2 and Lactate Dehydrogenase A Is an Effective Combination Strategy for the Treatment of Pancreatic Cancer. Cancers 2019, 11, 1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Cappello, P.; Tomaino, B.; Chiarle, R.; Ceruti, P.; Novarino, A.; Castagnoli, C.; Migliorini, P.; Perconti, G.; Giallongo, A.; Milella, M.; et al. An Integrated Humoral and Cellular Response Is Elicited in Pancreatic Cancer by α-Enolase, a Novel Pancreatic Ductal Adenocarcinoma-Associated Antigen. Int. J. Cancer 2009, 125, 639–648. [Google Scholar] [CrossRef]
- Principe, M.; Ceruti, P.; Shih, N.-Y.; Chattaragada, M.S.; Rolla, S.; Conti, L.; Bestagno, M.; Zentilin, L.; Yang, S.-H.; Migliorini, P.; et al. Targeting of Surface Alpha-Enolase Inhibits the Invasiveness of Pancreatic Cancer Cells. Oncotarget 2015, 6, 11098–11113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Wang, L.; Liu, H.-L. ENO1 Overexpression in Pancreatic Cancer Patients and Its Clinical and Diagnostic Significance. Gastroenterol. Res. Pract. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Bi, R.; Yin, H.; Liu, H.; Li, L. ENO1 Silencing Impaires Hypoxia-Induced Gemcitabine Chemoresistance Associated with Redox Modulation in Pancreatic Cancer Cells. Am. J. Transl. Res. 2019, 11, 4470–4480. [Google Scholar] [PubMed]
- Principe, M.; Borgoni, S.; Cascione, M.; Chattaragada, M.S.; Ferri-Borgogno, S.; Capello, M.; Bulfamante, S.; Chapelle, J.; Di Modugno, F.; Defilippi, P.; et al. Alpha-Enolase (ENO1) Controls Alpha v/Beta 3 Integrin Expression and Regulates Pancreatic Cancer Adhesion, Invasion, and Metastasis. J. Hematol. Oncol. J. Hematol. Oncol. 2017, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Cappello, P.; Rolla, S.; Chiarle, R.; Principe, M.; Cavallo, F.; Perconti, G.; Feo, S.; Giovarelli, M.; Novelli, F. Vaccination With ENO1 DNA Prolongs Survival of Genetically Engineered Mice With Pancreatic Cancer. Gastroenterology 2013, 144, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Cappello, P.; Tonoli, E.; Curto, R.; Giordano, D.; Giovarelli, M.; Novelli, F. Anti-α-Enolase Antibody Limits the Invasion of Myeloid-Derived Suppressor Cells and Attenuates Their Restraining Effector T Cell Response. OncoImmunology 2016, 5, e1112940. [Google Scholar] [CrossRef] [Green Version]
- Capello, M.; Ferri-Borgogno, S.; Riganti, C.; Chattaragada, M.S.; Principe, M.; Roux, C.; Zhou, W.; Petricoin, E.F.; Cappello, P.; Novelli, F. Targeting the Warburg Effect in Cancer Cells through ENO1 Knockdown Rescues Oxidative Phosphorylation and Induces Growth Arrest. Oncotarget 2015, 7, 5598–5612. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.; Daniels, I.; Symonds, P.; Pitt, T.; Gijon, M.; Xue, W.; Metheringham, R.; Durrant, L.; Brentville, V. Citrullinated α-Enolase Is an Effective Target for Anti-Cancer Immunity. OncoImmunology 2018, 7, e1390642. [Google Scholar] [CrossRef]
- Tomaino, B.; Cappello, P.; Capello, M.; Fredolini, C.; Sperduti, I.; Migliorini, P.; Salacone, P.; Novarino, A.; Giacobino, A.; Ciuffreda, L.; et al. Circulating Autoantibodies to Phosphorylated α-Enolase Are a Hallmark of Pancreatic Cancer. J. Proteome Res. 2011, 10, 105–112. [Google Scholar] [CrossRef]
- Niccolai, E.; Cappello, P.; Taddei, A.; Ricci, F.; D’Elios, M.M.; Benagiano, M.; Bechi, P.; Bencini, L.; Ringressi, M.N.; Coratti, A.; et al. Peripheral ENO1-Specific T Cells Mirror the Intratumoral Immune Response and Their Presence Is a Potential Prognostic Factor for Pancreatic Adenocarcinoma. Int. J. Oncol. 2016, 49, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amedei, A.; Niccolai, E.; Benagiano, M.; Della Bella, C.; Cianchi, F.; Bechi, P.; Taddei, A.; Bencini, L.; Farsi, M.; Cappello, P.; et al. Ex Vivo Analysis of Pancreatic Cancer-Infiltrating T Lymphocytes Reveals That ENO-Specific Tregs Accumulate in Tumor Tissue and Inhibit Th1/Th17 Effector Cell Functions. Cancer Immunol. Immunother. 2013, 62, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 Promotes Tumor Angiogenesis by Regulating HIF-1α through NF-ΚB Activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chu, L.; Yuan, S.; Yang, Y.; Yang, Y.; Xu, B.; Zhang, K.; Liu, X.-Y.; Wang, R.; Fang, L.; et al. RGD-Modified Oncolytic Adenovirus-Harboring ShPKM2 Exhibits a Potent Cytotoxic Effect in Pancreatic Cancer via Autophagy Inhibition and Apoptosis Promotion. Cell Death Dis. 2017, 8, e2835. [Google Scholar] [CrossRef] [PubMed]
- Hillis, A.L.; Lau, A.N.; Devoe, C.X.; Dayton, T.L.; Danai, L.V.; Di Vizio, D.; Vander Heiden, M.G. PKM2 Is Not Required for Pancreatic Ductal Adenocarcinoma. Cancer Metab. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Hu, L.; Chen, M.; Cao, W.; Chen, H.; He, T. Pyruvate Kinase M2 Overexpression and Poor Prognosis in Solid Tumors of Digestive System: Evidence from 16 Cohort Studies. OncoTargets Ther. 2016, 9, 4277–4288. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Deng, S.; Liu, M.; Jin, Y.; Zhu, S.; Deng, S.; Chen, J.; He, C.; Qin, Q.; Wang, C.; et al. The Responsively Decreased PKM2 Facilitates the Survival of Pancreatic Cancer Cells in Hypoglucose. Cell Death Dis. 2018, 9, 133. [Google Scholar] [CrossRef] [Green Version]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM Alternative Splicing by PTBP1 Promotes Gemcitabine Resistance in Pancreatic Cancer Cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.-Y.; Yang, Y.-C.; Wang, H.-P.; Tien, Y.-W.; Shun, C.-T.; Huang, H.-Y.; Hsiao, M.; Hua, K.-T. Pyruvate Kinase M2 Promotes Pancreatic Ductal Adenocarcinoma Invasion and Metastasis through Phosphorylation and Stabilization of PAK2 Protein. Oncogene 2018, 37, 1730–1742. [Google Scholar] [CrossRef]
- Masamune, A.; Hamada, S.; Yoshida, N.; Nabeshima, T.; Shimosegawa, T. Pyruvate Kinase Isozyme M2 Plays a Critical Role in the Interactions Between Pancreatic Stellate Cells and Cancer Cells. Dig. Dis. Sci. 2018, 63, 1868–1877. [Google Scholar] [CrossRef]
- Yu, L.; Teoh, S.T.; Ensink, E.; Ogrodzinski, M.P.; Yang, C.; Vazquez, A.I.; Lunt, S.Y. Cysteine Catabolism and the Serine Biosynthesis Pathway Support Pyruvate Production during Pyruvate Kinase Knockdown in Pancreatic Cancer Cells. Cancer Metab. 2019, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.S.; Bernier, M.; Wainer, I.W. Selective GPR55 Antagonism Reduces Chemoresistance in Cancer Cells. Pharmacol. Res. 2016, 111, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.D.; Richardson, D.A.; Oh, I.-W.; Sritangos, P.; Attard, T.; Barrett, L.; Bruce, J.I.E. Cutting off the Fuel Supply to Calcium Pumps in Pancreatic Cancer Cells: Role of Pyruvate Kinase-M2 (PKM2). Br. J. Cancer 2020, 122, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Park, Y.S.; Kang, M.G.; You, Y.-M.; Jung, Y.; Koo, H.; Kim, J.-A.; Kim, M.-J.; Hong, S.-M.; Lee, K.B.; et al. Pyruvate Kinase Isoenzyme M2 Is a Therapeutic Target of Gemcitabine-Resistant Pancreatic Cancer Cells. Exp. Cell Res. 2015, 336, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding Pancreatic Cancer Proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koleilat, M.K.; Kwong, L.N. Same Name, Different Game: EGFR Drives Intrinsic KRASG12C Inhibitor Resistance in Colorectal Cancer. Cancer Discov. 2020, 10, 1094–1096. [Google Scholar] [CrossRef]
- Kang, Y.W.; Lee, J.E.; Jung, K.H.; Son, M.K.; Shin, S.-M.; Kim, S.J.; Fang, Z.; Yan, H.H.; Park, J.H.; Han, B.; et al. KRAS Targeting Antibody Synergizes Anti-Cancer Activity of Gemcitabine against Pancreatic Cancer. Cancer Lett. 2018, 438, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Toubaji, A.; Achtar, M.; Provenzano, M.; Herrin, V.E.; Behrens, R.; Hamilton, M.; Bernstein, S.; Venzon, D.; Gause, B.; Marincola, F.; et al. Pilot Study of Mutant Ras Peptide-Based Vaccine as an Adjuvant Treatment in Pancreatic and Colorectal Cancers. Cancer Immunol. Immunother. 2008, 57, 1413–1420. [Google Scholar] [CrossRef]
- Quandt, J.; Schlude, C.; Bartoschek, M.; Will, R.; Cid-Arregui, A.; Schölch, S.; Reissfelder, C.; Weitz, J.; Schneider, M.; Wiemann, S.; et al. Long-Peptide Vaccination with Driver Gene Mutations in P53 and Kras Induces Cancer Mutation-Specific Effector as Well as Regulatory T Cell Responses. OncoImmunology 2018, 7, e1500671. [Google Scholar] [CrossRef]
- Cohn, A.; Morse, M.A.; Coeshott, C.; Ferraro, J.; Bellgrau, D.; Apelian, D.; Rodell, T.C. Whole Recombinant Saccharomyces Cerevisiae Yeast Expressing Ras Mutations as Treatment for Patients With Solid Tumors Bearing Ras Mutations: Results From a Phase 1 Trial. J. Immunother. 2018, 41, 10. [Google Scholar] [CrossRef] [Green Version]
- Butera, G.; Pacchiana, R.; Mullappilly, N.; Margiotta, M.; Bruno, S.; Conti, P.; Riganti, C.; Donadelli, M. Mutant P53 Prevents GAPDH Nuclear Translocation in Pancreatic Cancer Cells Favoring Glycolysis and 2-Deoxyglucose Sensitivity. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2018, 1865, 1914–1923. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. P53 Status Determines the Role of Autophagy in Pancreatic Tumour Development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with P53 Alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, P.; Quero, L.; Pacault, V.; Schlageter, M.-H.; Baruch-Hennequin, V.; Hennequin, C. Prognostic Significance of Anti-P53 and Anti-KRas Circulating Antibodies in Esophageal Cancer Patients Treated with Chemoradiotherapy. BMC Cancer 2012, 12, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suppiah, A.; Greenman, J. Clinical Utility of Anti-P53 Auto-Antibody: Systematic Review and Focus on Colorectal Cancer. World J. Gastroenterol. 2013, 19, 4651–4670. [Google Scholar] [CrossRef]
- Kumar, S.; Mohan, A.; Guleria, R. Prognostic Implications of Circulating Anti-P53 Antibodies in Lung Cancer--a Review. Eur. J. Cancer Care (Engl.) 2009, 18, 248–254. [Google Scholar] [CrossRef]
- Geng, J.; Yuan, X.; Wei, M.; Wu, J.; Qin, Z.-H. The Diverse Role of TIGAR in Cellular Homeostasis and Cancer. Free Radic. Res. 2018, 52, 1240–1249. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, Y.; Zhang, Y.; Yuan, M.; Li, X.; Gao, J.; Zhang, S.; Xing, C.; Qin, H.; Zhao, H.; et al. TIGAR Promotes Tumorigenesis and Protects Tumor Cells From Oxidative and Metabolic Stresses in Gastric Cancer. Front. Oncol. 2019, 9, 1258. [Google Scholar] [CrossRef] [Green Version]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, S1535610819305823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, I.S.; Trayhurn, P. Glucose Transporters (GLUT and SGLT): Expanded Families of Sugar Transport Proteins. Br. J. Nutr. 2003, 89, 3–9. [Google Scholar] [CrossRef]
- Carruthers, A.; DeZutter, J.; Ganguly, A.; Devaskar, S.U. Will the Original Glucose Transporter Isoform Please Stand Up! Am. J. Physiol. Endocrinol. Metab. 2009, 297, E836–E848. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.S.; Chun, Y.K.; Hur, M.H.; Lee, H.K.; Kim, Y.J.; Hong, S.R.; Lee, J.H.; Lee, S.G.; Park, Y.K. Clinical Significance of Glucose Transporter 1 (GLUT1) Expression in Human Breast Carcinoma. Jpn. J. Cancer Res. Gann 2002, 93, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and Cellular Regulation of Glucose Transporter (GLUT) Proteins in Cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 Glucose Transporters in Endometrial and Breast Cancers. Pathol. Oncol. Res. POR 2012, 18, 721–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, E.; Okuda, H.; Kobayashi, A.; Watabe, K. Metabolic Genes in Cancer: Their Roles in Tumor Progression and Clinical Implications. Biochim. Biophys. Acta 2010, 1805, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Chikamoto, A.; Inoue, R.; Komohara, Y.; Sakamaki, K.; Hashimoto, D.; Shiraishi, S.; Takamori, H.; Yamashita, Y.; Yoshida, N.; Yamanaka, T.; et al. Preoperative High Maximum Standardized Uptake Value in Association with Glucose Transporter 1 Predicts Poor Prognosis in Pancreatic Cancer. Ann. Surg. Oncol. 2017, 24, 2040–2046. [Google Scholar] [CrossRef]
- Saiyin, H.; Ardito-Abraham, C.M.; Wu, Y.; Wei, Y.; Fang, Y.; Han, X.; Li, J.; Zhou, P.; Yi, Q.; Maitra, A.; et al. Identification of Novel Vascular Projections with Cellular Trafficking Abilities on the Microvasculature of Pancreatic Ductal Adenocarcinoma. J. Pathol. 2015, 236, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Higashi, T.; Tamaki, N.; Torizuka, T.; Nakamoto, Y.; Sakahara, H.; Kimura, T.; Honda, T.; Inokuma, T.; Katsushima, S.; Ohshio, G.; et al. FDG Uptake, GLUT-1 Glucose Transporter and Cellularity in Human Pancreatic Tumors. J. Nucl. Med. 1998, 39, 1727–1735. [Google Scholar]
- Kishore, M.; Cheung, K.C.P.; Fu, H.; Bonacina, F.; Wang, G.; Coe, D.; Ward, E.J.; Colamatteo, A.; Jangani, M.; Baragetti, A.; et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 2017, 47, 875–889.e10. [Google Scholar] [CrossRef]
- Granville, C.A.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Handicapping the Race to Develop Inhibitors of the Phosphoinositide 3-Kinase/Akt/Mammalian Target of Rapamycin Pathway. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Hoensch, H.; Groh, B.; Edler, L.; Kirch, W. Prospective Cohort Comparison of Flavonoid Treatment in Patients with Resected Colorectal Cancer to Prevent Recurrence. World J. Gastroenterol. 2008, 14, 2187–2193. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 Is a Key Mediator of Aerobic Glycolysis and Promotes Tumor Growth in Human Glioblastoma Multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.-T.; Zhou, S.-H. Warburg Effect, Hexokinase-II, and Radioresistance of Laryngeal Carcinoma. Oncotarget 2016, 8, 14133–14146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botzer, L.E.; Maman, S.; Sagi-Assif, O.; Meshel, T.; Nevo, I.; Yron, I.; Witz, I.P. Hexokinase 2 Is a Determinant of Neuroblastoma Metastasis. Br. J. Cancer 2016, 114, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Thamrongwaranggoon, U.; Seubwai, W.; Phoomak, C.; Sangkhamanon, S.; Cha’on, U.; Boonmars, T.; Wongkham, S. Targeting Hexokinase II as a Possible Therapy for Cholangiocarcinoma. Biochem. Biophys. Res. Commun. 2017, 484, 409–415. [Google Scholar] [CrossRef]
- Nevo, I.; Oberthuer, A.; Botzer, E.; Sagi-Assif, O.; Maman, S.; Pasmanik-Chor, M.; Kariv, N.; Fischer, M.; Yron, I.; Witz, I.P. Gene-Expression-Based Analysis of Local and Metastatic Neuroblastoma Variants Reveals a Set of Genes Associated with Tumor Progression in Neuroblastoma Patients. Int. J. Cancer 2010, 126, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Kogure, K.; Yamamoto, K.; Majima, E.; Shinohara, Y.; Yamashita, K.; Terada, H. Alteration of Enzyme Function of the Type II Hexokinase C-Terminal Half on Replacements of Restricted Regions by Corresponding Regions of Glucokinase. J. Biol. Chem. 1996, 271, 15230–15236. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Li, J.; Luo, Y. Glucose Metabolism and Cancer. Biochemistry 2012. [Google Scholar] [CrossRef] [Green Version]
- Nowak, N.; Kulma, A.; Gutowicz, J. Up-Regulation of Key Glycolysis Proteins in Cancer Development. Open Life Sci. 2018, 13, 569–581. [Google Scholar] [CrossRef]
- Mazure, N.M. VDAC in Cancer. Biochim. Biophys. Acta BBA-Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-Mitochondria Interaction Mediated by Akt Is Required to Inhibit Apoptosis in the Presence or Absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef]
- Popescu, R.; Heiss, E.H.; Ferk, F.; Peschel, A.; Knasmueller, S.; Dirsch, V.M.; Krupitza, G.; Kopp, B. Ikarugamycin Induces DNA Damage, Intracellular Calcium Increase, P38 MAP Kinase Activation and Apoptosis in HL-60 Human Promyelocytic Leukemia Cells. Mutat. Res. Mol. Mech. Mutagen. 2011, 709–710, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Elkin, S.R.; Oswald, N.W.; Reed, D.K.; Mettlen, M.; MacMillan, J.B.; Schmid, S.L. Ikarugamycin: A Natural Product Inhibitor of Clathrin-Mediated Endocytosis. Traffic Cph. Den. 2016, 17, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, G.L.; Reig, A.; Viñas, O.; Mahler, M.; Wunsch, E.; Milkiewicz, P.; Swain, M.G.; Mason, A.; Stinton, L.M.; Aparicio, M.B.; et al. The Prevalence of Anti-Hexokinase-1 and Anti-Kelch-Like 12 Peptide Antibodies in Patients with Primary Biliary Cholangitis Is Similar in Europe and North America: A Large International, Multi-Center Study. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Reig, A.; Norman, G.L.; Garcia, M.; Shums, Z.; Ruiz-Gaspà, S.; Bentow, C.; Mahler, M.; Romera, M.A.; Vinas, O.; Pares, A. Novel Anti-Hexokinase 1 Antibodies Are Associated With Poor Prognosis in Patients With Primary Biliary Cholangitis. Am. J. Gastroenterol. 2020, 115, 1634–1641. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Yang, Y.-C.; Tien, C.-P.; Yang, C.-J.; Hsiao, M. Roles of Aldolase Family Genes in Human Cancers and Diseases. Trends Endocrinol. Metab. 2018, 29, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Mamczur, P.; Dzugaj, A. Aldolase A Is Present in Smooth Muscle Cell Nuclei. Acta Biochim. Pol. 2008, 55, 799–805. [Google Scholar] [CrossRef]
- Rose, I.A.; O’Connell, E.L. Studies on the Interaction of Aldolase with Substrate Analogues. J. Biol. Chem. 1969, 244, 126–134. [Google Scholar] [CrossRef]
- Lessa, R.C.; Campos, A.H.J.F.M.; de Freitas, C.E.; da Silva, F.R.; Kowalski, L.P.; Carvalho, A.L.; Vettore, A.L. Identification of Upregulated Genes in Oral Squamous Cell Carcinomas. Head Neck 2013, 35, 1475–1481. [Google Scholar] [CrossRef]
- Oparina, N.Y.; Snezhkina, A.V.; Sadritdinova, A.F.; Veselovskii, V.A.; Dmitriev, A.A.; Senchenko, V.N.; Mel’nikova, N.V.; Speranskaya, A.S.; Darii, M.V.; Stepanov, O.A.; et al. Differential expression of genes that encode glycolysis enzymes in kidney and lung cancer in humans. Genetika 2013, 49, 814–823. [Google Scholar] [CrossRef]
- Peng, Y.; Li, X.; Wu, M.; Yang, J.; Liu, M.; Zhang, W.; Xiang, B.; Wang, X.; Li, X.; Li, G.; et al. New Prognosis Biomarkers Identified by Dynamic Proteomic Analysis of Colorectal Cancer. Mol. Biosyst. 2012, 8, 3077–3088. [Google Scholar] [CrossRef]
- Kim, H.S.; Choi, J.Y.; Choi, D.W.; Lim, H.Y.; Lee, J.H.; Hong, S.P.; Cho, Y.S.; Lee, K.-H.; Kim, B.-T. Prognostic Value of Volume-Based Metabolic Parameters Measured by (18)F-FDG PET/CT of Pancreatic Neuroendocrine Tumors. Nucl. Med. Mol. Imaging 2014, 48, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Byun, B.H.; Moon, S.M.; Shin, U.S.; Lim, I.; Kim, B.I.; Choi, C.W.; Lim, S.M. Prognostic Value of 18F-FDG Uptake by Regional Lymph Nodes on Pretreatment PET/CT in Patients with Resectable Colorectal Cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2203–2211. [Google Scholar] [CrossRef]
- Park, S.Y.; Cho, A.; Yu, W.S.; Lee, C.Y.; Lee, J.G.; Kim, D.J.; Chung, K.Y. Prognostic Value of Total Lesion Glycolysis by 18F-FDG PET/CT in Surgically Resected Stage IA Non-Small Cell Lung Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, U.; Tatsumi, M.; Terauchi, T.; Ando, K.; Niitsu, N.; Kim, W.S.; Suh, C.; Ogura, M.; Tobinai, K. Prognostic Significance of Metabolic Tumor Burden by Positron Emission Tomography/Computed Tomography in Patients with Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Cancer Sci. 2015, 106, 186–193. [Google Scholar] [CrossRef]
- Shi, S.; Ji, S.; Qin, Y.; Xu, J.; Zhang, B.; Xu, W.; Liu, J.; Long, J.; Liu, C.; Liu, L.; et al. Metabolic Tumor Burden Is Associated with Major Oncogenomic Alterations and Serum Tumor Markers in Patients with Resected Pancreatic Cancer. Cancer Lett. 2015, 360, 227–233. [Google Scholar] [CrossRef]
- Takebayashi, Y.; Akiyama, S.; Akiba, S.; Yamada, K.; Miyadera, K.; Sumizawa, T.; Yamada, Y.; Murata, F.; Aikou, T. Clinicopathologic and Prognostic Significance of an Angiogenic Factor, Thymidine Phosphorylase, in Human Colorectal Carcinoma. J. Natl. Cancer Inst. 1996, 88, 1110–1117. [Google Scholar] [CrossRef] [Green Version]
- Takebayashi, Y.; Miyadera, K.; Akiyama, S.; Hokita, S.; Yamada, K.; Akiba, S.; Yamada, Y.; Sumizawa, T.; Aikou, T. Expression of Thymidine Phosphorylase in Human Gastric Carcinoma. Jpn. J. Cancer Res. 1996, 87, 288–295. [Google Scholar] [CrossRef]
- Imazano, Y.; Takebayashi, Y.; Nishiyama, K.; Akiba, S.; Miyadera, K.; Yamada, Y.; Akiyama, S.; Ohi, Y. Correlation between Thymidine Phosphorylase Expression and Prognosis in Human Renal Cell Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1997, 15, 2570–2578. [Google Scholar] [CrossRef]
- Chen, T.; Huang, Z.; Tian, Y.; Lin, B.; He, R.; Wang, H.; Ouyang, P.; Chen, H.; Wu, L. Clinical Significance and Prognostic Value of Triosephosphate Isomerase Expression in Gastric Cancer. Medicine 2017, 96, e6865. [Google Scholar] [CrossRef]
- Quan, M.; Wang, P.; Cui, J.; Gao, Y.; Xie, K. The Roles of FOXM1 in Pancreatic Stem Cells and Carcinogenesis. Mol. Cancer 2013, 12, 159. [Google Scholar] [CrossRef] [Green Version]
- Kalin, T.V.; Ustiyan, V.; Kalinichenko, V.V. Multiple Faces of FoxM1 Transcription Factor. Cell Cycle 2011, 10, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Wierstra, I.; Alves, J. FOXM1, a Typical Proliferation-Associated Transcription Factor. Biol. Chem. 2007, 388, 1257–1274. [Google Scholar] [CrossRef]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 Is Required for Execution of the Mitotic Programme and Chromosome Stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Liu, M.; Dai, B.; Kang, S.-H.; Ban, K.; Huang, F.-J.; Lang, F.F.; Aldape, K.D.; Xie, T.; Pelloski, C.E.; Xie, K.; et al. FoxM1B Is Overexpressed in Human Glioblastomas and Critically Regulates the Tumorigenicity of Glioma Cells. Cancer Res. 2006, 66, 3593–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.-T.; Lee, H.-T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 Promotes β-Catenin Nuclear Localization and Controls Wnt Target-Gene Expression and Glioma Tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macgregor-Das, A.M.; Iacobuzio-Donahue, C.A. Molecular Pathways in Pancreatic Carcinogenesis. J. Surg. Oncol. 2013, 107, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Tan, G.; Ding, M.; Dong, D.; Chen, T.; Meng, X.; Huang, X.; Tan, Y. Foxm1 Transcription Factor Is Required for Maintenance of Pluripotency of P19 Embryonal Carcinoma Cells. Nucleic Acids Res. 2010, 38, 8027–8038. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Xia, T.; Xie, D.; Gao, Y.; Jia, Z.; Wei, D.; Wang, L.; Huang, S.; Quan, M.; Xie, K. HGF/Met and FOXM1 Form a Positive Feedback Loop and Render Pancreatic Cancer Cells Resistance to Met Inhibition and Aggressive Phenotypes. Oncogene 2016, 35, 4708–4718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, P.D. The Enzymes; Academic Press: Cambridge, MA, USA, 1973; ISBN 978-0-08-086585-0. [Google Scholar]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 Promotes Liver Cancer Cell Proliferation and Tumorigenesis. Hepathology 2017, 65, 515–528. [Google Scholar] [CrossRef]
- Jiang, Y.; He, R.; Jiang, Y.; Liu, D.; Tao, L.; Yang, M.; Lin, C.; Shen, Y.; Fu, X.; Yang, J.; et al. Transcription Factor NFAT5 Contributes to the Glycolytic Phenotype Rewiring and Pancreatic Cancer Progression via Transcription of PGK1. Cell Death Dis. 2019, 10, 948. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Kim, K.-S.; Ramakrishna, S. NFAT5 Promotes in Vivo Development of Murine Melanoma Metastasis. Biochem. Biophys. Res. Commun. 2018, 505, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Li, C.; Guo, T.; Chen, J.; Wang, H.-T.; Wang, Y.-T.; Xiao, Y.-S.; Li, J.; Liu, P.; Liu, Z.-S.; et al. Upregulation of DARS2 by HBV Promotes Hepatocarcinogenesis through the MiR-30e-5p/MAPK/NFAT5 Pathway. J. Exp. Clin. Cancer Res. CR 2017, 36, 148. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Wang, Y.; Li, J.; Xiao, Y.; Liu, Z. NFAT5 Inhibits Invasion and Promotes Apoptosis in Hepatocellular Carcinoma Associated with Osmolality. Neoplasma 2017, 64, 502–510. [Google Scholar] [CrossRef]
- Papageorgis, P.; Cheng, K.; Ozturk, S.; Gong, Y.; Lambert, A.W.; Abdolmaleky, H.M.; Zhou, J.-R.; Thiagalingam, S. Smad4 Inactivation Promotes Malignancy and Drug Resistance of Colon Cancer. Cancer Res. 2011, 71, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Wang, Y.; Zhu, X. Baseline Serum Lactate Dehydrogenase Level Predicts Survival Benefit in Patients with Metastatic Colorectal Cancer Receiving Bevacizumab as First-Line Chemotherapy: A Systematic Review and Meta-Analysis of 7 Studies and 1,219 Patients. Ann. Transl. Med. 2019, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- He, G.-D.; Jiang, Y.; Zhang, B.; Wu, G.-H. The Effect of HIF-1α on Glucose Metabolism, Growth and Apoptosis of Pancreatic Cancerous Cells. Asia Pac. J. Clin. Nutr. 2014, 23, 174–180. [Google Scholar] [CrossRef]
- Li, X.; Jiang, F.; Ge, Z.; Chen, B.; Yu, J.; Xin, M.; Wang, J.; An, L.; Wei, J.; Wu, L. Fructose-Bisphosphate Aldolase A Regulates Hypoxic Adaptation in Hepatocellular Carcinoma and Involved with Tumor Malignancy. Dig. Dis. Sci. 2019, 64, 3215–3227. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Ding, H.; Liu, Y.; Pan, G.; Li, Q.; Yang, Z.; Liu, W. Expression of HMGB2 Indicates Worse Survival of Patients and Is Required for the Maintenance of Warburg Effect in Pancreatic Cancer. Acta Biochim. Biophys. Sin. 2017, 49, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Gao, Q.; Zhou, Y.; Dier, U.; Hempel, N.; Hochwald, S.N. Focal Adhesion Kinase-Promoted Tumor Glucose Metabolism Is Associated with a Shift of Mitochondrial Respiration to Glycolysis. Oncogene 2016, 35, 1926–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, B.; Kitagawa, T.; Nakamura, K.; Kuramitsu, Y. Isolation of a Growth Factor Stress-Induced Pancreatic Cancer Sub-Population: Insight into Changes Due to Micro-Environment. Cancer Genom. -Proteom. 2015, 12, 49–55. [Google Scholar]
- Zhou, W.; Capello, M.; Fredolini, C.; Racanicchi, L.; Dugnani, E.; Piemonti, L.; Liotta, L.A.; Novelli, F.; Petricoin, E.F. Mass Spectrometric Analysis Reveals O-Methylation of Pyruvate Kinase from Pancreatic Cancer Cells. Anal. Bioanal. Chem. 2013, 405, 4937–4943. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Liotta, L.A.; Petricoin, E.F. Cancer Metabolism and Mass Spectrometry-Based Proteomics. Cancer Lett. 2015, 356, 176–183. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, H.; Zhu, X.; Hu, X.; Zheng, L.; Zhu, X. Pyruvate Kinase M2 (PKM2) Expression Correlates with Prognosis in Solid Cancers: A Meta-Analysis. Oncotarget 2017, 8, 1628–1640. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Chen, G.; Zhuang, L.; Xu, L.; Lin, J.; Liu, L. ARHGAP4 Mediates the Warburg Effect in Pancreatic Cancer through the MTOR and HIF-1α Signaling Pathways. OncoTargets Ther. 2019, 12, 5003–5012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Ma, T.; Ge, Z.; Lin, J.; Ding, W.; Chen, H.; Zhu, W.; Zhou, S.; Tan, Y. PKM2 Gene Regulates the Behavior of Pancreatic Cancer Cells via Mitogen-Activated Protein Kinase Pathways. Mol. Med. Rep. 2015, 11, 2111–2117. [Google Scholar] [CrossRef]
- Jia, Y.; Li, H.-Y.; Wang, J.; Wang, Y.; Zhang, P.; Ma, N.; Mo, S.-J. Phosphorylation of 14-3-3ζ Links YAP Transcriptional Activation to Hypoxic Glycolysis for Tumorigenesis. Oncogenesis 2019, 8, 31. [Google Scholar] [CrossRef]
- Li, C.; Zhao, Z.; Zhou, Z.; Liu, R. PKM2 Promotes Cell Survival and Invasion Under Metabolic Stress by Enhancing Warburg Effect in Pancreatic Ductal Adenocarcinoma. Dig. Dis. Sci. 2016, 61, 767–773. [Google Scholar] [CrossRef]
- Tian, S.; Li, P.; Sheng, S.; Jin, X. Upregulation of Pyruvate Kinase M2 Expression by Fatty Acid Synthase Contributes to Gemcitabine Resistance in Pancreatic Cancer. Oncol. Lett. 2018, 15, 2211–2217. [Google Scholar] [CrossRef] [Green Version]
- Pandita, A.; Kumar, B.; Manvati, S.; Vaishnavi, S.; Singh, S.K.; Bamezai, R.N.K. Synergistic Combination of Gemcitabine and Dietary Molecule Induces Apoptosis in Pancreatic Cancer Cells and down Regulates PKM2 Expression. PLoS ONE 2014, 9, e107154. [Google Scholar] [CrossRef]
- Noguchi, T.; Inoue, H.; Tanaka, T. The M1- and M2-Type Isozymes of Rat Pyruvate Kinase Are Produced from the Same Gene by Alternative RNA Splicing. J. Biol. Chem. 1986, 261, 13807–13812. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Luo, M.; Zhou, Y.; Ren, W.; Wu, K.; Li, X.; Shen, J.; Hu, Y. The Pro-Apoptotic Role of the Regulatory Feedback Loop between MiR-124 and PKM1/HNF4α in Colorectal Cancer Cells. Int. J. Mol. Sci. 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Wu, J.; Qu, X.; Sheng, J.; Cui, M.; Liu, S.; Huang, X.; Xiang, Y.; Li, B.; Zhang, X.; et al. Glycometabolic Rearrangements--Aerobic Glycolysis in Pancreatic Cancer: Causes, Characteristics and Clinical Applications. J. Exp. Clin. Cancer Res. 2020, 39, 267. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ren, B.; Yang, G.; Wang, H.; Chen, G.; You, L.; Zhang, T.; Zhao, Y. The Enhancement of Glycolysis Regulates Pancreatic Cancer Metastasis. Cell. Mol. Life Sci. 2020, 77, 305–321. [Google Scholar] [CrossRef]
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET Enhances the Stem Cell-like Potential and Glycolysis of Pancreatic Cancer Cells via Activation of YAP/HIF-1α. Exp. Cell Res. 2018, 371, 63–71. [Google Scholar] [CrossRef]
- Cameron, M.E.; Yakovenko, A.; Trevino, J.G. Glucose and Lactate Transport in Pancreatic Cancer: Glycolytic Metabolism Revisited. J. Oncol. 2018. [Google Scholar] [CrossRef]
- Kong, S.C.; Nøhr-Nielsen, A.; Zeeberg, K.; Reshkin, S.J.; Hoffmann, E.K.; Novak, I.; Pedersen, S.F. Monocarboxylate Transporters MCT1 and MCT4 Regulate Migration and Invasion of Pancreatic Ductal Adenocarcinoma Cells. Pancreas 2016, 45, 1036–1047. [Google Scholar] [CrossRef]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-Associated Macrophages Promote Progression and the Warburg Effect via CCL18/NF-KB/VCAM-1 Pathway in Pancreatic Ductal Adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J.; Balaji, U.; Porembka, M.R.; Wachsmann, M.B.; McCue, P.A.; Knudsen, E.S.; Witkiewicz, A.K. Immunologic and Metabolic Features of Pancreatic Ductal Adenocarcinoma Define Prognostic Subtypes of Disease. Clin. Cancer Res. 2016, 22, 3606–3617. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis Controls the Induction of Human Regulatory T Cells by Modulating the Expression of FOXP3 Exon 2 Splicing Variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Green, D.R. Metabolic Checkpoints in Activated T Cells. Nat. Immunol. 2012, 13, 907–915. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.-C.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial Dysregulation and Glycolytic Insufficiency Functionally Impair CD8 T Cells Infiltrating Human Renal Cell Carcinoma. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Gemta, L.F.; Siska, P.J.; Nelson, M.E.; Gao, X.; Liu, X.; Locasale, J.W.; Yagita, H.; Slingluff, C.L.; Hoehn, K.L.; Rathmell, J.C.; et al. Impaired Enolase 1 Glycolytic Activity Restrains Effector Functions of Tumor Infiltrating CD8+ T Cells. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, K.E.; Hongo, R.; Ye, X.; Young, K.; Carbonell, K.; Healey, D.C.C.; Siska, P.J.; Barone, S.; Roe, C.E.; Smith, C.C.; et al. CD28 Costimulation Drives Tumor-Infiltrating T Cell Glycolysis to Promote Inflammation. JCI Insight 2020, 5, e138729. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Du, T.; Chauhan, D.; Anderson, K.C. Preclinical Validation of Alpha-Enolase (ENO1) as a Novel Immunometabolic Target in Multiple Myeloma. Oncogene 2020, 39, 2786–2796. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Target | Overexpression | Clinical Outcome | Targeting Approach | Correlation with Immune Response | |||
|---|---|---|---|---|---|---|---|
| Cell Line | Tissue | Survival | PD/M | CT Resistance | |||
| KRAS/TP53 | PCR [11] IHC [11,12] RNAseq [13] | ↓ [11,14] | ↑ M [11] | KRAS interference [11] KRAS/p53 peptide vaccination [11,15,16,17,18] RT11-i [19] | Chemokine secretion, macrophage recruitment and lymphocyte and myeloid cell infiltration [11] | ||
| GLUT1 | qPCR [20] WB [20,21] | RNAseq [15,16] IHC [17,18] | ↓ [22] | * ↑ M [21] | [22] | GLUT1 silencing [21] TWIST1 silencing [23] Apigenin [24,25] PON2 [26] | Positive correlation with PD1+ TILs [18] High GLUT1 expression in activated Tregs [27] |
| HK2 | qPCR [28] WB [28,29] | mRNA [30] IHC [30,31,32] Microarray [33] | ↓ [29,31] ↑ [32] | * ↑ M [28] | [28] | HK2 silencing [28] IKA [34] | |
| ALDOA | IHC [35] | ↓ [35] | * ↑ M [35] | ALDOA silencing [35] TDZD-8 [36] TX-2098 [37] | Circulating auto-Ab to ALDOA [10] | ||
| TPI | RNAseq [13] | RNAseq [13] IHC [38] | ↓ [13,38] | [13,39] | Circulating auto-Ab to TPI [9,10] | ||
| GAPDH | mRNA [40] WB [40] | 2-DE [41] IHC [41] mRNA [40] WB [40] | ↓ [39] | * ↑ M [42] | [39] | 2-DG [43] P1DG [44] AXP3009 [45] Genipin [45,46] KA [47] | Humoral and cellular responses to GAPDH [10,48] |
| FOXM1 | Microarray [49] mRNA [50] IHC [51] WB [51] | ↓ [50,52] | ↑ PD [53] ↑ M [50,53] | [51] * [54] | Genistein [55] FOXM1 siRNA [56,57] FOXM1 RNA interference [58] USP5 inhibition [59] Thiazole antibiotic [60] MG115 and MG132 [61,62] 5,7 Dimethoxyflavone [63] | DC maturation [64,65] T cell proliferation [65] | |
| PGK1 | 2-DE [66] | mRNA [67] 2-DE [66] | ↓ [67,68] | ↑ M [69] | [67] | Circulating auto-Ab [10,66,68] High concentration of PGK1 in serum [70] | |
| LDHA | WB [71,72,73,74] qPCR [72] | Microarray [71] IHC [71,74,75] mRNA [72] WB [72] | ↓ [71] | ↑ PD [75] | LDHA RNA interference [72] LDHA siRNA [73,74] EGCG [76] 2-DG [76] Graviola [77] FX11 [78,79] | Negative correlation with CD8+ TIL [71] * Low cytotoxic NK activity [80] * High MDSC activity [80] | |
| ENO1 | mRNA [40] WB [40] Microarray [81] Flow cytometry [82] | 2-DE [41] mRNA [40] WB [40] Microarray [81] IHC [83] | ↓ [39,83,84] | * ↑ M [85] | [39,84] | ENO1 DNA vaccination alone or in combination with GEM [10,86] ENO1 silencing [82,85] Anti-ENO1 mAb [82,87] PhAH [88] ENO1 knockdown [88] Citrullinated ENO1 peptides [89] | Increased level of anti-ENO1 Ab before and after GEM treatment [9,10,90,91] Increased T cell response in vitro and in vivo [81,91] Increased level of ENO1 in plasma patients [83] ENO1-specific Th17 and Treg response [92] |
| PKM2 | WB [71,93,94] qPCR [94] | IHC [31,71,93,94] ↓ IHC [95,96] Microarray [97] Oncomine database [97] | ↓ [31,71,97,98] | ↑ M [31,99] * ↑ PD [98] | [98] | PKM2 knockdown [94,99,100,101] PKM2 silencing [97] 2-DG [102,103] Shikonin [103] Betulinic acid and thymoquinone and GEM [104] | Increased level of anti-PKM2 Ab before and after GEM treatment [10] Negative correlation with CD8+ TIL [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curcio, C.; Brugiapaglia, S.; Bulfamante, S.; Follia, L.; Cappello, P.; Novelli, F. The Glycolytic Pathway as a Target for Novel Onco-Immunology Therapies in Pancreatic Cancer. Molecules 2021, 26, 1642. https://doi.org/10.3390/molecules26061642
Curcio C, Brugiapaglia S, Bulfamante S, Follia L, Cappello P, Novelli F. The Glycolytic Pathway as a Target for Novel Onco-Immunology Therapies in Pancreatic Cancer. Molecules. 2021; 26(6):1642. https://doi.org/10.3390/molecules26061642
Chicago/Turabian StyleCurcio, Claudia, Silvia Brugiapaglia, Sara Bulfamante, Laura Follia, Paola Cappello, and Francesco Novelli. 2021. "The Glycolytic Pathway as a Target for Novel Onco-Immunology Therapies in Pancreatic Cancer" Molecules 26, no. 6: 1642. https://doi.org/10.3390/molecules26061642