
Influenza Viruses: Harnessing the Crucial Role of the M2 Ion-Channel and Neuraminidase toward Inhibitor Design

 , , ,
, , ,  , and
, and 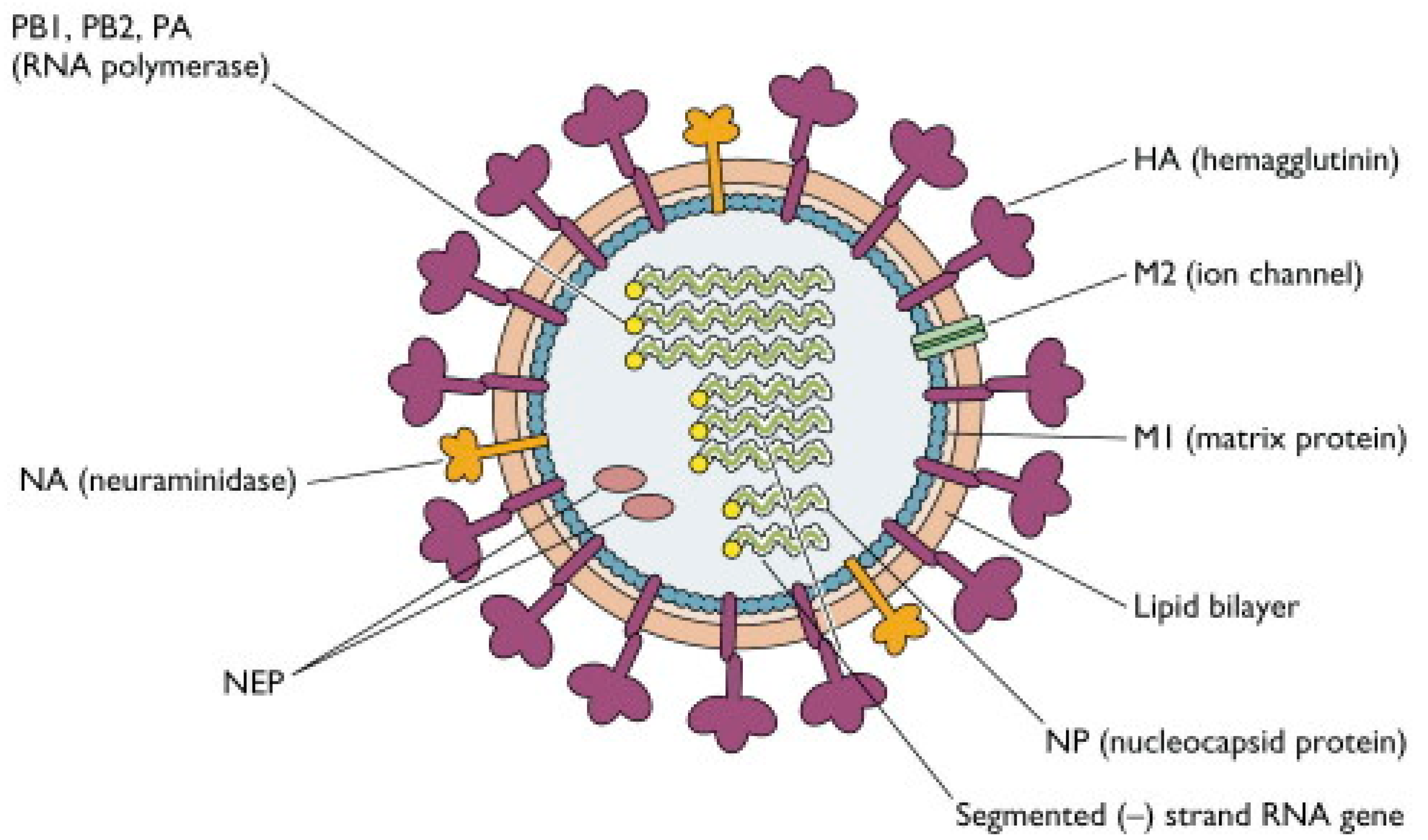{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Influenza Viruses
2.1. Structure of Influenza Viruses
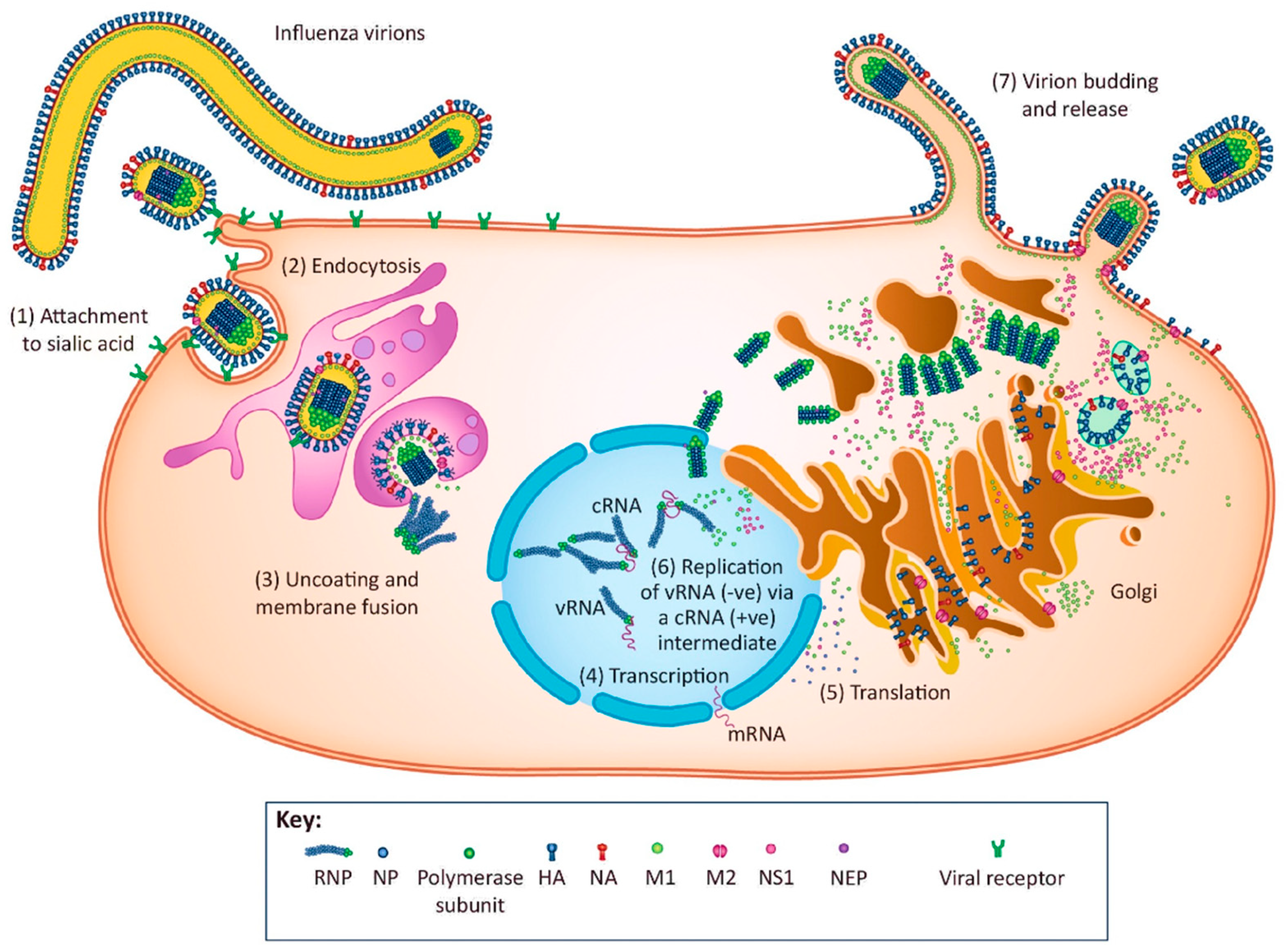
2.2. Replication Cycle of Influenza Virus
2.2.1. Virus Attachment
2.2.2. Endocytosis
2.2.3. Uncoating and Membrane Fusion
2.2.4. Transcription of the Viral RNA
2.2.5. Translation of Viral Proteins
2.2.6. Replication of the Viral RNA
2.2.7. Virion Budding and Release
3. The AM2 Ion Channel
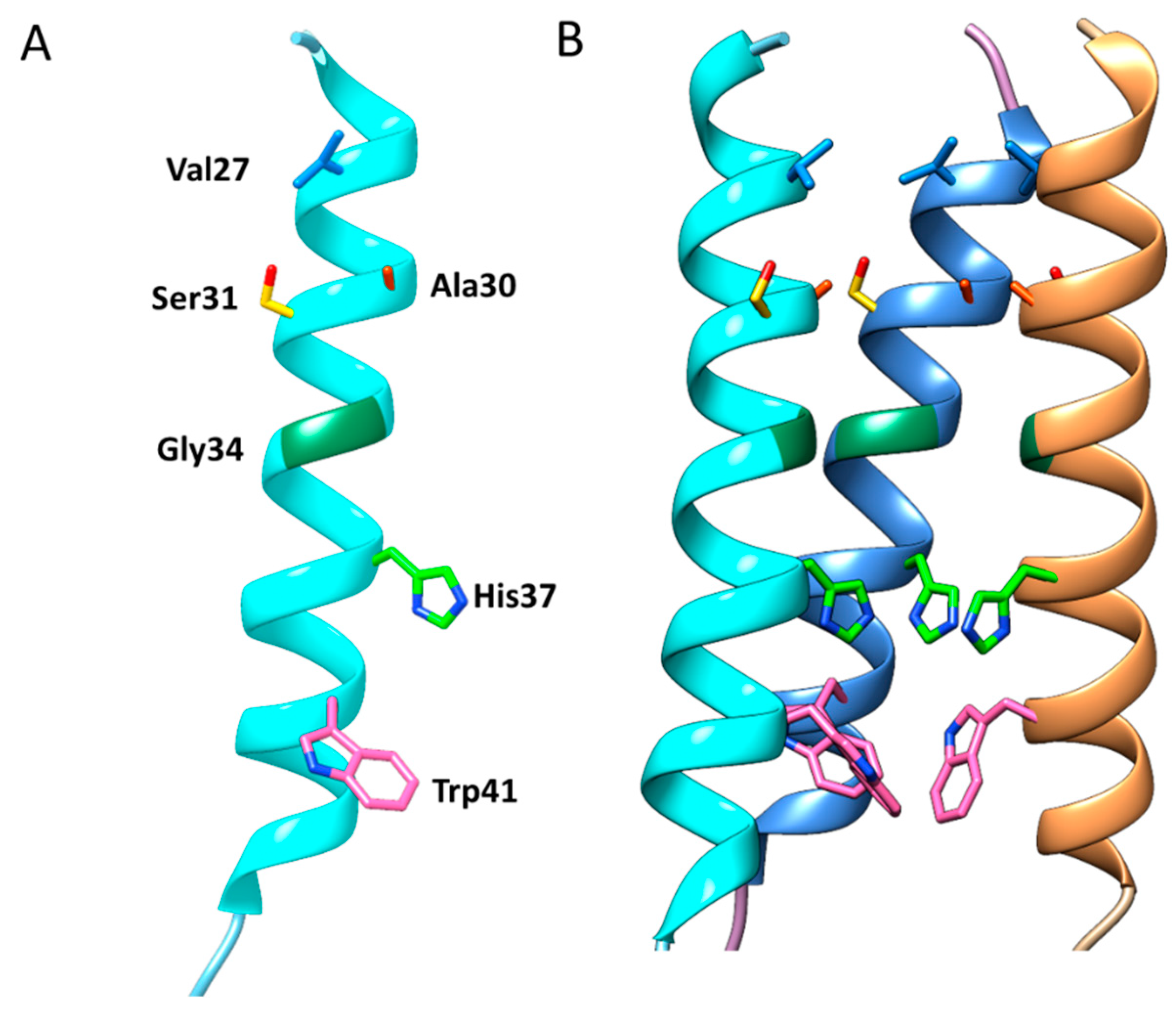
3.1. Structure and Function of the AM2 Ion Channel
3.2. Catalytic Mechanism of the AM2 Ion Channel
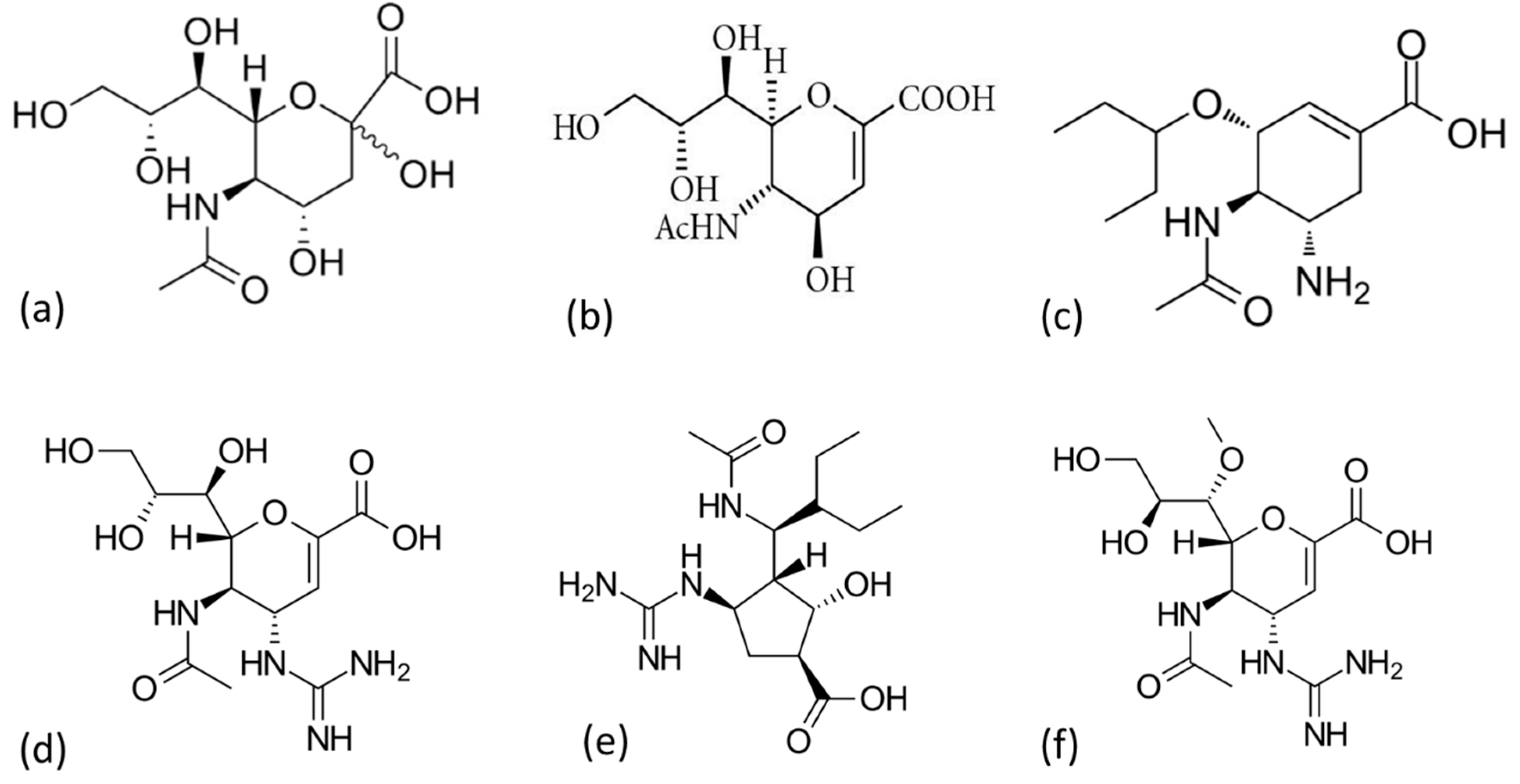
3.3. AM2 Channel Inhibitors
4. Neuraminidase (NA)
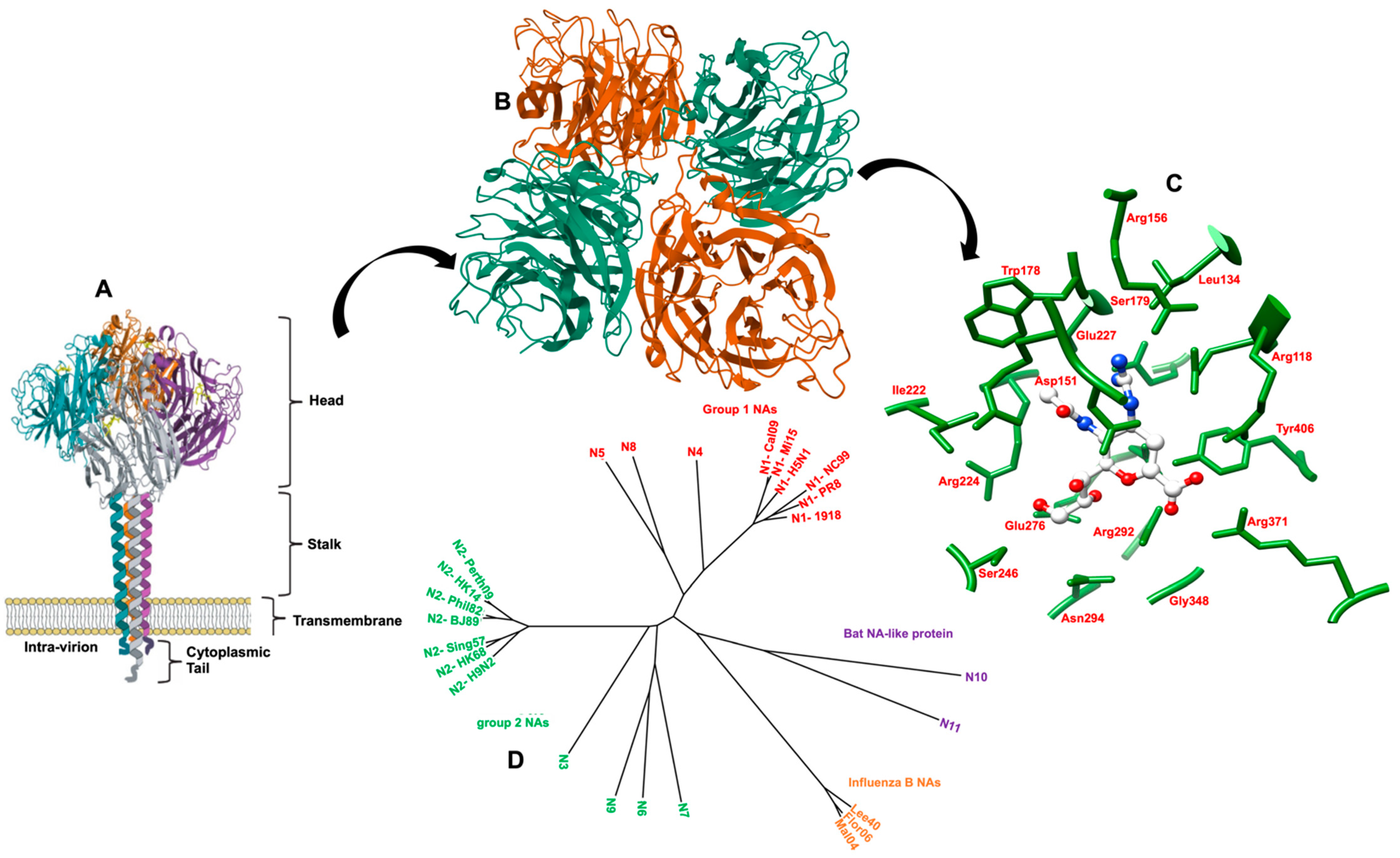
4.1. Structure and Function of NA
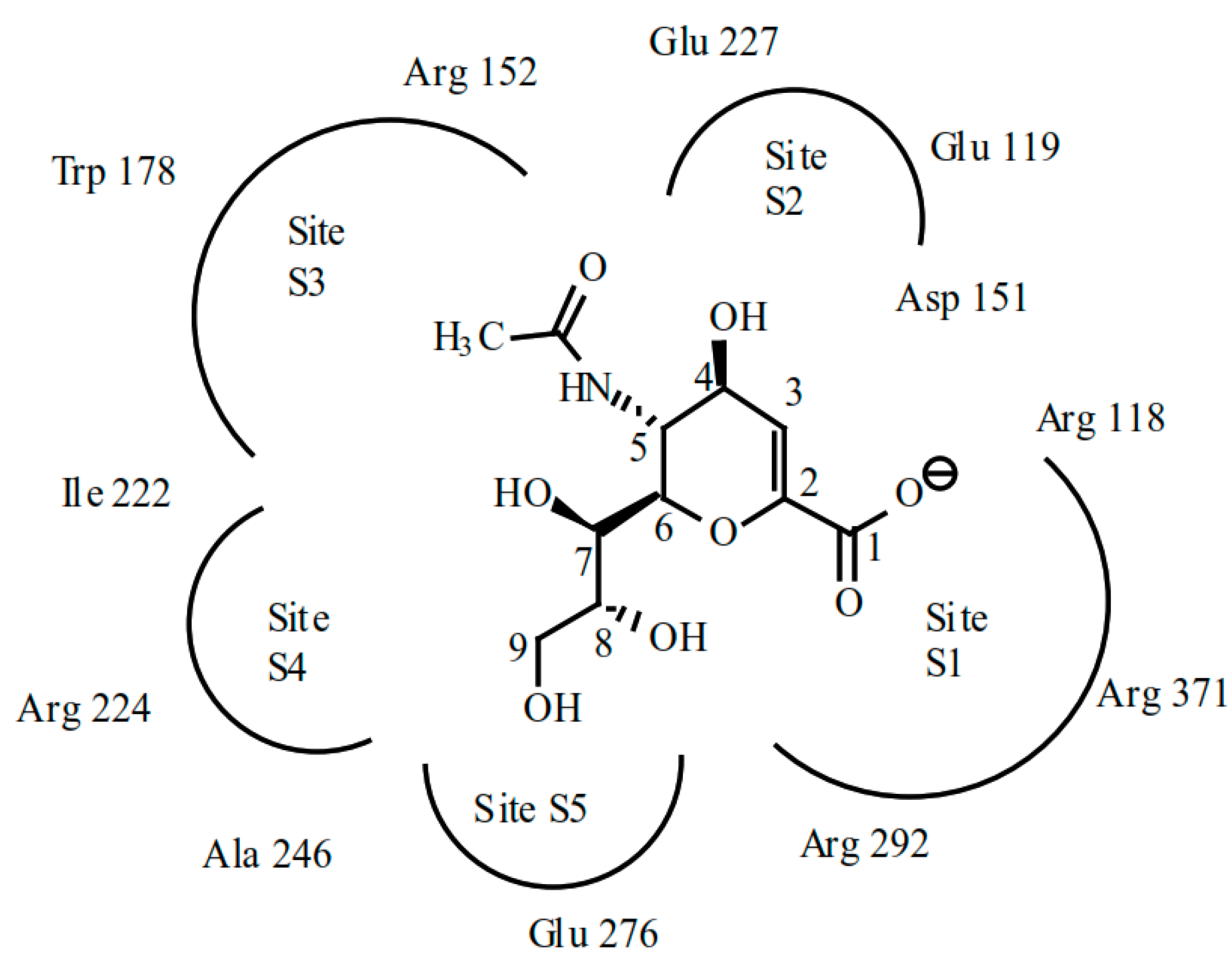
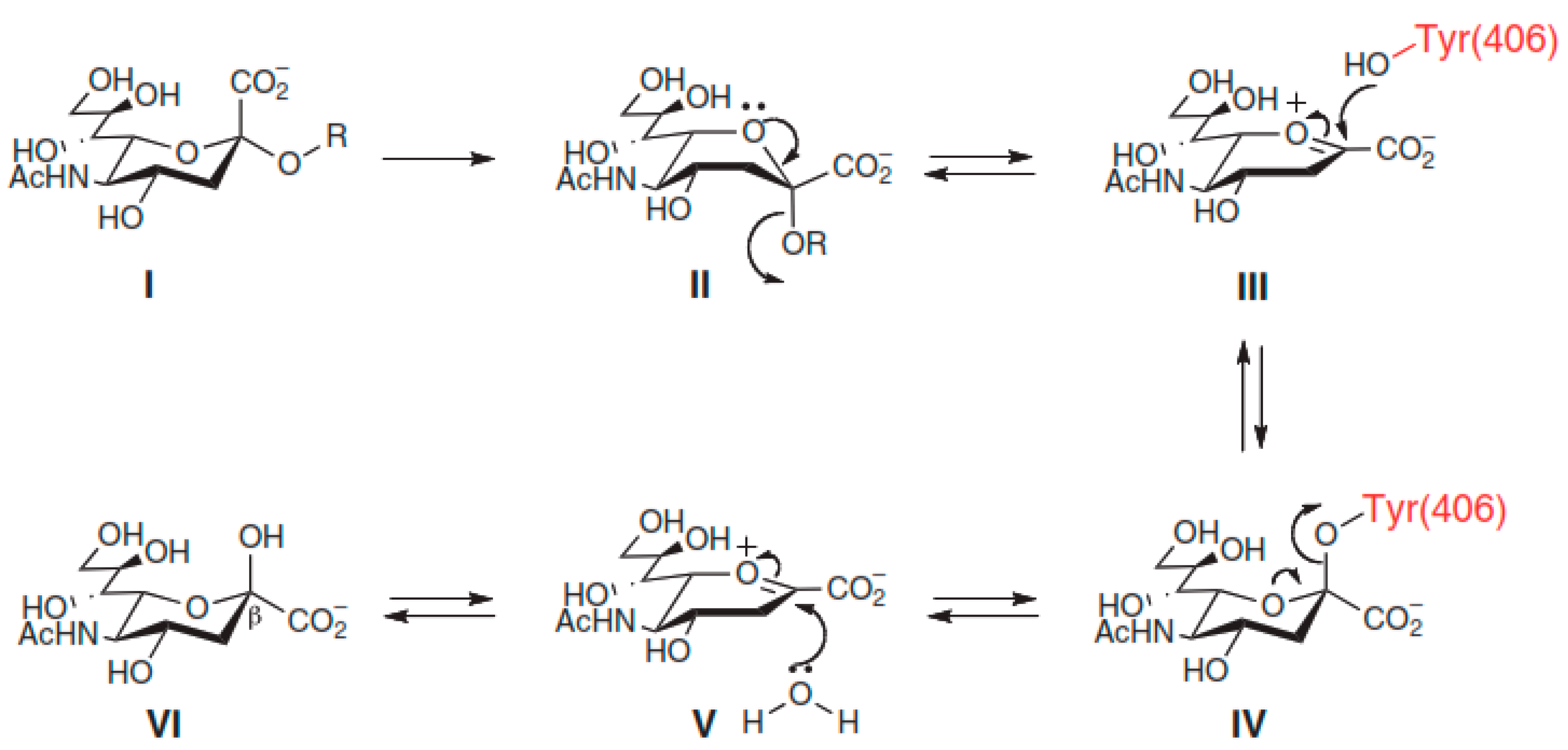
4.2. Catalytic Mechanism of NA
5. Conclusions and Future Perspectives
Author Contributions
Funding
Figure Disclosure
Acknowledgments
Conflicts of Interest
References
- Davidson, S. Treating influenza infection, from now and into the future. Front. Immunol. 2018, 9, 1946. [Google Scholar] [CrossRef] [PubMed]
- Paget, J.; Spreeuwenberg, P.; Charu, V.; Taylor, R.J.; Iuliano, A.D.; Bresee, J.; Simonsen, L.; Viboud, C.; GSIMCN and GlCT. Global mortality associated with seasonal influenza epidemics: New burden estimates and predictors from the GLaMOR Project. J. Glob. Health 2019, 9, 20421. [Google Scholar] [CrossRef] [PubMed]
- Disease Burden of Influenza; CDC: Atlanta, GA, USA, 2020.
- Das, K. Antivirals targeting influenza A virus. J. Med. Chem. 2012, 55, 6263–6277. [Google Scholar] [CrossRef]
- Hampson, A.W.; Mackenzie, J.S. The influenza viruses. Med. J. Aust. 2006, 185, S39–S43. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Camilloni, B.; Alunno, A.; Polinori, I.; Argentiero, A.; Esposito, S. Drugs for influenza treatment: Is there significant news? Front. Med. 2019, 6, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrjänen, R.K.; Jokinen, J.; Ziegler, T.; Sundman, J.; Lahdenkari, M.; Julkunen, I.; Kilpi, T.M. Effectiveness of pandemic and seasonal influenza vaccines in preventing laboratory-confirmed influenza in adults: A clinical cohort study during epidemic seasons 2009–2010 and 2010–2011 in Finland. PLoS ONE 2014, 9, e108538. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza virus: Dealing with a drifting and shifting pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- Wikramaratna, P.S.; Sandeman, M.; Recker, M.; Gupta, S. The antigenic evolution of influenza: Drift or thrift? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120200. [Google Scholar] [CrossRef] [Green Version]
- Lowen, A.C. Constraints, drivers, and implications of influenza A virus reassortment. Annu. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.-L. Evolution of influenza a virus by mutation and re-assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A. The persistent legacy of the 1918 influenza virus. N. Engl. J. Med. 2009, 361, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Taubenberger, J.K. The origin and virulence of the 1918 “Spanish” influenza virus. Proc. Am. Philos. Soc. 2006, 150, 86–112. [Google Scholar] [PubMed]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The mother of all pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef]
- Wilson, I.A.; Cox, N.J. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 1990, 8, 737–787. [Google Scholar] [CrossRef] [PubMed]
- Farrukee, R.; Hurt, A.C. Antiviral drugs for the treatment and prevention of influenza. Curr. Treat. Options Infect. Dis. 2017, 9, 318–332. [Google Scholar] [CrossRef]
- Tisdale, M. Influenza M2 ion-channel and neuraminidase inhibitors. In Antimicrobial Drug Resistance; Mayers, D.L., Ed.; Humana Press: Totowa, NJ, USA, 2009. [Google Scholar]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Balannik, V.; Pinto, L.H.; Lamb, R.A. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J. Virol. 2010, 84, 5078–5088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalily, P.H.; Duncan, M.C.; Fedida, D.; Wang, J.; Tietjen, I. Put a cork in it: Plugging the M2 viral ion channel to sink influenza. Antivir. Res. 2020, 178, 104780. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Takeuchi, K.; Pinto, L.H.; Lamb, R.A. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckimm-Breschkin, J.L. Influenza neuraminidase inhibitors: Anti-viral action and mechanisms of resistance. Influenza Other Respi. Viruses 2013, 7, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Takashita, E.; Daniels, R.S.; Fujisaki, S.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Lackenby, A.; Nguyen, H.T.; Pereyaslov, D.; et al. Global update on the susceptibilities of human influenza viruses to neuraminidase inhibitors and the cap-dependent endonuclease inhibitor baloxavir, 2017–2018. Antivir. Res. 2020, 175, 104718. [Google Scholar] [CrossRef]
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The viruses and their replication. In Fields’ Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1647–1689. [Google Scholar]
- Horimoto, T.; Kawaoka, Y. Influenza: Lessons from past pandemics, warnings from current incidents. Nat. Rev. Microbiol. 2005, 3, 591–600. [Google Scholar] [CrossRef]
- Kosik, I.; Yewdell, J.W. Influenza hemagglutinin and neuraminidase: Yin-Yang proteins coevolving to thwart immunity. Viruses 2019, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Gottschalk, A. Neuraminidase: The specific enzyme of influenza virus and Vibrio cholerae. Biochim. Biophys. Acta 1957, 23, 645–646. [Google Scholar] [CrossRef]
- Palese, P.; Schulman, J.L. Mapping of the influenza virus genome: Identification of the hemagglutinin and the neuraminidase genes. Proc. Natl. Acad. Sci. USA 1976, 73, 2142–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, G.N.; Herrler, G.; Paulson, J.C.; Klenk, H.D. Influenza C virus uses 9-O-acetyl-n-acetylneuraminic acid as a high affinity receptor determinant for attachment to cells. J. Biol. Chem. 1986, 261, 5947–5951. [Google Scholar] [CrossRef]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and swine: Proposal for a new genus in the orthomyxoviridae family. MBio 2014, 5, e00031-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Types of Influenza Viruses; CDC: Atlanta, GA, USA, 2019.
- Du, R.; Cui, Q.; Rong, L. Competitive cooperation of hemagglutinin and neuraminidase during influenza A virus entry. Viruses 2019, 11, 458. [Google Scholar] [CrossRef] [Green Version]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-W.; Webby, R.J.; Webster, R.G. Evolution and ecology of influenza A viruses. In Influenza Pathogenesis and Control; Compans, R.W., Oldstone, M.B.A., Eds.; Springer International Publishing: Midtown Manhattan, NY, USA, 2014. [Google Scholar]
- Fouchier, R.A.M.; Munster, V.; Wallensten, A.; Bestebroer, T.M.; Herfst, S.; Smith, D.; Rimmelzwaan, G.F.; Olsen, B.; Osterhaus, A.D.M.E. Characterization of a novel influenza a virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol. 2005, 79, 2814–2822. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.A.; Chen, L.-M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef] [Green Version]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Hurt, A.C.; Vijaykrishna, D.; Butler, J.; Baas, C.; Maurer-Stroh, S.; Silva-De-La-Fuente, M.C.; Medina-Vogel, G.; Olsen, B.; Kelso, A.; Barr, I.G.; et al. Detection of evolutionarily distinct avian influenza A viruses in antarctica. MBio 2014, 5, e01098-14. [Google Scholar] [CrossRef] [Green Version]
- Caini, S.; Kusznierz, G.; Garate, V.V.; Wangchuk, S.; Thapa, B.; de Paula Júnior, F.J.; de Ferreira Almeida, W.A.; Njouom, R.; Fasce, R.A.; Bustos, P.; et al. The epidemiological signature of influenza B virus and its B/Victoria and B/Yamagata lineages in the 21st century. PLoS ONE 2019, 14, e0222381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, R.G.; Shortridge, K.F.; Kawaoka, Y. Influenza: Interspecies transmission and emergence of new pandemics. FEMS Immunol. Med. Microbiol. 1997, 18, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Hampson, A.W. Influenza virus antigens and ‘antigenic drift’. Perspect. Med. Virol. 2002, 7, 49–85. [Google Scholar] [CrossRef]
- Garman, E.; Laver, G. The structure, function, and inhibition of influenza virus neuraminidase. In Viral Membrane Proteins: Structure, Function, and Drug Design; Fischer, W.B., Ed.; Springer: Boston, MA, USA, 2005. [Google Scholar]
- Rossman, J.S.; Lamb, R.A. Viral membrane scission. Annu. Rev. Cell Dev. Biol. 2013, 29, 551–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badham, M.D.; Rossman, J.S. Filamentous influenza viruses. Curr. Clin. Microbiol. Rep. 2016, 3, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racaniello, V. Structure of Influenza Virus. Available online: http://www.virology.ws/2009/04/30/structure-of-influenza-virus/ (accessed on 10 September 2020).
- Mcgeoch, D.; Fellner, P.; Newton, C. Influenza virus genome consists of eight distinct RNA species. Proc. Natl. Acad. Sci. USA 1976, 73, 3045–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.A.; Choppin, P.W.; Chanock, R.M.; Lai, C.J. Mapping of the two overlapping genes for polypeptides NS1 and NS2 on RNA segment 8 of influenza virus genome. Proc. Natl. Acad. Sci. USA 1980, 77, 1857–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briedis, D.J.; Lamb, R.A. Influenza B virus genome: Sequences and structural organization of RNA segment 8 and the mRNAs coding for the NS1 and NS2 proteins. J. Virol. 1982, 42, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Colacino, J.M.; Staschke, K.A.; Laver, W.G. Approaches and strategies for the treatment of influenza virus infections. Anti Viral Chem. Chemother. 1999, 10, 155–185. [Google Scholar] [CrossRef] [PubMed]
- Arranz, R.; Coloma, R.; Chichón, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortín, J.; Martín-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef]
- Moeller, A.; Kirchdoerfer, R.N.; Potter, C.S.; Carragher, B.; Wilson, I.A. Organization of the influenza virus replication machinery. Science 2012, 338, 1631–1634. [Google Scholar] [CrossRef] [Green Version]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, T.; Muraki, Y.; Noda, T.; Takashita, E.; Sho, R.; Sugawara, K.; Matsuzaki, Y.; Shimotai, Y.; Hongo, S. Role of the CM2 protein in the influenza C virus replication cycle. J. Virol. 2011, 85, 1322–1329. [Google Scholar] [CrossRef] [Green Version]
- Denney, L.; Ho, L.-P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef] [PubMed]
- van Riel, D.; Den Bakker, M.A.; Leijten, L.M.E.; Chutinimitkul, S.; Munster, V.J.; de Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.M.; Osterhaus, A.D.M.E.; Kuiken, T. Seasonal and pandemic human influenza viruses attach better to human upper respiratory tract epithelium than avian influenza viruses. Am. J. Pathol. 2010, 176, 1614–1618. [Google Scholar] [CrossRef]
- Hutchinson, E.C. Influenza virus. Trends Microbiol. 2018, 26, 809–810. [Google Scholar] [CrossRef]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, E.; Aoyama, T.; Kato, H.; Suzuki, Y.; Tateno, Y.; Nakajima, K. Comparison of complete amino acid sequences and receptor-binding properties among 13 serotypes of hemagglutinins of influenza a viruses. Virology 1991, 182, 475–485. [Google Scholar] [CrossRef]
- Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gambaryan, A.; Klimov, A.; Castrucci, M.R.; Donatelli, I.; Kawaoka, Y. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 2000, 74, 8502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, G.N.; Paulson, J.C. Receptor determinants of human and animal influenza virus isolates: Differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology 1983, 127, 361–373. [Google Scholar] [CrossRef]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.-D. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. USA 2004, 101, 4620–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couceiro, J.N.; Paulson, J.C.; Baum, L.G. Influenza virus strains selectively recognize sialyloligosaccharides on human respiratory epithelium; the role of the host cell in selection of hemagglutinin receptor specificity. Virus Res. 1993, 29, 155–165. [Google Scholar] [CrossRef]
- Matlin, K.S.; Reggio, H.; Helenius, A.; Simons, K. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 1981, 91, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004, 11, 567–573. [Google Scholar] [CrossRef] [Green Version]
- White, J.; Helenius, A.; Gething, M.-J. Haemagglutinin of influenza virus expressed from a cloned gene promotes membrane fusion. Nature 1982, 300, 658–659. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Ruigrok, R.W.; Cusack, S. The 2.2 A resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. EMBO J. 1992, 11, 49–56. [Google Scholar] [CrossRef]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, T. Membrane fusion mechanisms: The influenza hemagglutinin paradigm and its implications for intracellular fusion. Traffic 2000, 1, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Sieczkarski, S.B.; Whittaker, G.R. Viral entry. Curr. Top. Microbiol. Immunol. 2005, 285. [Google Scholar] [CrossRef]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [Green Version]
- Pinto, L.H.; Lamb, R.A. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006, 281, 8997–9000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.; Helenius, A. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 1991, 65, 232–244. [Google Scholar] [CrossRef] [Green Version]
- Bui, M.; Whittaker, G.; Helenius, A. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J. Virol. 1996, 70, 8391–8401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhauer, D.A.; Wharton, S.A.; Skehel, J.J.; Wiley, D.C.; Hay, A.J. Amantadine selection of a mutant influenza virus containing an acid-stable hemagglutinin glycoprotein: Evidence for virus-specific regulation of the pH of glycoprotein transport vesicles. Proc. Natl. Acad. Sci. USA 1991, 88, 11525–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Lamb, R.A. Influenza virus M2 protein ion channel activity stabilizes the native form of fowl plague virus hemagglutinin during intracellular transport. J. Virol. 1994, 68, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemler, I.; Whittaker, G.; Helenius, A. Nuclear import of microinjected influenza virus ribonucleoproteins. Virology 1994, 202, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- O’neill, R.E.; Jaskunas, R.; Blobel, G.; Palese, P.; Moroianu, J. Nuclear import of influenza virus rna can be mediated by viral nucleoprotein and transport factors required for protein import. J. Biol. Chem. 1995, 270, 22701–22704. [Google Scholar] [CrossRef] [Green Version]
- Cros, J.F.; Palese, P. Trafficking of viral genomic RNA into and out of the nucleus: Influenza, Thogoto and Borna disease viruses. Virus Res. 2003, 95, 3–12. [Google Scholar] [CrossRef]
- Fodor, E.; Seong, B.L.; Brownlee, G.G. Photochemical cross-linking of influenza a polymerase to its virion RNA promoter defines a polymerase binding site at residues 9 to 12 of the promoter. J. Gen. Virol. 1993, 74, 1327–1333. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Matsumae, H.; Katoh, M.; Eisfeld, A.J.; Neumann, G.; Hase, T.; Ghosh, S.; Shoemaker, J.E.; Lopes, T.J.S.; Watanabe, T.; et al. A comprehensive map of the influenza A virus replication cycle. BMC Syst. Biol. 2013, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Plotch, S.J.; Bouloy, M.; Ulmanen, I.; Krug, R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [Google Scholar] [CrossRef]
- Krug, R.M. Priming of influenza viral RNA transcription by capped heterologous RNAs. Curr. Top Microbiol. Immunol. 1981, 93, 125–149. [Google Scholar]
- Robertson, J.S.; Schubert, M.; Lazzarini, R.A. Polyadenylation sites for influenza virus mRNA. J. Virol. 1981, 38, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Palese, P. Characterization of the polyadenylation signal of influenza virus RNA. J. Virol. 1994, 68, 1245–1249. [Google Scholar] [CrossRef] [Green Version]
- Jorba, N.; Coloma, R.; Ortín, J. Genetic trans-complementation establishes a new model for influenza virus rna transcription and replication. PLoS Pathog. 2009, 5, e1000462. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E. The RNA polymerase of influenza A virus: Mechanisms of viral transcription and replication. Acta Virol. 2013, 57, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, L.L.; Kuo, R.-L.; Ye, Q.; Jiang, Y.; Tao, Y.J.; Krug, R.M. Interaction of the influenza A virus nucleocapsid protein with the viral rna polymerase potentiates unprimed viral rna replication. J. Virol. 2009, 83, 29. [Google Scholar] [CrossRef] [Green Version]
- York, A.; Hengrung, N.; Vreede, F.T.; Huiskonen, J.T.; Fodor, E. Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc. Natl. Acad. Sci. USA 2013, 110, E4238–E4245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, M.J.; Bruce, E.A.; Read, E.K.C.; Foeglein, A.; Mahen, R.; Stuart, A.D.; Digard, P. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 2011, 85, 4143–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisfeld, A.J.; Kawakami, E.; Watanabe, T.; Neumann, G.; Kawaoka, Y. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J. Virol. 2011, 85, 6117–6126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momose, F.; Sekimoto, T.; Ohkura, T.; Jo, S.; Kawaguchi, A.; Nagata, K.; Morikawa, Y. Apical transport of influenza A virus ribonucleoprotein requires Rab11-positive recycling endosome. PLoS ONE 2011, 6, e21123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enami, M.; Sharma, G.; Benham, C.; Palese, P. An influenza virus containing nine different RNA segments. Virology 1991, 185, 291–298. [Google Scholar] [CrossRef]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Gerl, M.J.; Sampaio, J.L.; Urban, S.; Kalvodova, L.; Verbavatz, J.-M.; Binnington, B.; Lindemann, D.; Lingwood, C.A.; Shevchenko, A.; Schroeder, C.; et al. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J. Cell Biol. 2012, 196, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Avalos, R.T.; Ponimaskin, E.; Nayak, D.P. Influenza virus assembly: Effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 2000, 74, 8709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnet, F.M.; Mccrea, J.F.; Stone, J.D. Modification of human red cells by virus action; the receptor gradient for virus action in human red cells. Br. J. Exp. Pathol. 1946, 27, 228–236. [Google Scholar] [PubMed]
- Webster, R.G.; Laver, W.G. Preparation and properties of antibody directed specifically against the neuraminidase of influenza virus. J. Immunol. 1967, 99, 49–55. [Google Scholar]
- Lamb, R.A.; Zebedee, S.L.; Richardson, C.D. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell 1985, 40, 627–633. [Google Scholar] [CrossRef]
- von Heijne, G.; Gavel, Y. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 1988, 174, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Chizhmakov, I.V.; Geraghty, F.M.; Ogden, D.C.; Hayhurst, A.; Antoniou, M.; Hay, A.J. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 1996, 494, 329–336. [Google Scholar] [CrossRef]
- Wang, C.; Lamb, R.A.; Pinto, L.H. Direct measurement of the influenza A virus M2 protein ion channel activity in mammalian cells. Virology 1994, 205, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Luo, G.; Hahnenberger, K.M.; Brooks, C.; Gecha, O.; Ingalls, K.; Numata, K.; Krystal, M. Growth impairment resulting from expression of influenza virus M2 protein in Saccharomyces cerevisiae: Identification of a novel inhibitor of influenza virus. Antimicrob. Agents Chemother. 1995, 39, 2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Q.; Pinto, L.H.; Luo, G.; Shaughnessy, M.A.; Mullaney, D.; Kurtz, S.; Krystal, M.; Lamb, R.A. Characterization of inhibition of M2 ion channel activity by BL-1743, an inhibitor of influenza A virus. J. Virol. 1996, 70, 4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helenius, A. Unpacking the incoming influenza virus. Cell 1992, 69, 577–578. [Google Scholar] [CrossRef]
- Ciampor, F.; Bayley, P.M.; Nermut, M.V.; Hirst, E.M.A.; Sugrue, R.J.; Hay, A.J. Evidence that the amantadine-induced, M2-mediated conversion of influenza A virus hemagglutinin to the low pH conformation occurs in an acidic trans golgi compartment. Virology 1992, 188, 14–24. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Leser, G.P.; Lamb, R.A. The ion channel activity of the influenza virus M2 protein affects transport through the Golgi apparatus. J. Cell Biol. 1996, 133, 733–747. [Google Scholar] [CrossRef]
- Moorthy, N.S.H.N.; Poongavanam, V.; Pratheepa, V. Viral M2 ion channel protein: A promising target for anti-influenza drug discovery. Mini Rev. Med. Chem. 2014, 14, 819–830. [Google Scholar]
- Deamer, D.W. Visualizing proton conductance in the gramicidin channel. Biophys. J. 1996, 71, 5. [Google Scholar] [CrossRef] [Green Version]
- Pinto, L.H.; Dieckmann, G.R.; Gandhi, C.S.; Papworth, C.G.; Braman, J.; Shaughnessy, M.A.; Lear, J.D.; Lamb, R.A.; Degrado, W.F. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl. Acad. Sci. USA 1997, 94, 11301–11306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zaitseva, F.; Lamb, R.A.; Pinto, L.H. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 2002, 277, 39880–39886. [Google Scholar] [CrossRef] [Green Version]
- Sansom, M.S.; Kerr, I.D.; Smith, G.R.; Son, H.S. The influenza A virus M2 channel: A molecular modeling and simulation study. Virology 1997, 233, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Q.; Husslein, T.; Moore, P.B.; Newns, D.M.; Pattnaik, P.; Klein, M.L. The M2 channel of influenza A virus: A molecular dynamics study. FEBS Lett. 1998, 434, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stouffer, A.L.; Acharya, R.; Salom, D.; Levine, A.S.; Di Costanzo, L.; Soto, C.S.; Tereshko, V.; Nanda, V.; Stayrook, S.; Degrado, W.F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Cady, S.D.; Schmidt-Rohr, K.; Wang, J.; Soto, C.S.; Degrado, W.F.; Hong, M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Yi, M.; Dong, H.; Qin, H.; Peterson, E.; Busath, D.D.; Zhou, H.-X.; Cross, T.A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Park, E.K.; Castrucci, M.R.; Portner, A.; Kawaoka, Y. The M2 ectodomain is important for its incorporation into influenza A virions. J. Virol. 1998, 72, 2449–2455. [Google Scholar] [CrossRef] [Green Version]
- Pinto, L.H.; Lamb, R.A. Controlling influenza virus replication by inhibiting its proton channel. Mol. Biosyst. 2007, 3, 18–23. [Google Scholar] [CrossRef]
- Acharya, R.; Carnevale, V.; Fiorin, G.; Levine, B.G.; Polishchuk, A.L.; Balannik, V.; Samish, I.; Lamb, R.A.; Pinto, L.H.; Degrado, W.F.; et al. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 15075–15080. [Google Scholar] [CrossRef] [Green Version]
- Thomaston, J.L.; Woldeyes, R.A.; Nakane, T.; Yamashita, A.; Tanaka, T.; Koiwai, K.; Brewster, A.S.; Barad, B.A.; Chen, Y.; Lemmin, T.; et al. XFEL structures of the influenza M2 proton channel: Room temperature water networks and insights into proton conduction. Proc. Natl. Acad. Sci. USA 2017, 114, 13357–13362. [Google Scholar] [CrossRef] [Green Version]
- Pielak, R.M.; Chou, J.J. Flu channel drug resistance: A tale of two sites. Protein Cell 2010, 1, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Holsinger, L.J.; Lamb, R.A. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology 1991, 183, 32–43. [Google Scholar] [CrossRef]
- Sugrue, R.J.; Hay, A.J. Structural characteristics of the M2 protein of influenza A viruses: Evidence that it forms a tetrameric channel. Virology 1991, 180, 617–624. [Google Scholar] [CrossRef]
- Okada, A.; Miura, T.; Takeuchi, H. Protonation of histidine and histidine-tryptophan interaction in the activation of the M2 ion channel from influenza A virus. Biochemistry 2001, 40, 6053–6060. [Google Scholar] [CrossRef]
- Betakova, T.; Ciampor, F.; Hay, A.J. Influence of residue 44 on the activity of the M2 proton channel of influenza A virus. J. Gen. Virol. 2005, 86, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Chou, J.J. Influenza M2 proton channels. Biochim. Biophys. Acta 2011, 1808, 522–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Gorman, O.T.; Kawaoka, Y.; Bean, W.J.; Webster, R.G. Evolutionary analysis of the influenza A virus M gene with comparison of the M1 and M2 proteins. J. Virol. 1991, 65, 5491–5498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Cho, K.J.; Fiers, W.; Saelens, X. M2e-based universal influenza A vaccines. Vaccines 2015, 3, 105–136. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lamb, R.A.; Pinto, L.H. Activation of the M2 ion channel of influenza virus: A role for the transmembrane domain histidine residuE. Biophys. J. 1995, 69, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, P.; Lamb, R.A.; Pinto, L.H. Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza A virus. J. Biol. Chem. 2005, 280, 21463–21472. [Google Scholar] [CrossRef] [Green Version]
- Kass, I.; Arkin, I.T. How pH opens a H+ channel: The gating mechanism of influenza A M2. Structure 2005, 13, 1789–1798. [Google Scholar] [CrossRef] [Green Version]
- Shimbo, K.; Brassard, D.L.; Lamb, R.A.; Pinto, L.H. Ion selectivity and activation of the M2 ion channel of influenza virus. Biophys. J. 1996, 70, 1335–1346. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Cross, T.A.; Zhou, H.-X. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc. Natl. Acad. Sci. USA 2009, 106, 13311–13316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, E.; Dal Peraro, M.; Devane, R.; Vemparala, S.; Degrado, W.F.; Klein, M.L. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc. Natl. Acad. Sci. USA 2009, 106, 1069–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mould, J.A.; Li, H.-C.; Dudlak, C.S.; Lear, J.D.; Pekosz, A.; Lamb, R.A.; Pinto, L.H. Mechanism for proton conduction of the M2 ion channel of influenza A virus. J. Biol. Chem. 2000, 275, 8592–8599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smondyrev, A.M.; Voth, G.A. Molecular dynamics simulation of proton transport through the influenza A virus M2 channel. Biophys. J. 2002, 83, 1987–1996. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Luo, W.; Hong, M. Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 2010, 330, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Cady, S.D.; Luo, W.; Hu, F.; Hong, M. Structure and function of the influenza A M2 proton channel. Biochemistry 2009, 48, 7356–7364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; Degrado, W.F. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 2012, 21, 1620–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Fu, R.; Nishimura, K.; Zhang, L.; Zhou, H.-X.; Busath, D.D.; Vijayvergiya, V.; Cross, T.A. Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity. Proc. Natl. Acad. Sci. USA 2006, 103, 6865–6870. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Swanson, J.M.J.; Madsen, J.J.; Hong, M.; Degrado, W.F.; Voth, G.A. Acid activation mechanism of the influenza A M2 proton channel. Proc. Natl. Acad. Sci. USA 2016, 113, E6955–E6964. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Schmidt-Rohr, K.; Hong, M. NMR detection of pH-dependent histidine-water proton exchange reveals the conduction mechanism of a transmembrane proton channel. J. Am. Chem. Soc. 2012, 134, 3703–3713. [Google Scholar] [CrossRef] [Green Version]
- Hay, A.J.; Wolstenholme, A.J.; Skehel, J.J.; Smith, M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985, 4, 3021–3024. [Google Scholar] [CrossRef]
- Balannik, V.; Carnevale, V.; Fiorin, G.; Levine, B.G.; Lamb, R.A.; Klein, M.L.; Degrado, W.F.; Pinto, L.H. Functional studies and modeling of pore-lining residue mutants of the influenza A virus M2 ion channel. Biochemistry 2010, 49, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.K.; Batsomboon, P.; Dai, J.; Hung, I.; Zhou, H.-X.; Dudley, G.B.; Cross, T.A. Differential binding of rimantadine enantiomers to influenza A M2 proton channel. J. Am. Chem. Soc. 2016, 138, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Hirst, G.K. Adsorption of influenza hemagglutinins and virus by red blood cells. J. Exp. Med. 1942, 76, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, J.A.; Basak, S.; Compans, R.W. Effects of hexose starvation and the role of sialic acid in influenza virus release. Virology 1983, 25, 324–334. [Google Scholar] [CrossRef]
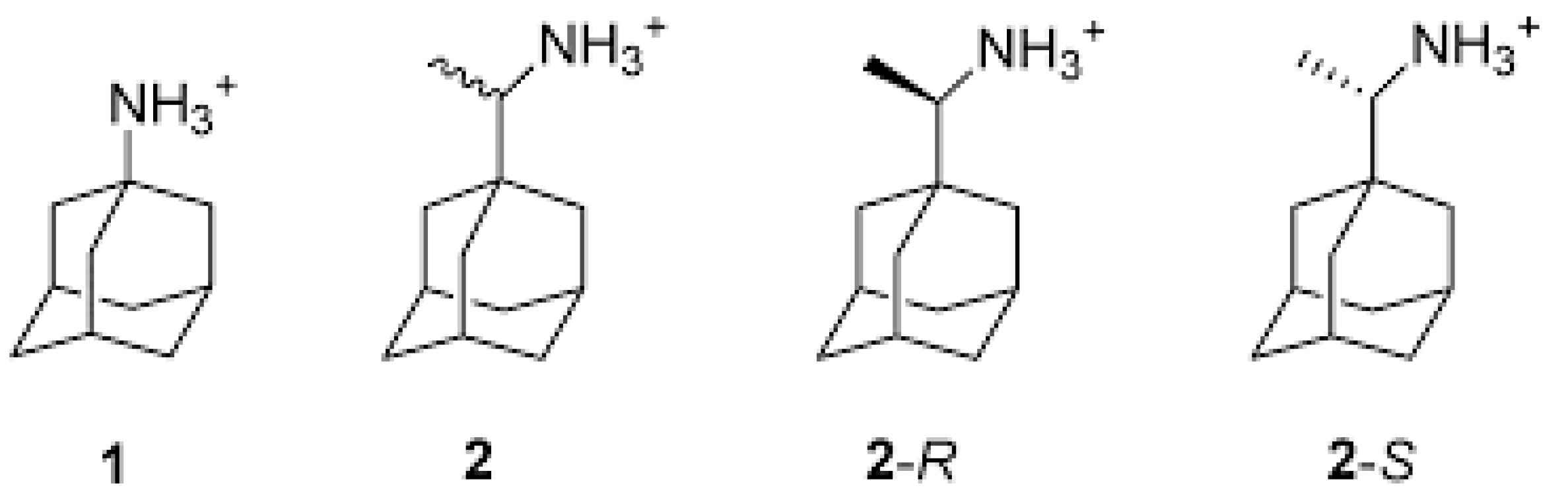
- Kolocouris, A.; Spearpoint, P.; Martin, S.R.; Hay, A.J.; López-Querol, M.; Sureda, F.X.; Padalko, E.; Neyts, J.; de Clercq, E. Comparisons of the influenza virus A M2 channel binding affinities, anti-influenza virus potencies and NMDA antagonistic activities of 2-alkyl-2-aminoadamantanes and analogues. Bioorg. Med. Chem. Lett. 2008, 18, 6156–6160. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cady, S.D.; Balannik, V.; Pinto, L.H.; Degrado, W.F.; Hong, M. Discovery of spiro-piperidine inhibitors and their modulation of the dynamics of the M2 proton channel from influenza A virus. J. Am. Chem. Soc. 2009, 131, 8066–8076. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Ma, C.; Ohigashi, Y.; Oliveira, F.A.; Jardetzky, T.S.; Pinto, L.H.; Lamb, R.A. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 10967–10972. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Hong, M. Conformational changes of an ion channel detected through water-protein interactions using solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010, 132, 2378–2384. [Google Scholar] [CrossRef] [Green Version]
- Laursen, N.S.; Wilson, I.A. Broadly neutralizing antibodies against influenza viruses. Antivir. Res. 2013, 98, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Zebedee, S.L.; Lamb, R.A. Influenza A virus M2 protein: Monoclonal antibody restriction of virus growth and detection of M2 in virions. J. Virol. 1988, 62, 2762–2772. [Google Scholar] [CrossRef] [Green Version]
- Padilla-Quirarte, H.O.; Lopez-Guerrero, D.V.; Gutierrez-Xicotencatl, L.; Esquivel-Guadarrama, F. Protective antibodies against influenza proteins. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Manzoor, R.; Eguchi, N.; Yoshida, R.; Ozaki, H.; Kondoh, T.; Okuya, K.; Miyamoto, H.; Takada, A. A novel mechanism underlying antiviral activity of an influenza virus M2-specific antibody. J. Virol. 2020, 95. [Google Scholar] [CrossRef]
- Okuya, K.; Eguchi, N.; Manzoor, R.; Yoshida, R.; Saito, S.; Suzuki, T.; Sasaki, M.; Saito, T.; Kida, Y.; Mori-Kajihara, A.; et al. Comparative analyses of the antiviral activities of IgG and IgA antibodies to influenza A virus M2 protein. Viruses 2020, 12, 780. [Google Scholar] [CrossRef] [PubMed]
- Kolpe, A.; Arista-Romero, M.; Schepens, B.; Pujals, S.; Saelens, X.; Albertazzi, L. Super-resolution microscopy reveals significant impact of M2e-specific monoclonal antibodies on influenza A virus filament formation at the host cell surface. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Skalickova, S.; Heger, Z.; Krejcova, L.; Pekarik, V.; Bastl, K.; Janda, J.; Kostolansky, F.; Vareckova, E.; Zitka, O.; Adam, V.; et al. Perspective of use of antiviral peptides against influenza virus. Viruses 2015, 7, 5428–5442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Kong, B.; Moon, S.; Yu, S.H.; Chung, J.; Ban, C.; Chung, W.J.; Kim, S.G.; Kweon, D.H. Envelope-deforming antiviral peptide derived from influenza virus M2 protein. Biochem. Biophys. Res. Commun. 2019, 517, 507–512. [Google Scholar] [CrossRef]
- Webster, R.G.; Laver, W.G.; Kilbourne, E.D. Reactions of antibodies with surface antigens of influenza virus. J. Gen. Virol. 1968, 3, 315–326. [Google Scholar] [CrossRef]
- Palese, P.; Tobita, K.; Ueda, M.; Compans, R.W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 1974, 61, 397–410. [Google Scholar] [CrossRef]
- Burnet, F.M.; Stone, J.D. The receptor-destroying enzyme of V. cholerae. Aust. J. Exp. Biol. Med. Sci. 1947, 25, 227–233. [Google Scholar] [CrossRef]
- Colman, P.M.; Tulip, W.R.; Varghese, J.N.; Tulloch, P.A.; Baker, A.T.; Laver, W.G.; Air, G.M.; Webster, R.G. Three-dimensional structures of influenza virus neuraminidase-antibody complexes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1989, 323, 511–518. [Google Scholar]
- Varghese, J.N.; Laver, W.G.; Colman, P.M. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature 1983, 303, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.J.; Gamblin, S.J.; Skehel, J.J. Influenza glycoproteins: Hemagglutinin and neuraminidase. Textb. Influenza 2013. [Google Scholar] [CrossRef]
- Vavricka, C.J.; Liu, Y.; Kiyota, H.; Sriwilaijaroen, N.; Qi, J.; Tanaka, K.; Wu, Y.; Li, Q.; Li, Y.; Yan, J.; et al. Influenza neuraminidase operates via a nucleophilic mechanism and can be targeted by covalent inhibitors. Nat. Commun. 2013, 4, 1491. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.J.; Haire, L.F.; Stevens, D.J.; Collins, P.J.; Lin, Y.P.; Blackburn, G.M.; Hay, A.J.; Gamblin, S.J.; Skehel, J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, X.; Wilson, I.A. Structure determination of the 1918 H1N1 neuraminidase from a crystal with lattice-translocation defects. Acta Crystallogr. D Biol. Crystallogr. 2008, D64, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Qi, J.; Liu, Y.; Vavricka, C.J.; Wu, Y.; Li, Q.; Gao, G.F. Influenza A virus N5 neuraminidase has an extended 150-cavity. J. Virol. 2011, 85, 8431–8435. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.T.; Varghese, J.N.; Laver, W.G.; Air, G.M.; Colman, P.M. Three-dimensional structure of neuraminidase of subtype N9 from an avian influenza virus. Proteins Struct. Funct. Bioinform. 1987, 2, 111–117. [Google Scholar] [CrossRef]
- Cheng, C.K.; Tsai, C.H.; Shie, J.J.; Fang, J.M. From neuraminidase inhibitors to conjugates: A step towards better anti-influenza drugs? Future Med. Chem. 2014, 6, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Bossart-Whitaker, P.; Carson, M.; Babu, Y.S.; Smith, C.D.; Laver, W.G.; Air, G.M. Three-dimensional structure of influenza a N9 neuraminidase and its complex with the inhibitor 2-deoxy 2,3-dehydro-N-acetyl neuraminic acid. J. Mol. Biol. 1993, 232, 1069–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, J.N.; Colman, P.M. Three-dimensional structure of the neuraminidase of influenza virus A/Tokyo/3/67 at 2·2 Å resolution. J. Mol. Biol. 1991, 221, 473–486. [Google Scholar] [CrossRef]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza virus neuraminidase structure and functions. Front. Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vavricka, C.J.; Li, Q.; Wu, Y.; Qi, J.; Wang, M.; Liu, Y.; Gao, F.; Liu, J.; Feng, E.; He, J.; et al. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog. 2011, 7, e1002249. [Google Scholar] [CrossRef]
- Krammer, F.; Fouchier, R.A.M.; Eichelberger, M.C.; Webby, R.J.; Shaw-Saliba, K.; Wan, H.; Wilson, P.C.; Compans, R.W.; Skountzou, I.; Monto, A.S. NAction! how can neuraminidase-based immunity contribute to better influenza virus vaccines? MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Colman, P.M.; Hoyne, P.A.; Lawrence, M.C. Sequence and structure alignment of paramyxovirus hemagglutinin-neuraminidase with influenza virus neuraminidase. J. Virol. 1993, 67, 2972–2980. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.U.; Chen, X.; Mendel, D.B. Neuraminidase inhibitors as anti-influenza virus agents. Anti Viral Chem. Chemother. 1999, 10, 141–154. [Google Scholar] [CrossRef]
- Gong, J.; Xu, W.; Zhang, J. Structure and functions of influenza virus neuraminidase. Curr. Med. Chem. 2007, 14, 113–122. [Google Scholar] [CrossRef]
- Stoll, V.; Stewart, K.D.; Maring, C.J.; Muchmore, S.; Giranda, V.; Gu, Y.-G.Y.; Wang, G.; Chen, Y.; Sun, M.; Zhao, C.; et al. Influenza neuraminidase inhibitors: Structure-based design of a novel inhibitor series. Biochemistry 2003, 42, 718–727. [Google Scholar] [CrossRef]
- von Itzstein, M.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Wang, Y.-F.; Xu, J.; Gu, Q.; Liu, H.-B.; Xiao, P.-G.; Zhou, J.; Liu, Y.; Yang, Z.; Su, H. Anti-influenza agents from Traditional Chinese Medicine. Nat. Prod. Rep. 2010, 27, 1758. [Google Scholar] [CrossRef]
- Meyer, E. Internal water molecules and H-bonding in biological macromolecules: A review of structural features with functional implications. Protein Sci. 1992, 1, 1543–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, A.K.J.; Pegg, M.S.; Taylor, N.R.; von Itzstein, M. Evidence for a sialosyl cation transition-state complex in the reaction of sialidase from influenza virus. Eur. J. Biochem. 1992, 207, 335–343. [Google Scholar] [CrossRef]
- Taylor, N.R.; von Itzstein, M. Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J. Med. Chem. 1994, 37, 616–624. [Google Scholar] [CrossRef]
- Meindl, P.; Bodo, G.; Palese, P.; Schulman, J.; Tuppy, H. Inhibition of neuraminidase activity by derivatives of 2-deoxy-2,3-dehydro-N-acetylneuraminic acid. Virology 1974, 58, 457–463. [Google Scholar] [CrossRef]
- Goodford, P. Multivariate characterization of molecules for QSAR analysis. J. Chemom. 1996, 10, 107–117. [Google Scholar] [CrossRef]
- Holzer, C.T.; von Itzstein, M.; Jin, B.; Pegg, M.S.; Stewart, W.P.; Wu, W.-Y. Inhibition of sialidases from viral, bacterial and mammalian sources by analogues of 2-deoxy-2,3-didehydro-N-acetylneuraminic acid modified at the C-4 position. Glycoconj. J. 1993, 10, 40–44. [Google Scholar] [CrossRef]
- von Itzstein, M.; Dyason, J.C.; Oliver, S.W.; White, H.F.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S. A study of the active site of influenza virus sialidase: An approach to the rational design of novel anti-influenza drugs. J. Med. Chem. 1996, 39, 388–391. [Google Scholar] [CrossRef]
- Phillips, D.C. The three-dimensional structure of an enzyme molecule. Sci. Am. 1966, 215, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y.; et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Escarpe, P.A.; Eisenberg, E.J.; Cundy, K.C.; Sweet, C.; Jakeman, K.J.; Merson, J.; Lew, W.; Williams, M.; Zhang, L.; et al. Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrob. Agents Chemother. 1998, 42, 647. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.U.; Lew, W.; Williams, M.A.; Wu, H.; Zhang, L.; Chen, X.; Escarpe, P.A.; Mendel, D.B.; Laver, W.G.; Stevens, R.C. Structure—Activity relationship studies of novel carbocyclic influenza neuraminidase inhibitors. J. Med. Chem. 1998, 41, 2451–2460. [Google Scholar] [CrossRef]
- Babu, Y.S.; Chand, P.; Bantia, S.; Kotian, P.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T.L.; Elliott, A.J.; Parker, C.D.; et al. BCX-1812 (RWJ-270201): Discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J. Med. Chem. 2000, 43, 3482–3486. [Google Scholar] [CrossRef]
- Bantia, S.; Arnold, C.S.; Parker, C.D.; Upshaw, R.; Chand, P. Anti-influenza virus activity of peramivir in mice with single intramuscular injection. Antivir. Res. 2006, 69, 39–45. [Google Scholar] [CrossRef]
- Mclaughlin, M.M.; Skoglund, E.W.; Ison, M.G. Peramivir: An intravenous neuraminidase inhibitor. Expert Opin. Pharmacother. 2015, 16, 1889–1900. [Google Scholar] [CrossRef]
- Scott, L.J. Peramivir: A review in uncomplicated influenza. Drugs 2018, 78, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M. Laninamivir and its prodrug, CS-8958: Long-acting neuraminidase inhibitors for the treatment of influenza. Anti-viral Chem. Chemother. 2010, 21, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Ikematsu, H.; Kawai, N. Laninamivir octanoate: A new long-acting neuraminidase inhibitor for the treatment of influenza. Expert Rev. Anti Infect. Ther. 2011, 9, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, H.; Toyama, K.; Yoshiba, S.; Okabe, H.; Furuie, H. Intrapulmonary distribution and pharmacokinetics of laninamivir, a neuraminidase inhibitor, after a single inhaled administration of its prodrug, laninamivir octanoate, in healthy volunteers. Antimicrob. Agents Chemother. 2012, 56, 3873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.X.; Cheng, L.P.; Li, M.; Pang, W.; Wu, F.H. Discovery of novel acylhydrazone neuraminidase inhibitors. Eur. J. Med. Chem. 2019, 173, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Cheng, L.P.; Li, M.; Pang, W. Discovery of novel neuraminidase inhibitors by structure-based virtual screening, structural optimization, and bioassay. ACS Med. Chem. Lett. 2019, 10, 1667–1673. [Google Scholar] [CrossRef]
- Li, M.; Cheng, L.P.; Pang, W.; Zhong, Z.J.; Guo, L.L. Design, synthesis, and biological evaluation of novel acylhydrazone derivatives as potent neuraminidase inhibitors. ACS Med. Chem. Lett. 2020, 11, 1745–1750. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Zhang, J.; Ai, W.; Ding, X.; Desta, S.; Sun, L.; Sun, Z.; Ma, X.; Li, Z.; Wang, D.; et al. Design, synthesis and biological evaluation of “Multi-Site”-binding influenza virus neuraminidase inhibitors. Eur. J. Med. Chem. 2019, 178, 64–80. [Google Scholar] [CrossRef]
- Ju, H.; Zhang, J.; Sun, Z.; Huang, Z.; Qi, W.; Huang, B.; Zhan, P.; Liu, X. Discovery of C-1 modified oseltamivir derivatives as potent influenza neuraminidase inhibitors. Eur. J. Med. Chem. 2018, 146, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, D.; Huang, B.; Ma, X.; Qi, W.; Shi, F.; Liu, X.; Zhang, Y.; Xu, W. Discovery of N-substituted oseltamivir derivatives as potent and selective inhibitors of H5N1 influenza neuraminidase. J. Med. Chem. 2014, 57, 8445–8458. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mtambo, S.E.; Amoako, D.G.; Somboro, A.M.; Agoni, C.; Lawal, M.M.; Gumede, N.S.; Khan, R.B.; Kumalo, H.M. Influenza Viruses: Harnessing the Crucial Role of the M2 Ion-Channel and Neuraminidase toward Inhibitor Design. Molecules 2021, 26, 880. https://doi.org/10.3390/molecules26040880
Mtambo SE, Amoako DG, Somboro AM, Agoni C, Lawal MM, Gumede NS, Khan RB, Kumalo HM. Influenza Viruses: Harnessing the Crucial Role of the M2 Ion-Channel and Neuraminidase toward Inhibitor Design. Molecules. 2021; 26(4):880. https://doi.org/10.3390/molecules26040880
Chicago/Turabian StyleMtambo, Sphamadla E., Daniel G. Amoako, Anou M. Somboro, Clement Agoni, Monsurat M. Lawal, Nelisiwe S. Gumede, Rene B. Khan, and Hezekiel M. Kumalo. 2021. "Influenza Viruses: Harnessing the Crucial Role of the M2 Ion-Channel and Neuraminidase toward Inhibitor Design" Molecules 26, no. 4: 880. https://doi.org/10.3390/molecules26040880