Novel Small-Molecule Scaffolds as Candidates against the SARS Coronavirus 2 Main Protease: A Fragment-Guided in Silico Approach



Abstract
:
1. Introduction
2. Results
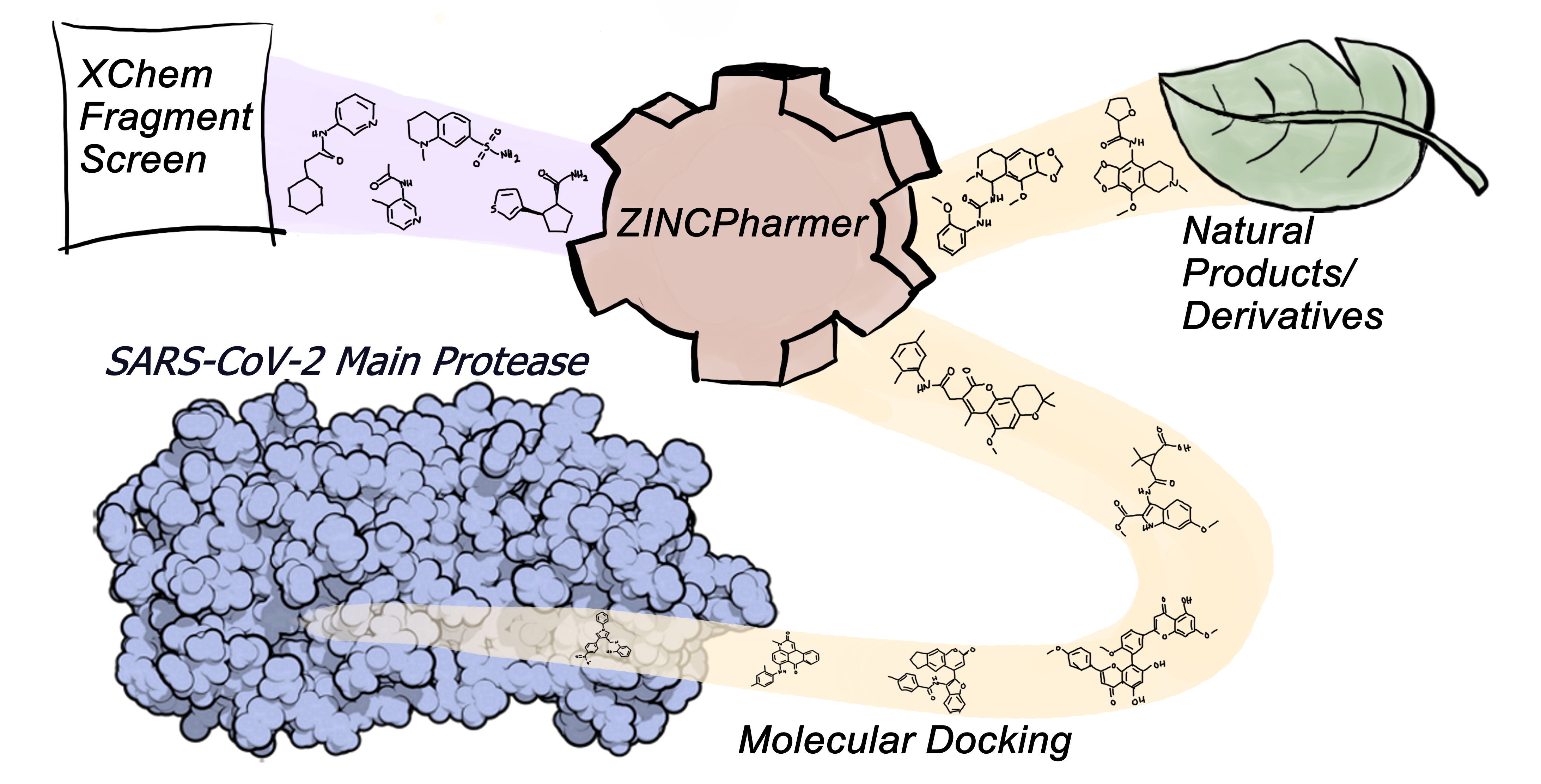
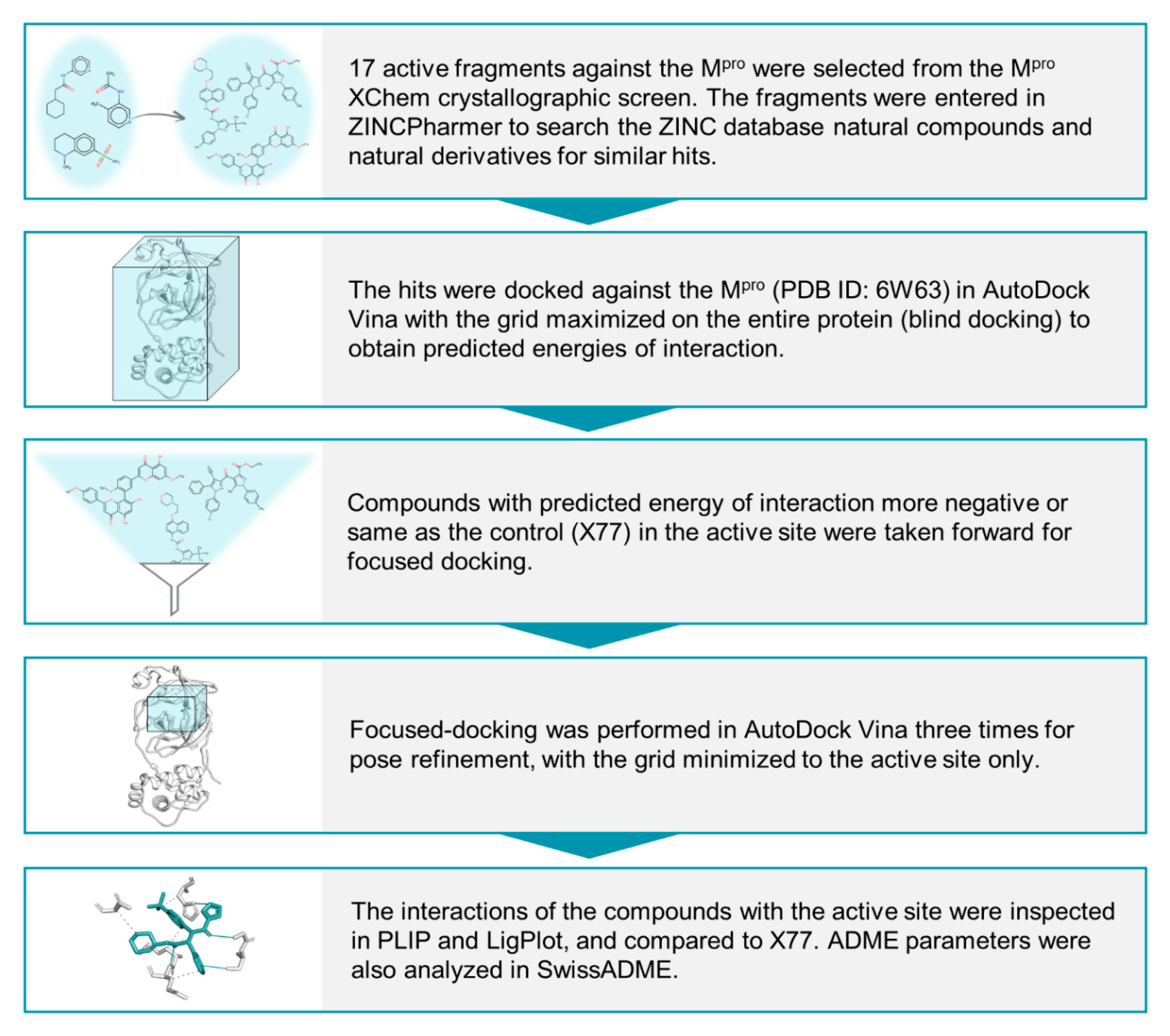
2.1. Identification of Initial Hits through Fragment-Derived Pharmacophore-Based Screening
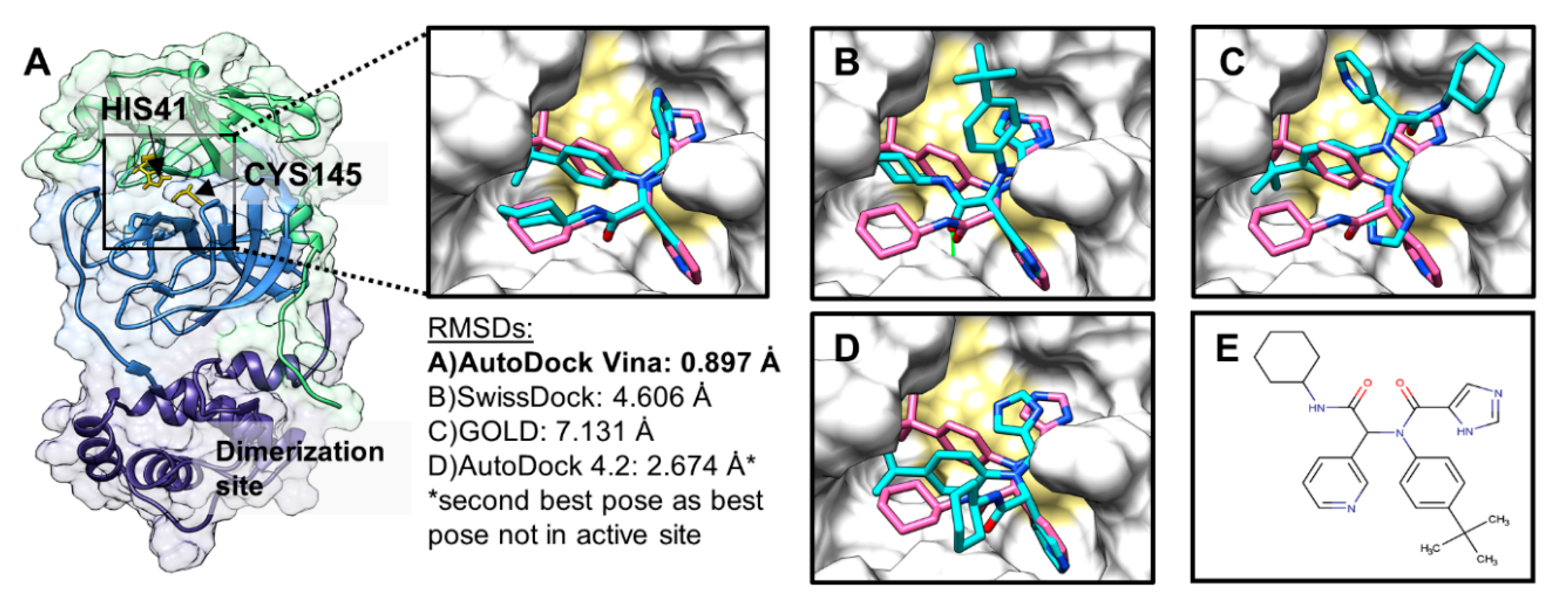
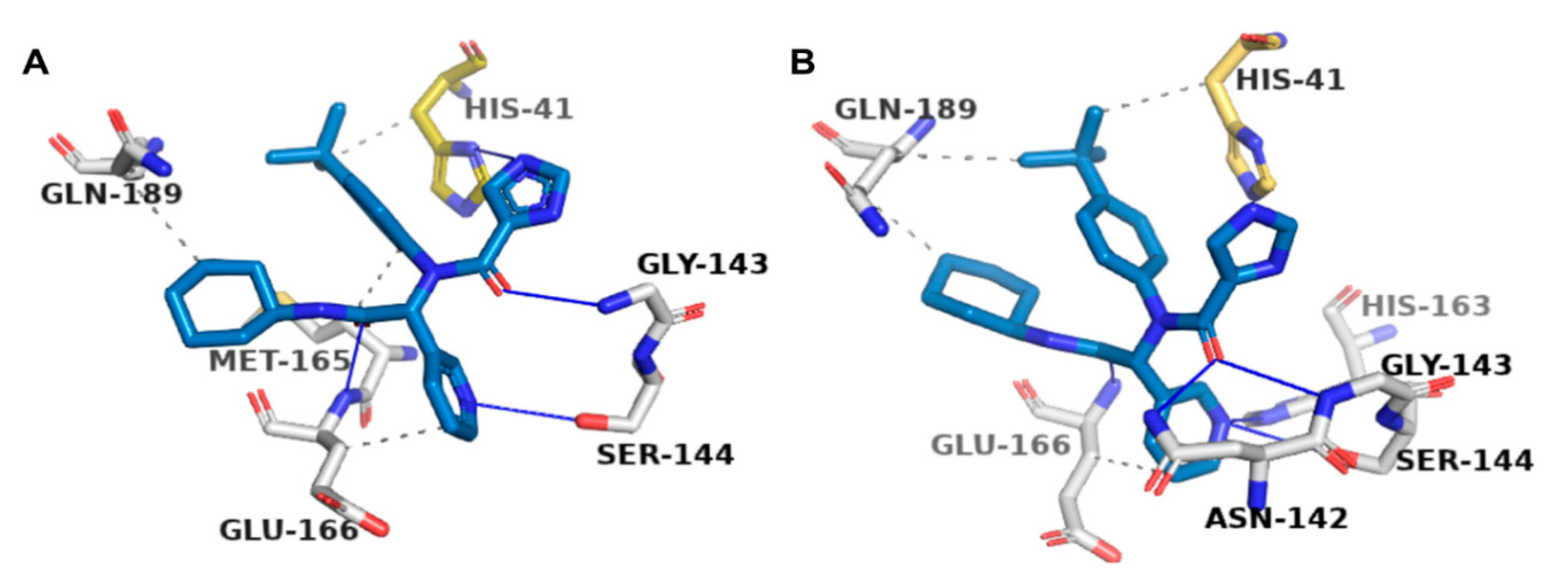
2.2. Docking-Protocol Validations
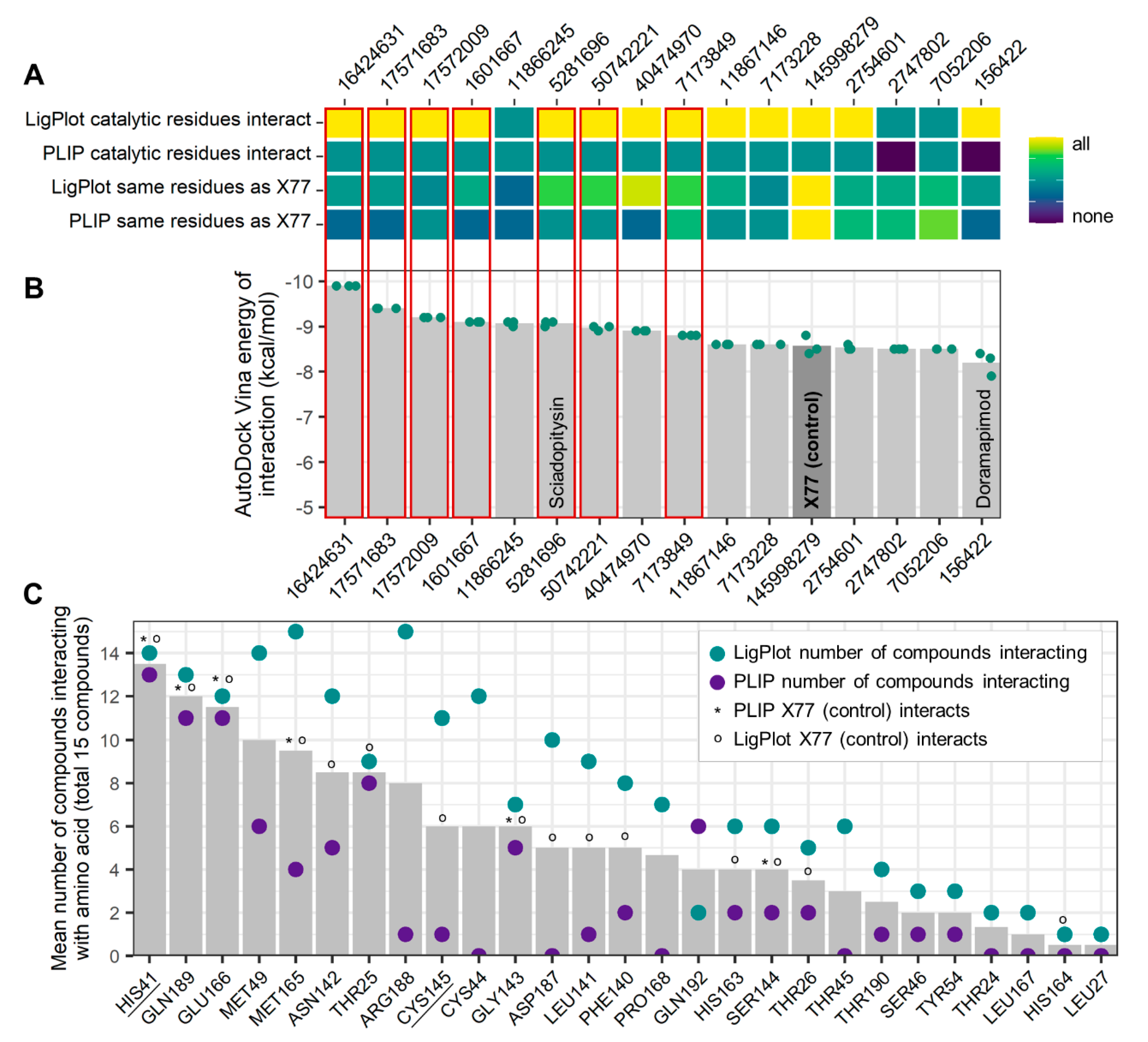
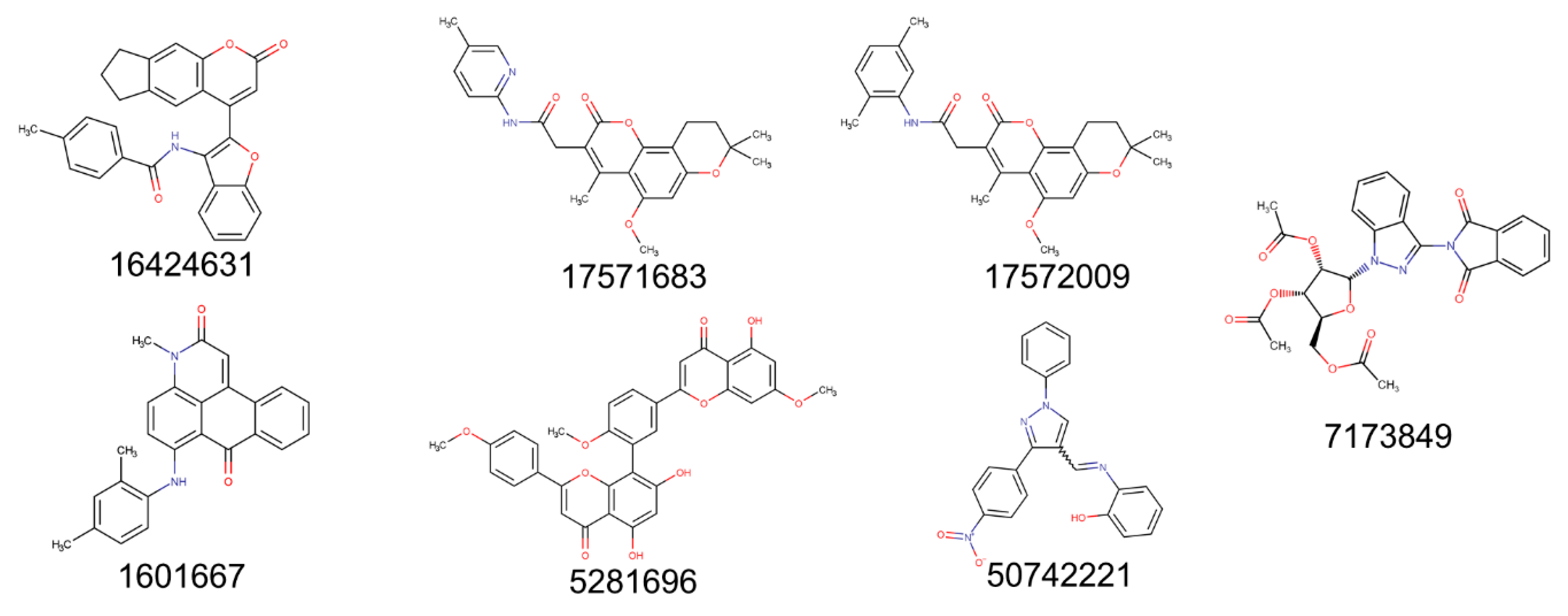
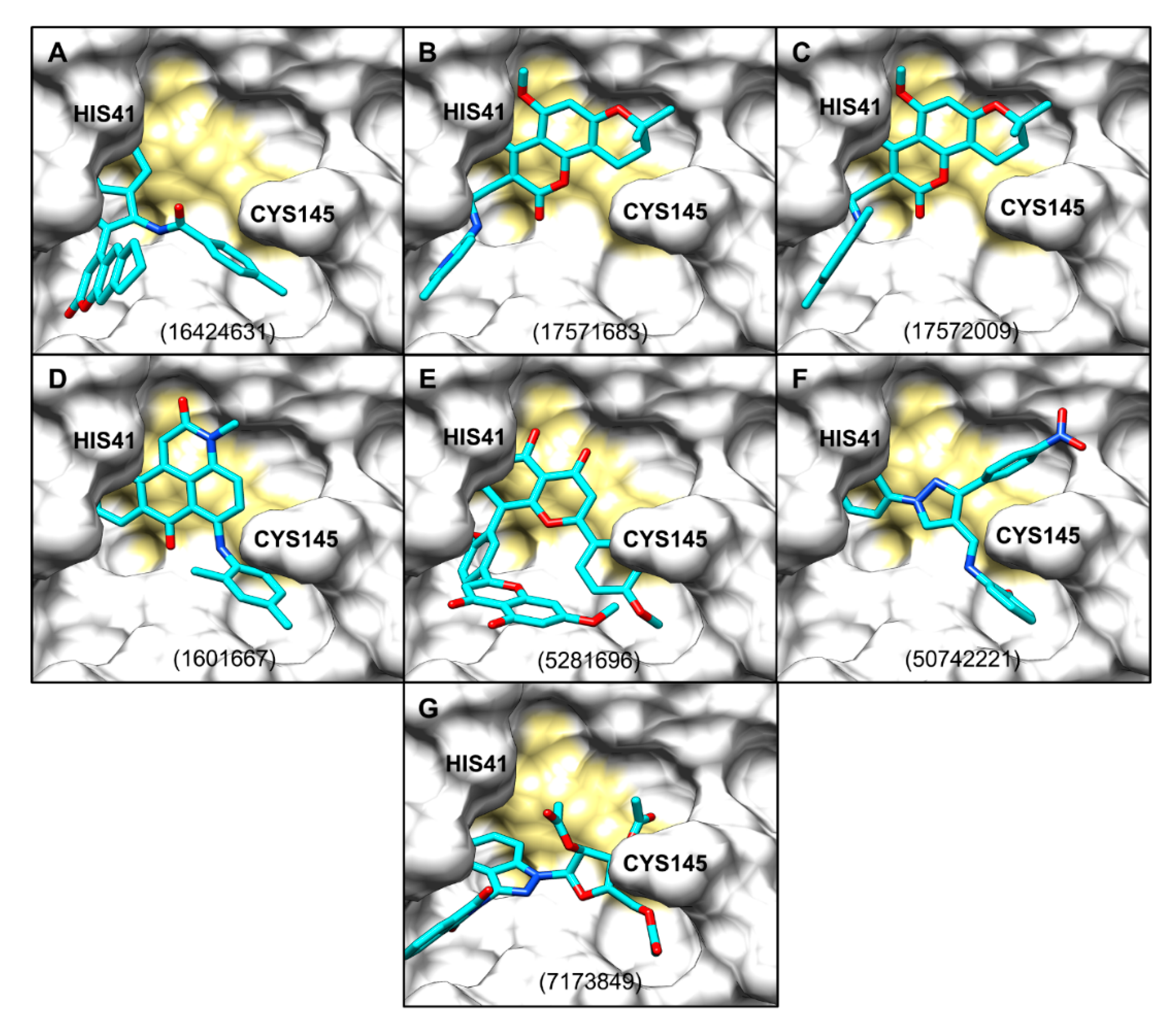
2.3. Evaluation of ZINCPharmer Hits Using Molecular Docking
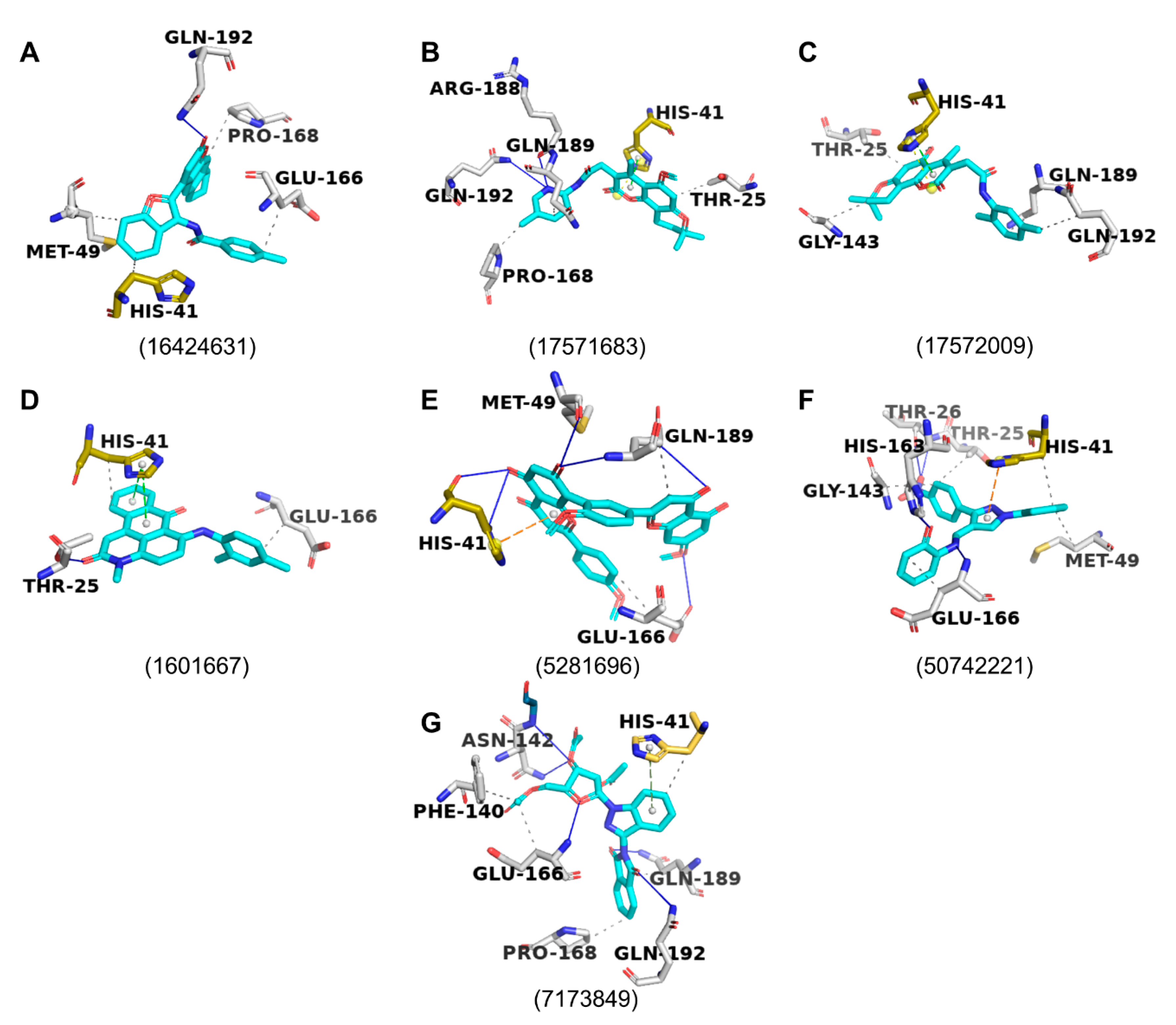
2.4. Protein–Ligand Interactions
2.5. Ligand Properties
3. Discussion
4. Materials and Methods
4.1. Virtual Screening with Fragment-Based Pharmacophores
4.2. Protein-Structure Preparation
4.3. Molecular Docking
4.4. Analysis of Protein–Ligand Interactions
4.5. Scaffold Novelty
4.6. Exploring Ligand Properties
4.7. Figure Creation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 30 September 2020).
- McKee, M.; Stuckler, D. If the world fails to protect the economy, COVID-19 will damage health not just now but also in the future. Nat. Med. 2020, 26, 640–642. [Google Scholar] [CrossRef] [Green Version]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, F. SARS-CoV-2 vaccines in development. Nat. Cell Biol. 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chem. Phys. Lett. 2020, 750, 137489. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Anand, K. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Ionescu, M.I. An Overview of the Crystallized Structures of the SARS-CoV-2. Protein J. 2020, 24, 1–19. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.-H. An Overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Zhang, S.; Zou, P.; Chen, J.; Kang, X.; Li, Z.; Liang, C.; Jin, C.; Xia, B. Without Its N-Finger, the Main Protease of Severe Acute Respiratory Syndrome Coronavirus Can Form a Novel Dimer through Its C-Terminal Domain. J. Virol. 2008, 82, 4227–4234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.P.; Elkhateeb, E.; Jo, H.; Natalie, N.; Lythgoe, E.; Tang, W.; Jamei, M.; Sharma, S.; Hodjegan, A.R. Pharmacokinetics under the COVID-19 storm! Drug Targets Potential Treat. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M.; Tomar, S.; et al. Discovery, Synthesis, and Structure-Based Optimization of a Series of N-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) Acetamides (ML188) as Potent Noncovalent Small Molecule Inhibitors of the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) 3CL Protease. J. Med. Chem. 2013, 56, 534–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Berry, M.; Fielding, B.; Gamieldien, J. Human coronavirus OC43 3CL protease and the potential of ML188 as a broad-spectrum lead compound: Homology modelling and molecular dynamic studies. BMC Struct. Biol. 2015, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mesecar, A.D. A taxonomically-driven approach to development of potent, broad-spectrum inhibitors of coronavirus main protease including SARS-CoV-2 (COVID-19). 2020. Unpublished work. [Google Scholar]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Jones, G.H.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking 1 Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Fang, Y.; Moreno, J.; Ramanujam, J.; Jarrell, M.; Brylinski, M. Assessing the similarity of ligand binding conformations with the Contact Mode Score. Comput. Biol. Chem. 2016, 64, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.; Ball, J.K.; Tsoleridis, T. Coronavirus (CoV) proteins (version 2020.5) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. CITE 2020, 2020. [Google Scholar] [CrossRef]
- PostEra COVID Moonshot. Available online: https://covid.postera.ai/covid (accessed on 13 September 2020).
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Ryu, Y.B.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, J.Y.; Kim, D.; Nguyen, T.T.; Park, S.J.; Chang, J.S.; Park, K.H.; et al. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg. Med. Chem. 2010, 18, 7940–7947. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Lee, A.A.; London, N.; Von Delft, F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 2020, 12, 581. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganisms 2020, 8, 1250. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Ke, Z.; Liu, C.; Wang, Z.; Liu, D.; Zhang, L.; Wang, J.; He, W.; Xu, Z.; Li, Y.; et al. Systemic in Silico Screening in Drug Discovery for Coronavirus Disease (COVID-19) with an Online Interactive Web Server. J. Chem. Inf. Model. 2020, 28. [Google Scholar] [CrossRef]
- Carlson, E.E. Natural Products as Chemical Probes. ACS Chem. Biol. 2010, 5, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Rana, S.; Sharma, S.; Ghosh, K. Virtual Screening of Naturally Occuring Antiviral Molecules for SARS-CoV-2 Mitigation Using Docking Tool on Multiple Molecular Targets. ChemRxiv 2020. Available online: https://chemrxiv.org/articles/preprint/Virtual_Screening_of_Naturally_Occurring_Antiviral_Molecules_for_SARS-CoV-2_Mitigation_Using_Docking_Tool_on_Multiple_Molecular_Targets/12403940/1 (accessed on 20 November 2020).
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nat. Cell Biol. 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Sharma, P.; Vijayan, V.; Pant, P.; Sharma, M.; Vikram, N.; Kaur, P.; Singh, T.P.; Sharma, S. Identification of potential drug candidates to combat COVID-19: A structural study using the main protease (mpro) of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Doiphode, S.; Vyas, R.; Swamy, K.V. Molecular docking and simulation studies on SARS-CoV-2 Mpro reveals Mitoxantrone, Leucovorin, Birinapant, and Dynasore as potent drugs against COVID-19. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Al-Khafaji, K.; Al-Duhaidahawi, D.L.; Tok, T.T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef]
- Mahanta, S.; Chowdhury, P.; Gogoi, N.; Goswami, N.; Borah, D.; Kumar, R.; Chetia, D.; Borah, P.; Buragohain, A.K.; Gogoi, B. Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: An in silico based approach. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Anwar, S.; Alajmi, M.F.; Hussain, A.; Rehman, T.; Islam, A.; Hassan, I. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: Possible implication in COVID-19 therapy. Biosci. Rep. 2020, 40, 6. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent Advances in Scaffold Hopping. J. Med. Chem. 2016, 60, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Grisoni, F.; Merk, D.; Byrne, R.; Schneider, G. Scaffold-Hopping from Synthetic Drugs by Holistic Molecular Representation. Sci. Rep. 2018, 8, 16469. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Koes, D.R.; Pabon, N.A.; Deng, X.; Phillips, M.A.; Camacho, C.J. A Teach-Discover-Treat Application of ZincPharmer: An Online Interactive Pharmacophore Modeling and Virtual Screening Tool. PLoS ONE 2015, 10, e0134697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kerdawy, A.M.; Osman, A.A.; Zaater, M. Receptor-based pharmacophore modeling, virtual screening, and molecular docking studies for the discovery of novel GSK-3β inhibitors. J. Mol. Model. 2019, 25, 171. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Jain, P.; Dikshit, S.N. Ligand-based pharmacophore detection, screening of potential gliptins and docking studies to get effective antidiabetic agents. Comb. Chem. High Throughput Screen. 2012, 15, 849–876. [Google Scholar] [CrossRef] [PubMed]
- Katarkar, A.; Haldar, P.K.; Chaudhuri, K. De novo design based pharmacophore query generation and virtual screening for the discovery of Hsp-47 inhibitors. Biochem. Biophys. Res. Commun. 2015, 456, 707–713. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Chen, P.; Ke, Y.; Lu, Y.; Du, Y.; Li, J.; Yan, H.; Zhao, H.; Zhou, Y.; Yang, Y. DLIGAND2: An improved knowledge-based energy function for protein–ligand interactions using the distance-scaled, finite, ideal-gas reference state. J. Cheminform. 2019, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Theerawatanasirikul, S.; Kuo, C.J.; Phetcharat, N.; Lekcharoensuk, P. In silico and in vitro analysis of small molecules and natural compounds targeting the 3CL protease of feline infectious peritonitis virus. Antivir. Res. 2020, 174, 104697. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kim, H.; Choi, J. In Silico Molecular Docking and In Vivo Validation with Caenorhabditis elegans to Discover Molecular Initiating Events in Adverse Outcome Pathway Framework: Case Study on Endocrine-Disrupting Chemicals with Estrogen and Androgen Receptors. Int. J. Mol. Sci. 2019, 20, 1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuccioloni, M.; Bonfili, L.; Cecarini, V.; Cocchioni, F.; Petrelli, D.; Crotti, E.; Zanchi, R.; Eleuteri, A.M.; Angeletti, M. Structure/activity virtual screening and in vitro testing of small molecule inhibitors of 8-hydroxy-5-deazaflavin:NADPH oxidoreductase from gut methanogenic bacteria. Sci. Rep. 2020, 10, 13150. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC-a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Callejo, G.; Pattison, L.A.; Greenhalgh, J.C.; Chakrabarti, S.; Andreopoulou, E.; Hockley, J.R.; Smith, E.S.J.; Rahman, T. In silico screening of GMQ-like compounds reveals guanabenz and sephin1 as new allosteric modulators of acid-sensing ion channel 3. Biochem. Pharmacol. 2020, 174, 113834. [Google Scholar] [CrossRef]
- Greenhalgh, J.C.; Chandran, A.; Harper, M.T.; Ladds, G.; Rahman, T. Proposed model of the Dictyostelium cAMP receptors bound to cAMP. J. Mol. Graph. Model. 2020, 100, 107662. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- ChemAxon. Marvin, 20.16; ChemAxon Ltd.: Budapest, Hungary, 2020. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PubChem CID (also Known as) | Druglikeness | Structural Alerts | Pharmacokinetics 4 | Water Solubility 5 |
|---|---|---|---|---|
| 16424631 | Meets Lipinski’s rules, except MLOGP 1 > 4.15 | PAINS 2: 0 alerts; Brenk 3: 1 alert-coumarin | High gastrointestinal (GI) absorption | Poorly soluble; Ilogp 6: 3.65 |
| 17571683 | Meets Lipinski’s rules | PAINS: 0 alerts; Brenk: 1 alert-coumarin | High GI absorption | Moderately soluble; iLOGP: 3.83 |
| 17572009 | Meets Lipinski’s rules | PAINS: 0 alerts; Brenk: 1 alert-coumarin | High GI absorption | Moderately soluble; iLOGP: 4.40 |
| 1601667 | Meets Lipinski’s rules | PAINS: 0 alerts; Brenk: 2 alerts-2 polycyclic aromatic hydrocarbons | High GI absorption | Moderately soluble; iLOGP: 3.66 |
| 5281696 (sciadopitysin) | Meets Lipinski’s rules except MW > 500 | PAINS: 0 alerts; Brenk: 0 alerts | Low GI absorption | Poorly soluble; iLOGP: 4.65 |
| 50742221 | Meets Lipinski’s rules | PAINS: 1 alert-imine phenol; Brenk: 3 alerts-imine, nitro group, oxygen–nitrogen single bond | High GI absorption | Poorly soluble; iLOGP: 3.15 |
| 7173849 | Meets Lipinski’s rules except MW > 500 and hydrogen bond acceptors > 10 | PAINS: 0 alerts; Brenk: 2 alerts-more than 2 esters, phthalimide | Low GI absorption | Moderately soluble; iLOGP: 2.85 |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Augustin, T.L.; Hajbabaie, R.; Harper, M.T.; Rahman, T. Novel Small-Molecule Scaffolds as Candidates against the SARS Coronavirus 2 Main Protease: A Fragment-Guided in Silico Approach. Molecules 2020, 25, 5501. https://doi.org/10.3390/molecules25235501
Augustin TL, Hajbabaie R, Harper MT, Rahman T. Novel Small-Molecule Scaffolds as Candidates against the SARS Coronavirus 2 Main Protease: A Fragment-Guided in Silico Approach. Molecules. 2020; 25(23):5501. https://doi.org/10.3390/molecules25235501
Chicago/Turabian StyleAugustin, Teresa L., Roxanna Hajbabaie, Matthew T. Harper, and Taufiq Rahman. 2020. "Novel Small-Molecule Scaffolds as Candidates against the SARS Coronavirus 2 Main Protease: A Fragment-Guided in Silico Approach" Molecules 25, no. 23: 5501. https://doi.org/10.3390/molecules25235501