
Molecular Modeling, Synthesis and Biological Evaluation of N-Phenyl-4-Hydroxy-6-Methyl-2-Quinolone-3-CarboxAmides as Anticancer Agents

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation of the Synthesized Compounds
2.3. Computational Studies
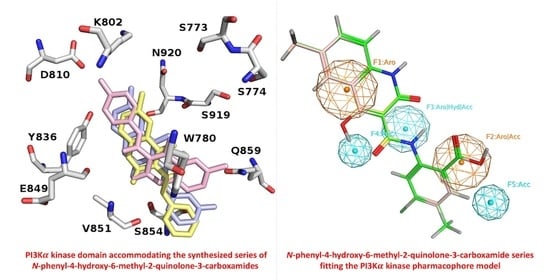
2.3.1. Molecular Docking
2.3.2. Descriptor Analysis
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis of Target Compounds
3.2.1. Ethyl 4-hydroxy-6-methyl-2-quinolone-3-carboxylate (5)
3.2.2. N-Phenyl-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (7)
3.2.3. N-(Pyridin-3-yl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (8)
3.2.4. N-Benzyl-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (9)
3.2.5. N-(Anilino)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (10)
3.2.6. N-(4-hydroxyphenyl) 4-hydroxy-6-methyl-2-quinolone-3-carboxamide (11)
3.2.7. N-(4-methoxyphenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (12)
3.2.8. N-(p-tolyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (13)
3.2.9. N-(2-fluorophenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (14)
3.2.10. N-(3-fluorophenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (15)
3.2.11. N-(4-fluorophenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (16)
3.2.12. N-(2-(trifluoromethyl)phenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (17)
3.2.13. N-(3-(trifluoromethyl)phenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (18)
3.2.14. N-(4-(trifluoromethyl)phenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (19)
3.2.15. N-(2-benzoic acid)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (20)
3.2.16. N-(3-benzoic acid)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (21)
3.2.17. N-(4-benzoic acid)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (22)
3.2.18. N-(2-methyl benzoate)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (23)
3.2.19. N-(4-chlorobenzoic acid)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (24)
3.2.20. N-(5-methylbenzoic acid)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (25)
3.2.21. N-(4-mercaptophenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (26)
3.2.22. N-(4-(methylthio) phenyl)-4-hydroxy-6-methyl-2-quinolone-3-carboxamide (27)
3.3. Biology
3.3.1. Culture Conditions
3.3.2. MTT Assay
3.3.3. Statistical Analysis
3.3.4. Quantitative Real-Time PCR
RNA Extraction
Complementary DNA (cDNA) Synthesis
Real-Time PCR
3.4. Computational Methods
3.4.1. Preparation of PI3Kα Structure
3.4.2. Preparation of Ligand Structures
3.4.3. Induced-Fit Docking (IFD)
3.4.4. Molecular Descriptors
3.4.5. Principal Component Analysis (PCA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vanhaesebroeck, B.; Waterfield, M.D. Signaling by distinct classes of phosphoinositide 3-kinases. Exp. Cell Res. 1999, 253, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Gabelli, S.B.; Amzel, L.M. The structure of a human p110 alpha/p85 alpha complex elucidates the effects of oncogenic PI3K alpha mutations. Science 2007, 318, 1744–1748. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.H.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, D.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; DeLong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110 alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Release 3; National Cancer Institute. Available online: http://cactus.nci.nih.gov/download/nci (accessed on 15 June 2012).
- Sabbah, D.A.; Simms, N.A.; Brattain, M.G.; Vennerstrom, J.L.; Zhong, H. Biological evaluation and docking studies of recently identified inhibitors of phosphoinositide-3-kinases. Bioorg. Med. Chem. Lett. 2012, 22, 876–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H. Docking studies on isoform-specific inhibition of phosphoinositide-3-kinases. J. Chem. Inf. Model. 2010, 50, 1887–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MOE. Version 2016; The Molecular Operating, Environment Chemical Computing Group, Inc.: Montreal, QC, Canada.
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H.A. Binding selectivity studies of phosphoinositide 3-kinases using free energy calculations. J. Chem. Inf. Model. 2012, 52, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Simms, N.A.; Wang, W.; Dong, Y.; Ezell, E.L.; Brattain, M.G.; Vennerstrom, J.L.; Zhong, H.A. N-Phenyl-4-hydroxy-2-quinolone-3-carboxamides as selective inhibitors of mutant H1047R phosphoinositide-3-kinase (PI3Kα). Bioorg. Med. Chem. 2012, 20, 7175–7183. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Hishmah, B.; Sweidan, K.; Bardaweel, S.; AlDamen, M.; Zhong, H.A.; Abu-Khalaf, R.; Hasan-Ibrahim, A.; Al-Qirim, T.; Abu-Sheikha, G. Structure-based design: Synthesis, X-ray crystallography, and biological evaluation of N-substituted-4-hydroxy-2-quinolone-3-carboxamides as potential cytotoxic agents. Anticancer Agents Med. Chem. 2018, 18, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Saada, M.; Khalaf, R.A.; Bardaweel, S.; Sweidan, K.; Al-Qirim, T.; Al-Zughier, A.; Halim, H.A.; Sheikha, G.A. Molecular modeling based approach, synthesis, and cytotoxic activity of novel benzoin derivatives targeting phosphoinostide 3-kinase (PI3Kα). Bioorg. Med. Chem. Lett. 2015, 25, 3120–3124. [Google Scholar] [CrossRef]
- Sweidan, K.; Sabbah, D.A.; Bardaweel, S.; Dush, K.A.; Sheikha, G.A.; Mubarak, M.S. Computer-aided design, synthesis, and biological evaluation of new indole-2-carboxamide derivatives as PI3Kα/EGFR inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2685–2690. [Google Scholar] [CrossRef]
- Sweidan, K.; Sabbah, D.A.; Bardaweel, S.; Abu-Sheikha, G.; Al-Qirim, T.; Salih, H.; El-Abadelah, M.M.; Mubarak, M.S.; Voelter, W. Facile synthesis, characterization, and cytotoxicity study of new 3-(indol-2-yl)bicyclotetrazatridecahexaens. Can. J. Chem. 2017, 95, 858–862. [Google Scholar] [CrossRef]
- Sweidan, K.; Zalloum, H.; Sabbah, D.A.; Idris, G.; Abudosh, K.; Mubarak, M.S. Synthesis, characterization, and anticancer evaluation of some new N1-(anthraquinon-2-yl) amidrazone derivatives. Can. J. Chem. 2018, 96, 1123–1128. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Al-Tarawneh, F.; Talib, W.H.; Sweidan, K.; Bardaweel, S.K.; Al-Shalabi, E.; Zhong, H.A.; Abu Sheikha, G.; Abu Khalaf, R.; Mubarak, M.S. Benzoin schiff bases: Design, synthesis, and biological evaluation as potential antitumor agents. Med. Chem. 2018, 14, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Ibrahim, A.H.; Talib, W.H.; Alqaisi, K.M.; Sweidan, K.; Bardaweel, S.K.; Sheikha, G.A.; Zhong, H.A.; Al-Shalabi, E.; Khalaf, R.A. Ligand-based drug design: Synthesis and biological evaluation of substituted benzoin derivatives as potential antitumor agents. Med. Chem. 2019, 15, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, X.; Hernandez, A.; Kim, S.; Evers, B.M. Inhibition of the phosphatidylinositol 3-kinase pathway contributes to HT29 and Caco-2 intestinal cell differentiation. Gastroenterology 2001, 120, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Sambuy, Y.; de Angelis, I.; Ranaldi, G.; Scarino, M.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Shao, J.; Townsend, C.M.; Evers, B.M. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut 2003, 52, 1472–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brattain, M.G.; Levine, A.E.; Chakrabarty, S.; Yeoman, L.C.; Willson, J.K.V.; Long, B. Heterogeneity of human colon carcinoma. Cancer Metastasis Rev. 1984, 3, 177–191. [Google Scholar] [CrossRef]
- Mandelker, D.; Gabelli, S.B.; Schmidt-Kittler, O.; Zhu, J.; Cheong, I.; Huang, C.-H.; Kinzler, K.W.; Vogelstein, B.; Amzel, L.M. A frequent kinase domain mutation that changes the interaction between PI3K alpha and the membrane. Proc. Natl. Acad. Sci. USA 2009, 106, 16996–17001. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger. Protein Preparation Wizard, Maestro, Macromodel, QPLD-dock, and Pymol; Schrödinger, LLC: Portland, OR, USA, 2016. [Google Scholar]
- Zhao, Y.; Zhang, X.; Chen, Y.; Lu, S.; Peng, Y.; Wang, X.; Guo, C.; Zhou, A.; Zhang, J.; Luo, Y.; et al. Crystal structures of PI3Kalpha complexed with PI103 and its derivatives: New directions for inhibitors design. ACS Med. Chem. Lett. 2013, 5, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Sweidan, K.; Sabbah, D.A.; Engelmann, J.; Halim, H.A.; Sheikha, G.A. Computational Docking Studies of Novel Heterocyclic Carboxamides as Potential PI3Kα Inhibitors. Lett. Drug Des. Discov. 2015, 12, 856–863. [Google Scholar] [CrossRef]
- alvaDesc. Available online: https://chm.kode-solutions.net/products_alvadesc.php (accessed on 30 September 2020).
- Monge, A.; Arrault, A.; Marot, C.; Morin-Allory, L. Managing, profiling and analyzing a library of 2.6 million compounds gathered from 32 chemical providers. Mol. Divers. 2006, 10, 389–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Chen, G.; Zheng, S.; Luo, X.; Shen, J.; Zhu, W.; Liu, H.; Gui, C.; Zhang, J.; Zheng, M.; Puah, C.M. Focused combinatorial library design based on structural diversity, druglikeness and binding affinity score. J. Comb. Chem. 2005, 7, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Luo, X.; Chen, G.; Zhu, W.; Shen, J.; Chen, K.; Jiang, H. A new rapid and effective chemistry space filter in recognizing a druglike database. J. Chem. Inf. Model. 2005, 45, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Kode. Available online: https://chm.kode-solutions.net/products_dragon.php (accessed on 10 September 2020).
- Bardaweel, S.K.; Abu-Dahab, R.; Almomani, N.F. An in vitro based investigation into the cytotoxic effects of D-amino acids. Acta Pharm. 2013, 63, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelsohn, L.D. ChemDraw 8 ultra, windows and macintosh versions. J. Chem. Inf. Model. 2004, 44, 2225–2226. [Google Scholar] [CrossRef]
- Hajjo, R.; Setola, V.; Roth, B.L.; Tropsha, A. Chemocentric informatics approach to drug discovery: Identification and experimental validation of selective estrogen receptor modulators as ligands of 5-hydroxytryptamine-6 receptors and as potential cognition enhancers. J. Med. Chem. 2012, 55, 5704–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | Ar | Compound | Ar |
|---|---|---|---|
| 7 |  | 18 |  |
| 8 |  | 19 |  |
| 9 |  | 20 |  |
| 10 |  | 21 |  |
| 11 |  | 22 |  |
| 12 |  | 23 |  |
| 13 |  | 24 |  |
| 14 |  | 25 |  |
| 15 |  | 26 |  |
| 16 |  | 27 |  |
| 17 |  | ||
| Compound’s Number | IC50 (µM) ± SD | Compound’s Number | IC50 (µM) ± SD | ||
|---|---|---|---|---|---|
| Caco-2 | HCT-116 | Caco-2 | HCT-116 | ||
| LY294002 | 7.4 | 6.5 | 17 | 2321 | 615 |
| 5 | 338.5 | 346.8 | 18 | 486.6 | 422.5 |
| 7 | 264 | 212.2 | 19 | 402.3 | 304.2 |
| 8 | 98 | 337 | 20 | 1559 | 789.4 |
| 9 | 356 | 383.7 | 21 | 664.5 | 16.04 |
| 10 | 275 | 390.4 | 22 | 458.2 | 184.6 |
| 11 | 204 | 92.61 | 23 | 261.4 | 46.38 |
| 12 | 260 | 207.4 | 24 | 298 | 241.1 |
| 13 | 687 | 178.6 | 25 | 302 | 140.0 |
| 14 | 354 | 441.2 | 26 | 282.5 | 405.7 |
| 15 | 212 | 282.2 | 27 | 289.2 | 462.7 |
| 16 | 13 | 240.2 | |||
| 2RD0 | 3HHM | |||
|---|---|---|---|---|
| Compound | Docking Score | Binding Residues | Docking Score | Binding Residues |
| 5 | −7.58 | K802, D933 | −7.77 | V851 |
| 7 | −8.04 | V851 | −8.91 | S774, D933, N920 |
| 8 | −7.84 | S774, D933 | −8.73 | S774, D933, S919 |
| 9 | −8.98 | E849, V851 | −8.71 | S774, D933 |
| 10 | −9.39 | W780, E849, V851, S854 | −8.72 | S774, D933, S919, N920 |
| 11 | −9.40 | V851 | −9.68 | S774, D933, S919, F1059 |
| 12 | −8.69 | V851, Q859 | −9.32 | Y836, V851 |
| 13 | −8.59 | V851 | −7.92 | S774, D933 |
| 14 | −8.37 | Y836, D933 | −8.09 | S774, S919, D933 |
| 15 | −8.79 | V851 | −8.99 | S774, S919, D933 |
| 16 | −9.20 | W780, V851 | −8.41 | Y836 |
| 17 | −8.44 | S854, Q859 | −8.39 | N920, D933 |
| 18 | −8.65 | W780, V851 | −8.33 | S774, D933, N920 |
| 19 | −8.42 | K802, D933 | −8.95 | S773, D933 |
| 20 | −8.89 | S774, K802, D933 | −9.98 | S774, A775, K776, D933, N920 |
| 21 | −9.15 | S774, K776, D805, D933 | −9.08 | S774, D915, H917, N920, D933 |
| 22 | −10.32 | K802, D810, V851 | −9.58 | S774, E849, D933 |
| 23 | −8.11 | S774, K802, D933 | −8.47 | S774, N920 |
| 24 | −8.39 | W780, S854, Q859 | −9.46 | S774, H917, N920 |
| 25 | −9.83 | V851, S854, D933 | −10.04 | S774, K776, N920, D933 |
| 26 | −8.59 | D933 | −8.52 | S773, N920 |
| 27 | −8.81 | V851 | −10.0 | D810, Y836, D933 |
| Target | Forward Primers (5′→3′) | Reverse Primers (5′→3′) |
|---|---|---|
| β-actin | ACGGGGTCACCCACACTGTGC | CTAGAAGCATTTGCGGTGGACGATG |
| BAD | CCTCAGGCCTATGCAAAAAG | AAACCCAAAACTTCCGATGG |
| PI3K | ACCCAGCAACAGAAAAATGG | GCGCTGTGAATTTAGCCTTC |
| AKT | AACCTGTGCTCCATGACCTC | CCCTTCTACAACCAGGACCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabbah, D.A.; Hasan, S.E.; Abu Khalaf, R.; Bardaweel, S.K.; Hajjo, R.; Alqaisi, K.M.; Sweidan, K.A.; Al-Zuheiri, A.M. Molecular Modeling, Synthesis and Biological Evaluation of N-Phenyl-4-Hydroxy-6-Methyl-2-Quinolone-3-CarboxAmides as Anticancer Agents. Molecules 2020, 25, 5348. https://doi.org/10.3390/molecules25225348
Sabbah DA, Hasan SE, Abu Khalaf R, Bardaweel SK, Hajjo R, Alqaisi KM, Sweidan KA, Al-Zuheiri AM. Molecular Modeling, Synthesis and Biological Evaluation of N-Phenyl-4-Hydroxy-6-Methyl-2-Quinolone-3-CarboxAmides as Anticancer Agents. Molecules. 2020; 25(22):5348. https://doi.org/10.3390/molecules25225348
Chicago/Turabian StyleSabbah, Dima A., Shaima’ E. Hasan, Reema Abu Khalaf, Sanaa K. Bardaweel, Rima Hajjo, Khalid M. Alqaisi, Kamal A. Sweidan, and Aya M. Al-Zuheiri. 2020. "Molecular Modeling, Synthesis and Biological Evaluation of N-Phenyl-4-Hydroxy-6-Methyl-2-Quinolone-3-CarboxAmides as Anticancer Agents" Molecules 25, no. 22: 5348. https://doi.org/10.3390/molecules25225348