3.1. Chemistry
3.1.1. General Information
All starting materials were commercially available and used without further purification unless otherwise stated. TLC analysis was performed using pre-coated glass plates. Column chromatography was performed using silica gel (300–400 mesh) or octadecylsilyl silica gel (75 μm) for separation and purification.
1H-NMR and
13C-NMR spectra were recorded on DRX-400 spectrometer (Bruker, Billerica, MA, USA) with CDCl
3, CD
3OD, acetone-
d6 or DMSO-
d6 as solvents. Resonances (
δ) are given in parts per million relatives to tetramethylsilane or a residual solvent peak (CDCl
3:
1H:
δ = 7.26 ppm,
13C:
δ = 77.00 ppm; CD
3OD:
1H:
δ = 3.31 ppm,
13C:
δ = 49.00 ppm; acetone-
d6:
1H:
δ = 2.05 ppm,
13C:
δ = 206.26 ppm, 29.84 ppm; DMSO-
d6:
1H:
δ = 2.50 ppm,
13C:
δ = 39.50 ppm). Data are reported as follows: chemical shift; multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet); coupling constants (Hz); and integration. HRMS were obtained on an Triple TOF
®® 5600
+ (AB Sciex, Foster City, CA, USA). More spectral information can be found in
Supplementary Materials.
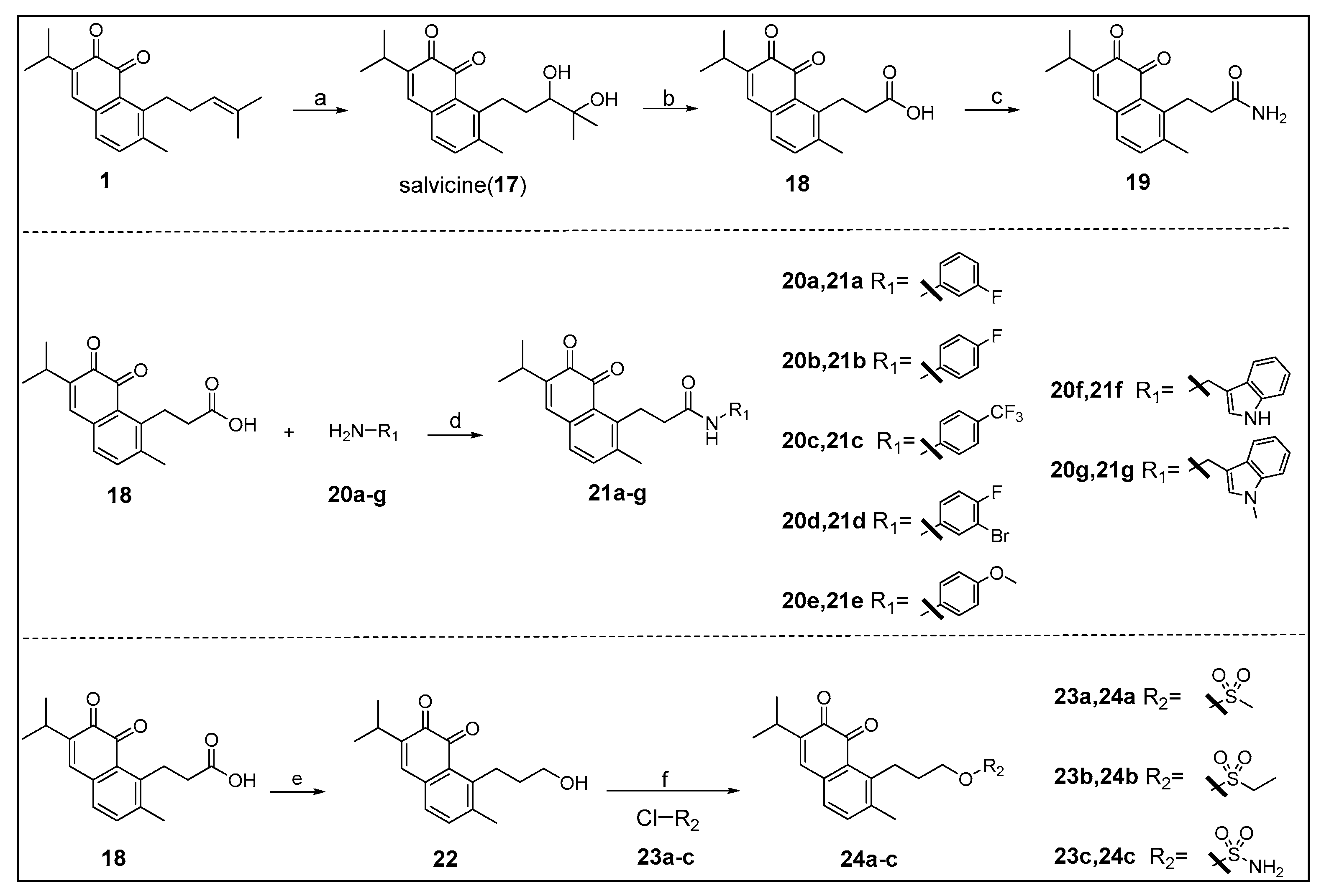
3.1.2. Synthesis of 8-(3,4-Dihydroxy-4-methylpentyl)-3-isopropyl-7-methylnaphthalene-1,2-dione (17)
To a stirred solution of m-chloroperbenzoic acid (1.45 g, 8.40 mmol) in dichloromethane (24 mL) held at 0 °C was added a solution of compound 1 (2 g, 6.75 mmol) dissolved in dichloromethane (20 mL) during a period of 30 min. The mixture was stirred overnight at room temperature, washed with saturated solution of NaHCO3 and dried over anhydrous MgSO4. The solvent was concentrated in vacuo and dissolved in tetrahydrofuran (35 mL) was added 6.7 mL of water. The solution was stirred and 4 mL of 8% perchloric acid was added. After stirring for 6 h under N2 at room temperature, saturated solution of NaCl was added and the mixture was extracted several times with ethyl acetate. The organic phase was washed with dilute sodium bicarbonate and dried over anhydrous MgSO4, evaporated under reduced pressure and purified by silica gel column chromatography using petroleum ether/ethyl acetate mixture (4:1 to 1:1) as eluent to afford 17 as an orange solid in 92% yield (2.05 g). 1H-NMR (400 MHz, CDCl3) δ 7.38 (d, J = 7.6 Hz, 1H, Ar-H), 7.10 (s, 1H, Ar-H), 7.09 (d, J = 7.6 Hz, 1H, Ar-H), 3.36–3.27 (m, 1H, CH of hydroxymethine), 3.15–3.05 (m, 1H, one of Ar-CH2 protons), 3.05–2.99 (m, 1H, CH of isopropyl group), 2.98–2.93 (m, 1H, one of Ar-CH2 protons), 2.39 (s, 3H, Ar-CH3), 1.90–1.78 (m, 1H, one of CH2CO protons), 1.75–1.64 (m, 1H, one of CH2CO protons), 1.33 (s, 3H, CH3), 1.28 (s, 3H, CH3), 1.18 (d, J = 6.9 Hz, 3H, CH3 of isopropyl group), 1.17 (d, J = 6.9 Hz, 3H, CH3 of isopropyl group). ESI-MS m/z: 353.2 [M + Na]+.
3.1.3. Synthesis of 3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanoic acid (18)
Compound 17 (505.2 mg, 1.53 mmol) was dissolved in dichloromethane (5 mL), pyridiniumchlorochromate (3.5 g, 16.24 mmol) was added and 1.5 mL of water was added dropwise. The mixture was stirred overnight at room temperature. The crude material was filtered through kieselguhr and purified by silica gel column chromatography using petroleum ether/ethyl acetate mixture (3:1 to 1:2) as eluent to afford 18 as an orange solid in 47% yield (204.9 mg). 1H-NMR (400 MHz, CDCl3) δ 7.38 (d, J = 7.6 Hz, 1H, Ar-H), 7.09 (s, 1H, Ar-H), 7.08 (d, J = 7.6 Hz, 1H, Ar-H), 3.35 (t, J = 7.7 Hz, 2H, Ar-CH2), 3.00 (septet, J = 6.9 Hz, 1H, CH), 2.61 (t, J = 7.7 Hz, 2H, CH2CO), 2.39 (s, 3H, Ar-CH3), 1.15 (d, J = 6.9 Hz, 6H, 2CH3). ESI-MS m/z: 287.0 [M + H]+.
3.1.4. Synthesis of 3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydro- naphthalen-1-yl)propanamide (19)
To a solution of compound 18 (100.0 mg, 0.35 mmol) in dichloromethane (5 mL) held at 0 °C was added thionylchloride (100 μL) and 1H-benzotriazole (42.9 mg, 0.36 mmol), and the mixture was stirred at reflux temperature for 2 h. The solvent was concentrated in vacuo and dissolved in dichloromethane (1 mL). Ammonium hydroxide solution was added at 0 °C and the mixture was stirred at room temperature for 1 h. Water was added and the mixture was extracted several times with DCM. The organic phase was dried over anhydrous MgSO4, evaporated under reduced pressure and purified by octadecylsilyl silica gel column chromatography using methanol/water (2:3) as eluent to afford 19 as an orange solid in 40% yield (40.0 mg). 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 7.1 Hz, 1H, Ar-H), 7.12 (d, J = 7.1 Hz, 2H, Ar-H), 6.39 (s, 1H, one of CONH2 protons), 5.45 (s, 1H, one of CONH2 protons), 3.33 (m, 2H, Ar-CH2), 3.03 (septet, J = 5.9 Hz, 1H, CH), 2.43 (s, 3H, Ar-CH3), 2.39 (m, 2H, CH2CO), 1.18 (d, J = 5.9 Hz, 6H, 2CH3). 13C NMR (100 MHz, CDCl3) δ 182.67, 181.09, 174.87, 146.50, 144.98, 140.32, 140.26, 137.47, 135.31, 128.87, 128.14, 35.27, 27.88, 26.99, 21.48, 19.74. ESI-MS m/z: 308.0 [M + Na]+. HRESI-MS calcd. for C17H20NO3+ ([M + H]+) 286.1438, found 286.1440.
3.1.5. General Procedure A: Synthesis of Compounds 21a–21g and 31a–31d
Compound 18 or 30a–30d was dissolved in 1 mL of dry N,N-dimethylformamide, 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) and N-ethyl-diisopropylamine (DIPEA) were added. After stirring uniformly, amine 20a–20g or 26a was added. The mixture was stirred overnight at room temperature. dichloromethane was added and the mixture was washed with saturated solution of NaHCO3,the organic phase was purified by column chromatography to afford 21a–21g or 31a–31d.
N-(3-Fluorophenyl)-3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamide (21a): Compound 18 (40.1 mg, 0.14 mmol), HATU (64.6 mg, 0.17 mmol), DIPEA (45.3 μL, 0.26 mmol) and 20a (16.3 μL, 0.17 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (3:1) as eluent to afford 21a as an orange solid in 72% yield (38.4 mg). 1H-NMR (400 MHz, CDCl3) δ 8.42 (s, 1H, CONH), 7.64 (d, J = 11.3 Hz, 1H, Ar-H of phenyl group), 7.44 (d, J = 7.5 Hz, 1H, Ar-H of naphthoquinone), 7.31–7.22 (m, 2H, Ar-H of phenyl group), 7.15–7.10 (m, 2H, Ar-H of naphthoquinone), 6.88–6.71 (m, 1H, Ar-H of phenyl group), 3.42–3.32 (m, 2H, Ar-CH2), 3.03 (septet, J = 6.9 Hz, 1H, CH), 2.55–2.48 (m, 2H, CH2CO), 2.45 (s, 3H, Ar-CH3), 1.19 (d, J = 6.9 Hz, 6H, 2CH3).13C-NMR (100 MHz, CDCl3) δ 182.76, 181.04, 170.93, 162.99 (d, J = 244.3 Hz), 146.31, 144.96, 140.38, 140.32, 139.74 (d, J = 10.9 Hz), 137.64, 135.40, 129.94 (d, J = 9.3 Hz), 129.02, 128.11, 115.11, 110.71 (d, J = 21.3 Hz), 107.29 (d, J = 26.4 Hz), 36.96, 27.83, 27.06, 21.45, 19.75. ESI-MS m/z: 380.0 [M + H]+. HRESI-MS calcd. for C23H23FNO3+ ([M + H]+) 380.1656, found 380.1659.
N-(4-Fluorophenyl)-3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamide (21b): Compound 18 (28.6 mg, 0.10 mmol), HATU (49.4 mg, 0.13 mmol), DIPEA (34.8 μL, 0.20 mmol) and 20b (12.3 μL, 0.13 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (3:1) as eluent to afford 21b as an orange solid in 53% yield (20.1 mg). 1H-NMR (400 MHz, CDCl3) δ8.25 (s, 1H, CONH), 7.63–7.56 (m, 2H, Ar-H of phenyl group), 7.43 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 7.12 (s, 1H, Ar-H of naphthoquinone), 7.12 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 7.06–6.99 (m, 2H, Ar-H of phenyl group), 3.43–3.34 (m, 2H, Ar-CH2), 3.03 (septet, J = 6.9 Hz, 1H, CH), 2.55–2.45 (m, 2H, CH2CO), 2.45 (s, 3H, Ar-CH3), 1.19 (d, J = 6.9 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.82, 181.08, 170.70, 159.30 (d, J = 243.3 Hz), 146.37, 145.02, 140.36, 140.24, 137.59, 135.41, 134.17, 128.95, 128.15, 121.75 (d, J = 7.8 Hz), 115.51 (d, J = 22.4 Hz), 36.88, 27.97, 27.08, 21.44, 19.71. ESI-MS m/z: 380.0 [M + H]+. HRESI-MS calcd. for C23H23FNO3+ ([M + H]+) 380.1656, found 380.1660.
3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)-N-(4-(trifluoromethyl)phenyl)propanamide (21c): Compound 18 (28.6 mg, 0.10 mmol), HATU (49.4 mg, 0.13 mmol), DIPEA (34.8 μL, 0.20 mmol) and 20c (20.9 mg, 0.13 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (3:1) as eluent to afford 21c as an orange solid in 85% yield (36.5 mg). 1H-NMR (400 MHz, CDCl3) δ 8.58 (s, 1H, CONH), 7.79 (d, J = 8.3 Hz, 2H, Ar-H of phenyl group), 7.59 (d, J = 8.3 Hz, 2H, Ar-H of phenyl group), 7.44 (d, J = 7.6 Hz, 1H, Ar-H of naphthoquinone), 7.14 (d, J = 7.6 Hz, 1H, Ar-H of naphthoquinone), 7.13 (s, 1H, Ar-H of naphthoquinone), 3.43–3.34 (m, 2H, Ar-CH2), 3.03 (septet, J = 6.8 Hz, 1H, CH), 2.57–2.48 (m, 2H, CH2CO), 2.45 (s, 3H, Ar-CH3), 1.19 (d, J = 6.8 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.82, 181.05, 171.16, 146.26, 144.97, 141.36, 140.36, 137.70, 135.43, 129.07, 128.10, 126.15 (q, J = 3.4 Hz), 125.69 (q, J = 32.1 Hz), 122.84, 119.42, 36.95, 27.76, 27.07, 21.44, 19.74. ESI-MS m/z: 430.0 [M + H]+. HRESI-MS calcd. for C24H23F3NO3+ ([M + H]+) 430.1625, found 430.1625.
N-(3-Bromo-4-fluorophenyl)-3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamide (21d): Compound 18 (40.1 mg, 0.14 mmol), HATU (64.6 mg, 0.17 mmol), DIPEA (45.3 μL, 0.26 mmol) and 20d (32.1 mg, 0.17 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (4:1) as eluent to afford 21d as an orange solid in 53% yield (34.0 mg).1H-NMR (400 MHz, CDCl3) δ 8.33 (s, 1H, CONH), 8.02–7.95 (m, 1H, Ar-H of phenyl group), 7.56–7.49 (m, 1H, Ar-H of phenyl group), 7.45 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 7.14 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 7.13 (s, 1H, Ar-H of naphthoquinone), 7.12–7.05 (m, 1H, Ar-H of phenyl group), 3.41–3.33 (m, 2H, Ar-CH2), 3.04 (septet, J = 6.9 Hz, 1H, CH), 2.52–2.47 (m, 2H, CH2CO), 2.45 (s, 3H, Ar-CH3), 1.19 (d, J = 6.9 Hz, 6H, 2CH3). 13C-NMR (100 MHz, DMSO-d6) δ 182.11, 180.85, 171.27, 154.46 (d, J = 242.1 Hz), 145.79, 144.13, 140.46, 140.44, 137.16, 135.15, 134.20 (d, J = 12.4 Hz), 129.33, 128.96, 123.68, 120.44 (d, J = 6.7 Hz), 117.14 (d, J = 22.8 Hz), 107.95 (d, J = 21.6 Hz), 35.47, 26.96, 25.74, 21.79, 19.84. ESI-MS m/z: 458.8 [M + H]+. HRESI-MS calcd. for C23H22BrFNO3+ ([M + H]+) 458.0762, found 458.0765.
3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)-N-(4-methoxyphenyl)propanamide (21e): Compound 18 (28.6 mg, 0.10 mmol), HATU (49.4 mg, 0.13 mmol), DIPEA (34.8 μL, 0.20 mmol) and 20e (16.0 mg, 0.13 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (3:1) as eluent to afford 21e as an orange solid in 26% yield (10.3 mg).1H-NMR (400 MHz, CDCl3) δ 8.06 (s, 1H, CONH), 7.52 (d, J = 8.8 Hz, 2H, Ar-H of phenyl group), 7.43 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 7.13 (s, 1H, Ar-H of naphthoquinone), 7.12 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 6.88 (d, J = 8.8 Hz, 2H, Ar-H of phenyl group), 3.80 (s, 3H, OCH3), 3.44–3.35 (m, 2H, Ar-CH2), 3.04 (septet, J = 6.9 Hz, 1H, CH), 2.52–2.47 (m, 2H, CH2CO), 2.45 (s, 3H, Ar-CH3), 1.19 (d, J = 6.9 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.75, 181.11, 170.51, 156.34, 146.52, 145.00, 140.41, 140.25, 137.54, 135.36, 131.26, 128.90, 128.16, 121.85, 114.11, 55.49, 36.90, 28.11, 27.05, 21.47, 19.78. ESI-MS m/z: 392.0 [M + H]+. HRESI-MS calcd. for C24H26NO4+ ([M + H]+) 392.1856, found 392.1861.
N-((1H-indol-3-yl)methyl)-3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamide (21f): Compound 18 (28.6 mg, 0.10 mmol), HATU (49.4 mg, 0.13 mmol), DIPEA (34.8 μL, 0.20 mmol) and 20f (19.0 mg, 0.13 mmol) were used in general procedure A. The crude product was purified by flash chromatography on silica gel then octadecylsilyl silica gel column chromatography using methanol/water (3:1) as eluent to afford 21f as an orange solid in 52% yield (21.5 mg). 1H-NMR (400 MHz, CDCl3) δ 8.36 (s, 1H, CONH), 7.65 (d, J = 7.8 Hz, 1H, Ar-H of indole), 7.37 (d, J = 7.8 Hz, 1H, Ar-H of naphthoquinone), 7.23–7.01 (m, 5H, Ar-H, 3H of indole and 2H of naphthoquinone), 6.45–6.36 (m, 1H, Ar-H of indole), 4.67 (d, J = 5.3 Hz, 2H, CH2 by indole), 3.34–3.26 (m, 2H, CH2 bynaphthoquinone), 2.99 (septet, J = 6.9Hz, 1H, CH), 2.42 (s, 3H, Ar-CH3), 2.41–2.31 (m, 2H, CH2CO), 1.15 (d, J = 6.9 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.39, 181.08, 172.29, 146.66, 144.79, 140.50, 140.37, 137.35, 136.41, 135.12, 128.76, 128.13, 126.59, 123.27, 122.30, 119.76, 118.99, 112.94, 111.31, 35.67, 35.19, 27.79, 26.95, 21.47, 19.84. ESI-MS m/z: 415.2 [M + H]+. HRESI-MS calcd. for C26H27N2O3+ ([M + H]+) 415.2016, found 415.2026.
3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)-N-((1-methyl-1H-indol-3-yl)methyl)-propanamide (21g): Compound 18 (28.6 mg, 0.10 mmol), HATU (49.4 mg, 0.13 mmol), DIPEA (34.8 μL, 0.20 mmol) and 20g (20.8 mg, 0.13 mmol) were used in general procedure A. The crude product was purified by flash chromatography on silica gel then octadecylsilyl silica gel column chromatography using methanol/water (4:1) as eluent to afford 21g as an orange solid in 44% yield (19.0 mg). 1H-NMR (400 MHz, CDCl3) δ 7.65 (d, J = 7.9 Hz, 1H, Ar-H of indole), 7.37 (d, J = 7.6 Hz, 1H, Ar-H of naphthoquinone), 7.32–7.04 (m, 6H, CONH and 3Ar-H of indole and 2Ar-H of naphthoquinone), 6.40–6.29 (m, 1H, Ar-H of indole), 4.65 (d, J = 5.2 Hz, 2H, CH2 by indole), 3.77 (s, 3H, NCH3), 3.34–3.26 (m, 2H, CH2 by naphthoquinone), 3.00 (septet, J = 6.9 Hz, 1H, CH), 2.43 (s, 3H, Ar-CH3), 2.39–2.31 (m, 2H, CH2CO), 1.15 (d, J = 6.9 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3)δ 182.42, 181.08, 172.17, 146.65, 144.81, 140.48, 140.28, 137.32, 137.14, 135.13, 128.73, 128.16, 127.93, 127.06, 121.89, 119.33, 119.12, 111.44, 109.32, 35.73, 35.03, 32.73, 27.82, 26.95, 21.47, 19.84. ESI-MS m/z: 429.2 [M + H]+. HRESI-MS calcd. for C27H29N2O3+ ([M + H]+) 429.2173, found 429.2175.
tert-Butyl-(3-((2-((tert-butoxycarbonyl)amino)phenyl)amino)-3-oxopropyl)carbamate (31a): Compound 30a (39.7 mg, 0.21 mmol), HATU (95.1 mg, 0.25 mmol), DIPEA (73.2 μL, 0.42 mmol) and 26a (52.0 mg, 0.25 mmol) were used in general procedure A. The crude product was purified by silica gel column chromatography using petroleum ether/ethyl acetate mixture (3:1 to 1:1) as eluent to afford 31a as a colorless oil in 84% yield (66.5 mg). 1H-NMR (400 MHz, CDCl3) δ8.59 (s, 1H,Ph-NHCO), 7.54–7.44 (m, 1H, Ar-H), 7.38 (d, J = 7.0 Hz, 1H, Ar-H), 7.25 (s, 1H, Ph-NHCO), 7.21–7.06 (m, 2H, Ar-H), 5.32 (s, 1H, CONH), 3.58–3.29 (m, 2H, NH-CH2), 2.64–2.36 (m, 2H, CH2CO), 1.51 (s, 9H, 3CH3), 1.43 (s, 9H, 3CH3).ESI-MS m/z: 402.2 [M + Na]+.
tert-Butyl-(2-(4-((tert-butoxycarbonyl)amino)butanamido)phenyl)carbamate (31b): Compound 30b (99.5 mg, 0.49 mmol), HATU (224.2 mg, 0.59 mmol), DIPEA (170.7 μL, 0.98 mmol) and 26a (122.8 mg, 0.59 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (3:2) as eluent to afford 31b as a colorless oil in 41% yield (79.2 mg). 1H-NMR (400 MHz, CDCl3) δ8.98 (s, 1H,Ph-NHCO), 7.57 (d, J = 7.0 Hz, 1H, Ar-H), 7.47 (s, 1H,Ph-NHCO), 7.35 (d, J = 7.0 Hz, 1H, Ar-H), 7.15 (t, J = 7.0 Hz, 1H, Ar-H), 7.07 (t, J = 7.0 Hz, 1H, Ar-H), 5.03 (s, 1H, CONH), 3.21–3.08 (m, 2H, NH-CH2), 2.37–2.26 (m, 2H, CH2CO), 1.87–1.75 (m, 2H, CH2), 1.50 (s, 9H, 3CH3), 1.45 (s, 9H, 3CH3). m/z: 416.2 [M + Na]+.
tert-Butyl(2-(5-((tert-butoxycarbonyl)amino)pentanamido)phenyl)carbamate (31c): Compound 30c (99.9 mg, 0.46 mmol), HATU (209.1 mg, 0.55 mmol), DIPEA (160.2 μL, 0.92 mmol) and 26a (114.5 mg, 0.55 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (3:2) as eluent to afford 31c as a colorless oil in 53% yield (99.0 mg).1H-NMR (400 MHz, CDCl3) δ 8.74 (s, 1H,Ph-NHCO), 7.50 (d, J = 7.5 Hz, 1H, Ar-H), 7.44 (s, 1H,Ph-NHCO), 7.30 (d, J = 7.5 Hz, 1H, Ar-H), 7.14 (t, J = 7.5 Hz, 1H, Ar-H), 7.06 (t, J = 7.5 Hz, 1H, Ar-H), 4.86 (s, 1H, CONH), 3.10–2.99 (m, 2H, NH-CH2), 2.36–2.21 (m, 2H, CH2CO), 1.70–1.56 (m, 2H, CH2), 1.50 (s, 9H, 3CH3), 1.47–1.33 (m, 11H, CH2 and 3CH3). ESI-MS m/z: 430.2 [M + Na]+.
tert-Butyl(2-(6-((tert-butoxycarbonyl)amino)hexanamido)phenyl)carbamate (31d): Compound 30d (99.4 mg, 0.43 mmol), HATU (178.7 mg, 0.47 mmol), DIPEA (149.8 μL, 0.86 mmol) and 26a (97.8 mg, 0.47 mmol) were used in general procedure A. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (3:2) as eluent to afford 31d as a colorless oil in 50% yield (91.2 mg). 1H-NMR (400 MHz, CDCl3) δ 8.61 (s, 1H,Ph-NHCO), 7.45 (d, J = 7.3 Hz, 1H, Ar-H), 7.36 (d, J = 7.3 Hz, 1H, Ar-H), 7.35 (s, 1H,Ph-NHCO), 7.14 (t, J = 7.3 Hz, 1H, Ar-H), 7.08 (t, J = 7.3 Hz, 1H, Ar-H), 4.76 (s, 1H, CONH), 3.17–2.92 (m, 2H, NH-CH2), 2.46–2.16 (m, 2H, CH2CO), 1.75–1.57 (m, 2H, CH2), 1.51 (s, 9H, 3CH3), 1.47–1.37 (m, 11H, CH2 and 3CH3), 1.36–1.24 (m, 2H, CH2). ESI-MS m/z: 444.2 [M + Na]+.
3.1.6. Synthesis of 8-(3-Hydroxypropyl)-3-isopropyl-7-methylnaphthalene-1,2-dione (22)
Compound 18 (80.1 mg, 0.28 mmol) was dissolved in 4 mL of dry tetrahydrofuran, and a solution of 1M lithium aluminum hydride in tetrahydrofuran (560 μL, 0.56 mmol) was added portionwise over a period of 30 min. After refluxing for 1 h, 24 μL of water was added dropwise, followed by an equal volume of 15% sodium hydroxide and 3 times the amount of water. The crude material was filtered through kieselguhr and purified by silica gel column chromatography using petroleum ether/ethyl acetate mixture (3:1 to 1:1) as eluent to afford 22 as an orange oil (40.7mg, 53% yield). 1H-NMR (400 MHz, CDCl3) δ 7.36 (d, J = 7.6 Hz, 1H, Ar-H), 7.08 (s, 1H, Ar-H), 7.05 (d, J = 7.6 Hz, 1H, Ar-H), 3.79 (t, J = 6.1 Hz, 2H, OH-CH2), 3.14–3.07 (m, 2H, Ar-CH2), 3.01 (septet, J = 6.9 Hz, 1H, CH), 2.38 (s, 3H, Ar-CH3), 1.80–1.71 (m, 2H, CH2), 1.15 (d, J = 6.9 Hz, 6H, 2CH3). ESI-MS m/z: 295.2 [M + Na]+.
3.1.7. General Procedure B: Synthesis of Compounds 24a–24c
To a solution of compound 22 (32.7 mg, 0.12 mmol) in dry dichloromethane (5 mL) held at 0 °C was added dry triethylamine (0.1 mL, 0.74 mmol) and 23a–23c. The mixture was stirred 2 h at room temperature. Saturated solution of NaHCO3 was added and the mixture was extracted several times with dichloromethane. The organic phase was dried over anhydrous MgSO4, evaporated under reduced pressure and purified by octadecylsilyl silica gel column chromatography using methanol/water (7:3) as eluent to afford compounds 24a–24c.
3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propyl methanesulfonate (24a): Compound 23a (53.8 mg, 0.47 mmol) were used in general procedure B to afford 24a as an orange solid in 38% yield (16.2 mg). 1H-NMR (400 MHz, CDCl3) δ 7.40 (d, J = 7.6 Hz, 1H, Ar-H), 7.11 (s, 1H, Ar-H), 7.10 (d, J = 7.6 Hz, 1H, Ar-H), 4.43 (t, J = 6.1 Hz, 2H, CH2OSO2), 3.18–3.10 (m, 2H, Ar-CH2), 3.07 (s, 3H, SO2CH3), 3.01 (septet, J = 6.8 Hz, 1H, CH), 2.39 (s, 3H, Ar-CH3), 2.02–1.91 (m, 2H, CH2), 1.17 (d, J = 6.8 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.45, 181.18, 146.44, 144.92, 140.22, 140.13, 137.16, 135.16, 128.67, 128.35, 70.46, 37.38, 28.28, 26.95, 26.48, 21.48, 19.78. ESI-MS m/z: 373.0 [M + Na]+. HRESI-MS calcd. for C18H22NaO5S+ ([M + Na]+) 373.1080, found 373.1086.
3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propyl ethanesulfonate (24b): Compound 23b (46.3 mg, 0.36 mmol) were used in general procedure B to afford 24b as an orange solid in 18% yield (8.0 mg). 1H-NMR (400 MHz, CDCl3) δ 7.41 (d, J = 7.6 Hz, 1H, Ar-H), 7.11 (s, 1H, Ar-H), 7.10 (d, J = 7.6 Hz, 1H, Ar-H), 4.43 (t, J = 6.2 Hz, 2H, CH2OSO2), 3.19 (q, J = 7.4 Hz, 2H, SO2CH2), 3.16–3.11 (m, 2H, Ar-CH2), 3.03 (septet, J = 6.9 Hz, 1H, CH), 2.40 (s, 3H, Ar-CH3), 2.02–1.92 (m, 2H, CH2), 1.46 (t, J = 7.4 Hz, 3H, CH3), 1.17 (d, J = 6.9 Hz, 6H, 2CH3 of isopropyl group). 13C-NMR (100 MHz, CDCl3) δ182.41, 181.18, 146.49, 144.88, 140.24, 140.13, 137.14, 135.13, 128.66, 128.33, 70.13, 44.88, 28.41, 26.94, 26.51, 21.48, 19.77, 8.24. ESI-MS m/z: 387.0 [M + Na]+. HRESI-MS calcd. for C19H24NaO5S+ ([M + Na]+) 387.1237, found 387.1241.
3-(6-Isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propyl sulfamate (24c): Compound 23c (32.7 mg, 0.12 mmol) were used in general procedure B to afford 24c as an orange solid in 40% yield (17.0 mg). 1H-NMR (400 MHz, CD3OD) δ 7.48 (d, J = 7.9 Hz, 1H, Ar-H), 7.31 (s, 1H, Ar-H), 7.24 (d, J = 7.9 Hz, 1H, Ar-H), 4.28 (t, J = 6.3 Hz, 2H, CH2OSO2), 3.24–3.11 (m, 2H, Ar-CH2), 2.96 (septet, J = 6.9 Hz, 1H, CH), 2.41 (s, 3H, Ar-CH3), 2.00–1.84 (m, 2H, CH2), 1.18 (d, J = 6.9 Hz, 6H, 2CH3).13C-NMR (100 MHz, acetone-d6) δ 182.41, 181.01, 146.03, 144.42, 140.02, 139.90, 136.82, 135.32, 128.73, 69.78, 28.04, 26.87, 26.10, 20.82, 18.85. ESI-MS m/z: 374.0 [M + Na]+. HRESI-MS calcd. for C17H21NNaO5S+ ([M + Na]+) 374.1033, found 374.1038.
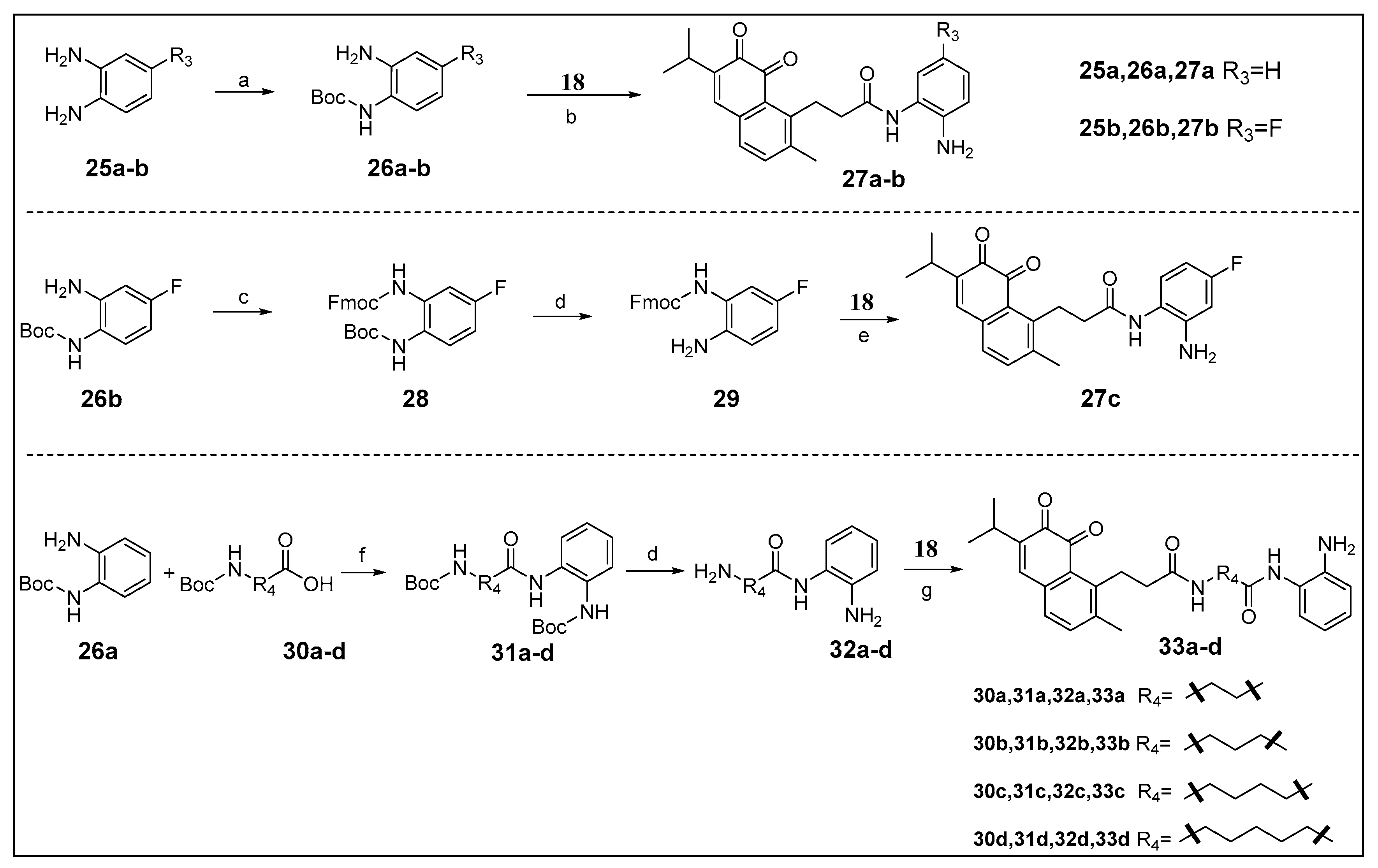
3.1.8. General Procedure C: Synthesis of Compounds 26a–26b
The reactant was dissolved in 6 mL of dichloromethane, and 0.1 mL of triethylamine was added. Di-tert-butyl dicarbonate was added at 0 °C and the mixture was stirred overnight at room temperature. The mixture was washed with water to give the crude product, which was used in the next reaction directly.
tert-Butyl (2-aminophenyl)carbamate (26a): Compound 25a (198.8 mg, 1.84 mmol) was used in general procedure C to afford 26a as a light yellow oil in 76% yield (292.5 mg). ESI-MS m/z: 231.0 [M + Na]+.
tert-Butyl (2-amino-4-fluorophenyl)carbamate (26b): Compound 25b (99.6 mg, 0.79 mmol) was used in general procedure C to afford 26b as a light yellow oil in 66% yield (117.6 mg). ESI-MS m/z: 249.0 [M + Na]+.
3.1.9. General Procedure D: Synthesis of Compounds 27a–27b
Compound 18 was dissolved in 1 mL of dry N,N-dimethylformamide, HATU and DIPEA were added. After stirring uniformly, amine 26a–26b was added. The mixture was stirred overnight at room temperature. Dichloromethane was added and the mixture was washed with saturated solution of NaHCO3, the organic phase was purified by octadecylsilyl silica gel column chromatography using methanol/water as eluent. The solvent was concentrated in vacuo and dissolved in 2.5 mL dichloromethane, 1 mL of trifluoroacetic acid was added at 0 °C and the mixture was stirred 1 h at room temperature. Saturated solution of NaHCO3 was added and the mixture was extracted several times with dichloromethane. The organic phase was dried over anhydrous MgSO4, evaporated under reduced pressure to afford compound 27a–27b.
N-(2-Aminophenyl)-3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamide (27a): Compound 18 (40.1 mg, 0.14 mmol), HATU (64.6 mg, 0.17 mmol), DIPEA (45.3 μL, 0.26 mmol) and 26a (33.3 mg, 0.16 mmol) were used in general procedure D. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (4:1) as eluent. Finally affording 27a as an orange solid in 27% yield (14.1 mg). 1H-NMR (400 MHz, CDCl3) δ 8.04 (s, 1H, CONH), 7.43 (d, J = 7.6 Hz, 1H, Ar-H of phenyl group), 7.31 (d, J = 7.9 Hz, 1H, Ar-H of naphthoquinone), 7.12 (s, 1H, Ar-H of naphthoquinone), 7.12 (d, J = 7.9 Hz, 1H, Ar-H of naphthoquinone), 7.05 (t, J = 7.6 Hz, 1H, Ar-H of phenyl group), 6.84–6.75 (m, 2H, Ar-H of phenyl group), 3.49–3.37 (m, 2H, Ar-CH2), 3.02 (septet, J = 6.8 Hz, 1H, CH), 2.61–2.49 (m, 2H, CH2CO), 2.44 (s, 3H, Ar-CH3), 1.18 (d, J = 6.8 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.75, 181.04, 171.18, 146.44, 144.99, 140.59, 140.39, 140.20, 137.51, 135.37, 128.91, 128.22, 126.95, 125.27, 124.31, 119.30, 117.83, 36.27, 27.98, 27.05, 21.47, 19.82. ESI-MS m/z: 377.2 [M + H]+. HRESI-MS calcd. for C23H25N2O3+ ([M + H]+) 377.1860, found 377.1865.
N-(2-Amino-5-fluorophenyl)-3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamide (27b): Compound 18 (85.8 mg, 0.30 mmol), HATU (136.8 mg, 0.36 mmol), DIPEA (104.5 μL, 0.60 mmol) and 26b (81.4 mg, 0.36 mmol) were used in general procedure D. The crude product was purified by octadecylsilyl silica gel column chromatography using methanol/water (4:1) as eluent. Finally affording 27b as an orange solid in 10% yield (12.0 mg). 1H-NMR (400 MHz, CDCl3) δ 8.18 (s, 1H, CONH), 7.48–7.38 (m, 2H, Ar-H, 1H of phenyl group and 1H of naphthoquinone), 7.13 (d, J = 6.6 Hz, 1H, Ar-H of naphthoquinone), 7.12 (s, 1H, Ar-H of naphthoquinone), 6.79–6.72 (m, 2H, Ar-H of phenyl group), 3.46–3.34 (m, 2H, Ar-CH2), 3.02 (septet, J = 6.7 Hz, 1H, CH), 2.60–2.50 (m, 2H, CH2CO), 2.45 (s, 3H, Ar-CH3), 1.18 (d, J = 6.7 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.84, 180.88, 171.03, 162.50 (d, J = 266.9 Hz), 146.36, 145.00, 140.34, 140.22, 137.64, 135.43, 134.77, 129.01, 128.16, 126.39 (d, J = 10.1 Hz), 118.83 (d, J = 8.6 Hz), 112.45 (d, J = 22.4 Hz), 110.83 (d, J = 26.0 Hz), 36.57, 28.02, 27.05, 21.48, 19.84. ESI-MS m/z: 395.2 [M + H]+. HRESI-MS calcd. for C23H24FN2O3+ ([M + H]+) 395.1765, found 395.1775.
3.1.10. Synthesis of (9H-fluoren-9-yl) methyl tert-butyl (4-fluoro-1,2-phenylene)dicarbamate (28)
Compound 26b (169.6 mg, 0.75 mmol) was dissolved in 3 mL of dichloromethane, and 6 mL of saturated solution of NaHCO3 was added. The mixture was stirred and (9H-fluoren-9-ylmethoxy)carbonyl chloride (271.6 mg, 1.05 mmol) was added. 3 h later, the mixture was washed by water and was extracted several times with dichloromethane. The organic phase was dried over anhydrous MgSO4, and purified by octadecylsilyl silica gel column chromatography using methanol/water (1:1) as eluent to afford 28 as a brown oil in 36% yield (121.1 mg). ESI-MS m/z: 471.0 [M + Na]+.
3.1.11. General Procedure E: Synthesis of Compounds 29 and 32a–32d
Compound 28 or 31a–31d was dissolved in 2.5 mL dichloromethane, 1 mL of trifluoroacetic acid was added at 0 °C and the mixture was stirred 1 h at room temperature. Saturated solution of NaHCO3 was added and the mixture was extracted several times with dichloromethane. The organic phase was dried over anhydrous MgSO4, evaporated under reduced pressure to afford compound 29 and 32a–32d, which was used in the next reaction directly.
3.1.12. Synthesis of N-(2-amino-4-fluorophenyl)-3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamide (27c)
Compound 18 (60.1 mg, 0.21 mmol) was dissolved in 1 mL of dry N,N-dimethylformamide, HATU (95.1 mg, 0.25 mmol) and DIPEA (73.2 μL, 0.42 mmol) were added. After stirring uniformly, 29 (88.5 mg, 0.25 mmol) was added. The mixture was stirred overnight at room temperature. Dichloromethane was added and the mixture was washed with saturated solution of NaHCO3, the organic phase was purified by silica gel column chromatography using petroleum ether/ethyl acetate mixture (4:1 to 1:1) as eluent. The solvent was concentrated in vacuo and dissolved in 2 mL dichloromethane, 2 mL diethylamine was added and the mixture was stirred 1 h at room temperature. The mixture was evaporated under reduced pressure and purified by octadecylsilyl silica gel column chromatography using methanol/water (3:2) as eluent to afford 27c as an orange solid in 4% Yield (4.3 mg). 1H-NMR (400 MHz, CDCl3) δ 8.14 (s, 1H, CONH), 7.44 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 7.26–7.22 (m, 1H, Ar-H of phenyl group), 7.13 (s, 1H, Ar-H of naphthoquinone), 7.13 (d, J = 7.7 Hz, 1H, Ar-H of naphthoquinone), 6.63–6.51 (m, 2H, Ar-H of phenyl group), 3.46–3.38 (m, 2H, Ar-CH2), 3.02 (septet, J = 6.8 Hz, 1H, CH), 2.60–2.52 (m, 2H, CH2CO), 2.45 (s, 3H, Ar-CH3), 1.18 (d, J = 6.8 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ182.83, 181.02, 171.48, 160.55 (d, J = 264.4 Hz), 146.38, 145.04, 142.93 (d, J = 12.2 Hz), 140.34, 140.23, 137.63, 135.44, 129.00, 128.16, 127.27 (d, J = 10.1 Hz), 119.65, 105.50 (d, J = 22.6 Hz), 103.91 (d, J = 25.6 Hz), 36.18, 28.14, 27.06, 21.48, 19.82. ESI-MS m/z: 395.0 [M + H]+. HRESI-MS calcd. for C23H24FN2O3+ ([M + H]+) 395.1765, found 395.1769.
3.1.13. General Procedure F: Synthesis of Compounds 33a–33d
Compound 18 was dissolved in 1 mL of dry N,N-dimethylformamide, 3-(3-dimethylaminopropyl)-1-ethylcarbodiimide hydrochloride (EDCI), 4-dimethylaminopyridine (DMAP) and DIPEA were added. After stirring uniformly, amine 32a–32d was added. The mixture was stirred overnight at room temperature. Dichloromethane was added and the mixture was washed with saturated solution of NaHCO3,the organic phase was purified by column chromatography to afford 33a–33d.
N-(2-aminophenyl)-3-(3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamido)-propanamide (33a): Compound 18 (28.6 mg, 0.10 mmol), EDCI (23.0 mg, 0.12 mmol), DMAP (1 mg), DIPEA (104.5 μL, 0.60 mmol) and 32a were used in general procedure F. The crude product was purified by flash chromatography on silica gel then octadecylsilyl silica gel column chromatography using methanol/water (3:2) as eluent to afford 33a as an orange solid in 12% yield (5.7 mg).1H-NMR (400 MHz, CDCl3) δ 8.21 (s, 1H, Ph-NHCO), 7.38 (d, J = 7.4 Hz, 1H, Ar-H of phenyl group), 7.24 (d, J = 7.8 Hz, 1H, Ar-H of naphthoquinone), 7.09 (s, 1H, Ar-H of naphthoquinone), 7.08 (d, J = 7.8 Hz, 1H, Ar-H of naphthoquinone), 7.02 (t, J = 7.4 Hz, 1H, Ar-H of phenyl group), 6.90–6.83 (m, 1H, Ar-H of phenyl group), 6.81–6.70 (m, 2H, CONH and Ar-H of phenyl group), 3.71–3.59 (m, 2H, NH-CH2), 3.36–3.23 (m, 2H, Ar-CH2), 3.00 (septet, J = 6.2 Hz, 1H, CH), 2.71–2.61 (m, 2H, CH2CO), 2.38 (s, 3H, Ar-CH3), 2.37–2.29 (m, 2H, CH2CO), 1.17 (d, J = 6.2 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.55, 181.20, 172.97, 170.37, 146.53, 144.77, 140.88, 140.55, 140.32, 137.32, 135.11, 128.82, 128.34, 127.10, 125.58, 124.07, 119.12, 117.82, 36.66, 35.79, 35.40, 27.11, 26.96, 21.48, 19.76. ESI-MS m/z: 448.2 [M + H]+. HRESI-MS calcd. for C26H30N3O4+ ([M + H]+) 448.2231, found 448.2238.
N-(2-Aminophenyl)-4-(3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamido)-butanamide (33b): Compound 18 (51.5 mg, 0.18 mmol), EDCI (38.3 mg, 0.20 mmol), DMAP (1.7 mg), DIPEA (188.1 μL, 1.08 mmol) and 32b were used in general procedure F. The crude product was purified by flash chromatography on silica gel then octadecylsilyl silica gel column chromatography using methanol/water (7:3) as eluent to afford 33b as an orange solid in 11% yield (10.2 mg). 1H-NMR (400 MHz, CDCl3) δ 8.65 (s, 1H, Ph-NHCO), 7.42 (d, J = 7.5 Hz, 1H, Ar-H of phenyl group), 7.37 (d, J = 7.8 Hz, 1H, Ar-H of naphthoquinone), 7.12 (s, 1H, Ar-H of naphthoquinone), 7.11 (d, J = 7.8 Hz, 1H, Ar-H of naphthoquinone), 7.02 (t, J = 7.5 Hz, 1H, Ar-H of phenyl group), 6.82–6.70 (m, 3H, CONH and 2Ar-H of phenyl group), 3.53–3.44 (m, 2H, NH-CH2), 3.33–3.24 (m, 2H, Ar-CH2), 3.02 (septet, J = 6.9 Hz, 1H, CH), 2.51–2.45 (m, 2H, CH2CO), 2.41 (s, 3H, Ar-CH3), 2.39–2.32 (m, 2H, CH2CO), 2.03–1.93 (m, 2H, CH2), 1.17 (d, J = 6.9 Hz, 6H, 2CH3). 13C-NMR (100 MHz, CDCl3) δ 182.75, 181.06, 173.78, 171.45, 146.58, 144.93, 140.58, 140.39, 140.33, 137.56, 135.34, 128.94, 128.10, 126.62, 125.16, 124.54, 118.96, 117.64, 38.40, 35.83, 33.96, 28.07, 27.04, 26.79, 21.46, 19.75. ESI-MS m/z: 462.2 [M + H]+. HRESI-MS calcd. for C27H32N3O4+ ([M + H]+) 462.2387, found 462.2398.
N-(2-Aminophenyl)-5-(3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamido)-pentanamide (33c): Compound 18 (57.2 mg, 0.20 mmol), EDCI (46.0 mg, 0.24 mmol), DMAP (1.9 mg), DIPEA (209.0 μL, 1.20 mmol) and 32c were used in general procedure F. The crude product was purified by flash chromatography on silica gel then octadecylsilyl silica gel column chromatography using methanol/water (7:3) as eluent to afford 33c as an orange solid in 9% yield (10.6 mg).1H-NMR (400 MHz, CDCl3) δ 7.83 (s, 1H, Ph-NHCO), 7.40 (d, J = 7.4 Hz, 1H, Ar-H of naphthoquinone), 7.11 (s, 1H, Ar-H of naphthoquinone), 7.09 (d, J = 7.4 Hz, 1H, Ar-H of naphthoquinone), 7.03 (t, J = 7.3 Hz, 1H, Ar-H of phenyl group), 6.83–6.53 (m, 3H, CONH and 2Ar-H of phenyl group), 3.43–3.36 (m, 2H, NH-CH2), 3.32–3.23 (m, 2H, Ar-CH2), 3.01 (septet, J = 6.8 Hz, 1H, CH), 2.56–2.47 (m, 2H, CH2CO), 2.38 (s, 3H, Ar-CH3), 2.36–2.29 (m, 2H, CH2CO), 1.89–1.79 (m, 2H, CH2), 1.74–1.63 (m, 2H, CH2), 1.17 (d, J = 6.8 Hz, 6H, 2CH3). 13C-NMR (100 MHz, DMSO-d6) δ 182.01, 180.86, 171.76, 171.48, 146.30, 144.02, 142.30, 140.53, 140.48, 137.06, 135.08, 129.19, 128.83, 126.12, 125.68, 124.02, 116.60, 116.33, 38.77, 35.89, 34.66, 29.28, 26.94, 26.16, 23.27, 21.79, 19.91. ESI-MS m/z: 476.2 [M + H]+. HRESI-MS calcd. for C28H34N3O4+ ([M + H]+) 476.2544, found 476.2548.
N-(2-Aminophenyl)-6-(3-(6-isopropyl-2-methyl-7,8-dioxo-7,8-dihydronaphthalen-1-yl)propanamido) hexanamide (33d): Compound 18 (57.2 mg, 0.20 mmol), EDCI (46.0 mg, 0.24 mmol), DMAP (1.9 mg), DIPEA (209.0 μL, 1.20 mmol) and 32d were used in general procedure F. The crude product was purified by flash chromatography on silica gel then octadecylsilyl silica gel column chromatography using methanol/water (7:3) as eluent to afford 33d as an orange solid in 25% yield (25.7 mg).1H-NMR (400 MHz, CDCl3) δ 7.97 (s, 1H, Ph-NHCO), 7.39 (d, J = 7.5 Hz, 1H, Ar-H of phenyl group), 7.19 (d, J = 7.6 Hz, 1H, Ar-H of naphthoquinone), 7.11 (s, 1H, Ar-H of naphthoquinone), 7.09 (d, J = 7.6 Hz, 1H, Ar-H of naphthoquinone), 7.00 (t, J = 7.5 Hz, 1H, Ar-H of phenyl group), 6.80–6.57 (m, 3H, CONH and 2Ar-H of phenyl group). 3.37–3.30 (m, 2H, NH-CH2), 3.30–3.21 (m, 2H, Ar-CH2), 3.00 (septet, J = 6.8 Hz, 1H, CH), 2.49–2.43 (m, 2H, CH2CO), 2.39 (s, 3H, Ar-CH3), 2.34–2.27 (m, 2H, CH2CO), 1.85–1.74 (m, 2H, CH2), 1.67–1.57 (m, 2H, CH2), 1.56–1.46 (m, 2H, CH2), 1.17 (d, J = 6.8 Hz, 6H, 2CH3). 13C- NMR (100 MHz, CDCl3) δ 182.59, 181.24, 172.62, 171.96, 146.84, 144.80, 140.90, 140.61, 140.49, 137.42, 135.17, 128.85, 128.12, 126.90, 125.29, 124.51, 119.21, 118.03, 39.06, 36.88, 35.79, 29.06, 28.08, 26.99, 26.22, 25.28, 21.47, 19.72. ESI-MS m/z: 490.2 [M + H]+. HRESI-MS calcd. for C29H36N3O4+ ([M + H]+) 490.2700, found 490.2706.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













