
[99mTc]Tc-DB1 Mimics with Different-Length PEG Spacers: Preclinical Comparison in GRPR-Positive Models

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Radiolabeling and Quality Control
2.2. In Vitro Assays in PC-3 Cells
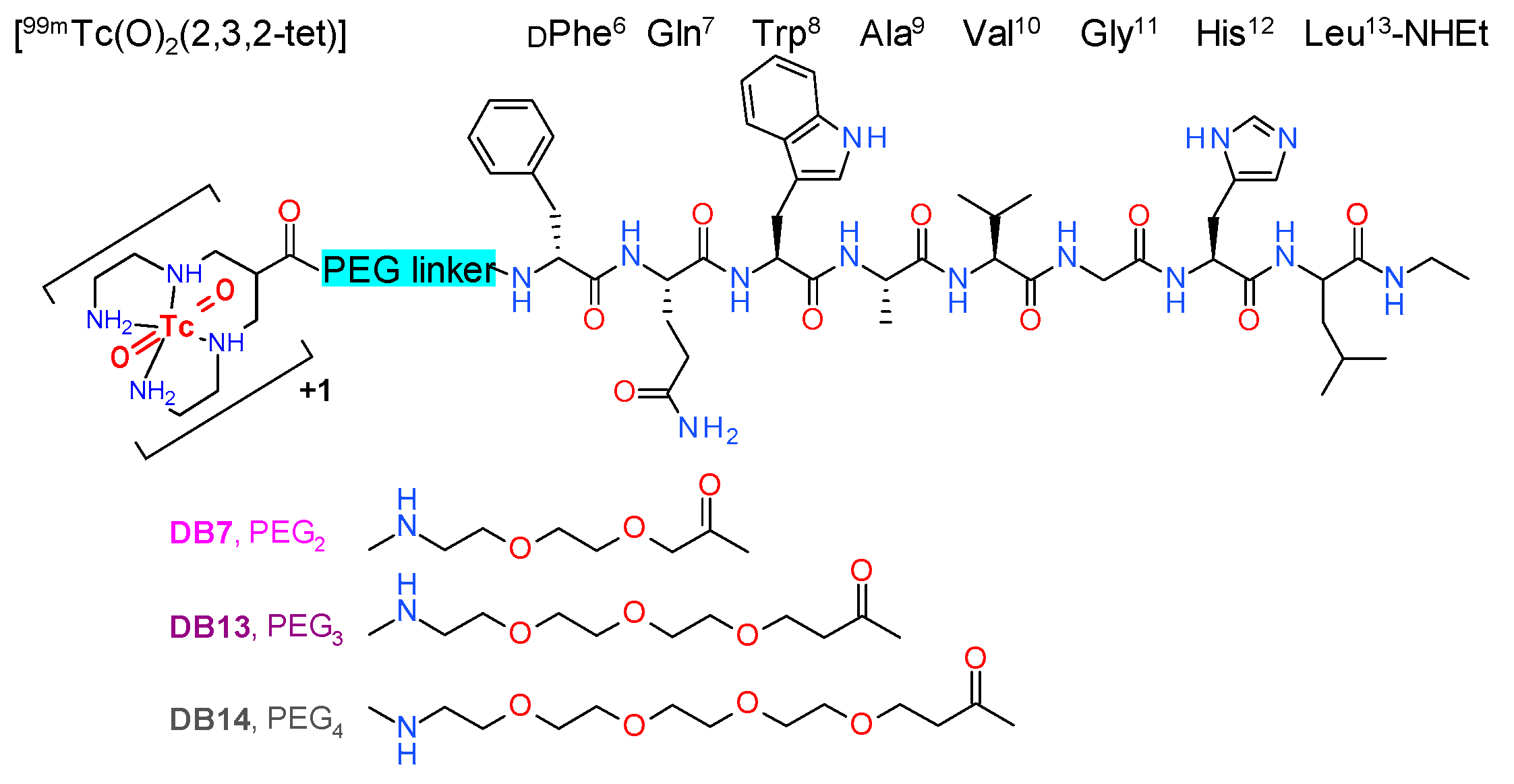
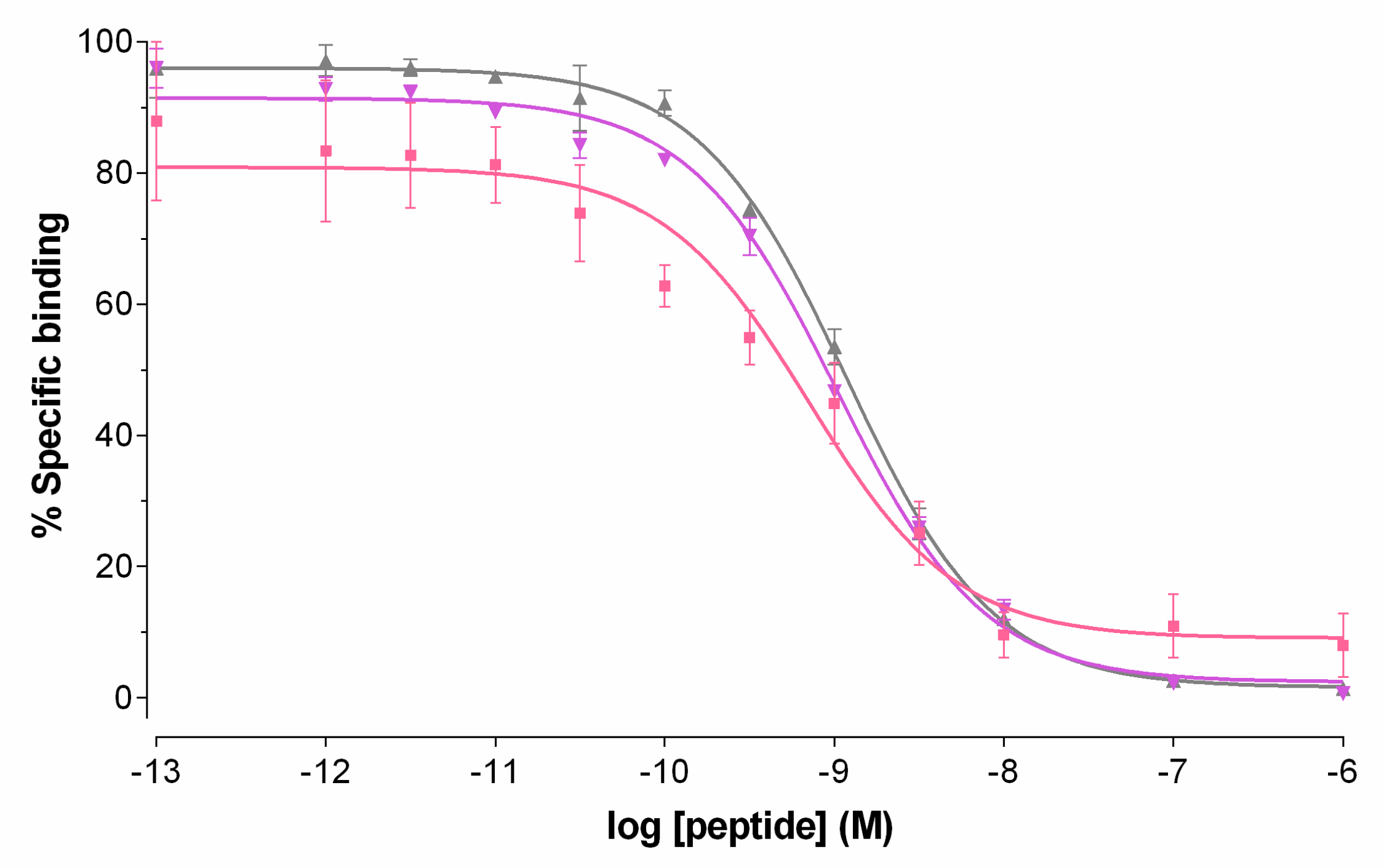
2.2.1. GRPR Affinity of Peptide Conjugates
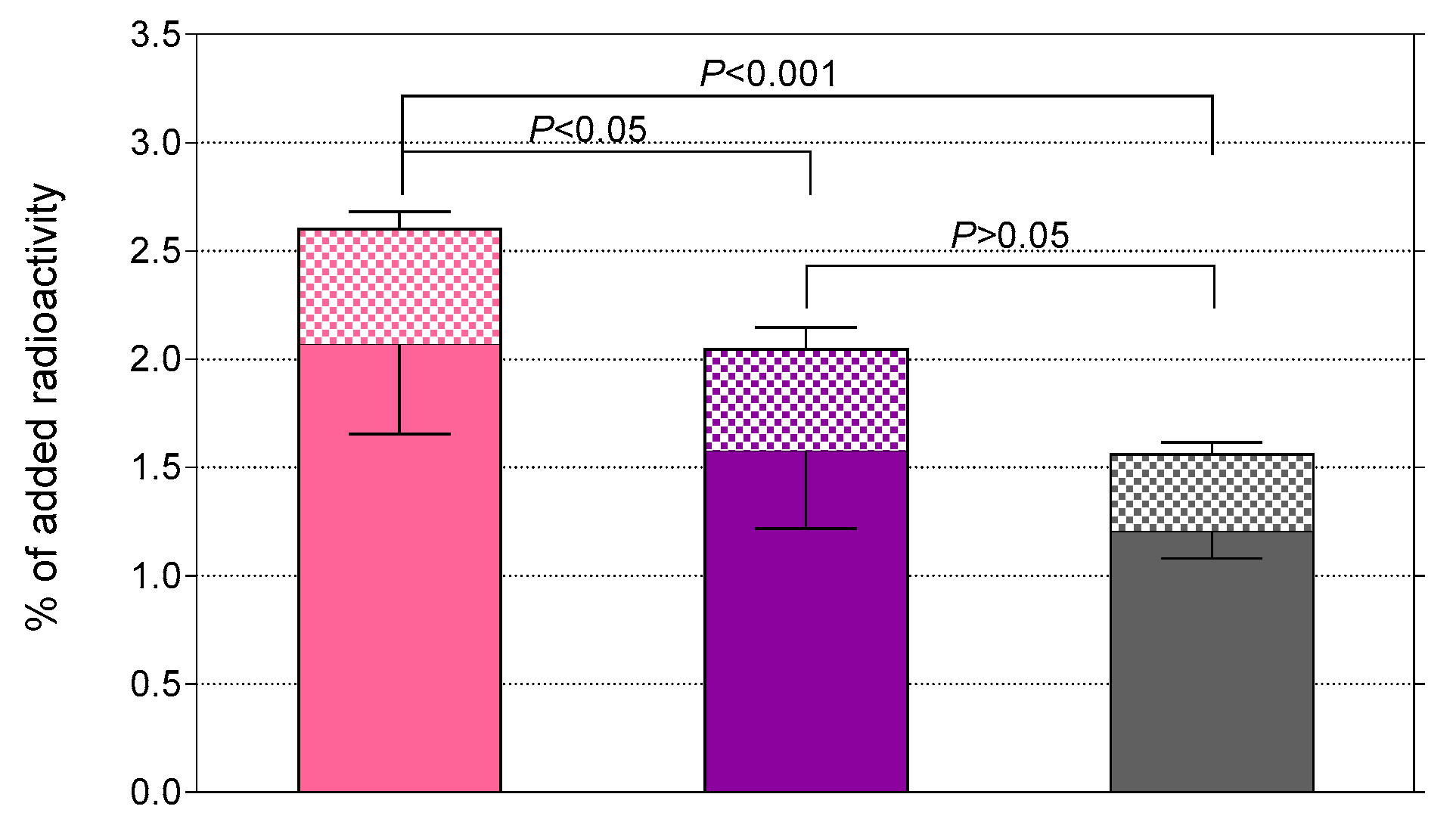
2.2.2. Radiotracer Uptake in PC-3 Cells
2.3. In Vivo Comparison of [99mTc]Tc-DB7, [99mTc]Tc-DB13, and [99mTc]Tc-DB14
2.3.1. Metabolic Studies in Mice
2.3.2. Biodistribution in PC-3 Tumor-Bearing Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals and Radionuclides
4.1.1. Radiolabeling
4.1.2. Quality Control
4.2. In Vitro Assays
4.2.1. Cell Lines and Culture
4.2.2. Competitive Binding in PC-3 Cell Membranes
4.2.3. Internalization of [99mTc]Tc Radiotracers in PC-3 Cells
4.3. Animal Studies
4.3.1. Metabolic Studies in Mice
4.3.2. Biodistribution in SCID Mice Bearing PC-3 Xenografts
4.3.3. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Markwalder, R.; Reubi, J.C. Gastrin-releasing peptide receptors in the human prostate: Relation to neoplastic transformation. Cancer Res. 1999, 59, 1152–1159. [Google Scholar] [PubMed]
- Körner, M.; Waser, B.; Rehmann, R.; Reubi, J.C. Early over-expression of GRP receptors in prostatic carcinogenesis. Prostate 2014, 74, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Beer, M.; Montani, M.; Gerhardt, J.; Wild, P.J.; Hany, T.F.; Hermanns, T.; Muntener, M.; Kristiansen, G. Profiling gastrin-releasing peptide receptor in prostate tissues: Clinical implications and molecular correlates. Prostate 2012, 72, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Gugger, M.; Reubi, J.C. Gastrin-releasing peptide receptors in non-neoplastic and neoplastic human breast. Am. J. Pathol. 1999, 155, 2067–2076. [Google Scholar] [CrossRef] [Green Version]
- Halmos, G.; Wittliff, J.L.; Schally, A.V. Characterization of bombesin/gastrin-releasing peptide receptors in human breast cancer and their relationship to steroid receptor expression. Cancer Res. 1995, 55, 280–287. [Google Scholar]
- Mattei, J.; Achcar, R.D.; Cano, C.H.; Macedo, B.R.; Meurer, L.; Batlle, B.S.; Groshong, S.D.; Kulczynski, J.M.; Roesler, R.; Dal Lago, L.; et al. Gastrin-releasing peptide receptor expression in lung cancer. Arch. Pathol. Lab. Med. 2014, 138, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Reubi, J.C.; Körner, M.; Waser, B.; Mazzucchelli, L.; Guillou, L. High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 803–810. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.A. From bench to bed: New gastrin-releasing peptide receptor-directed radioligands and their use in prostate cancer. PET Clin. 2017, 12, 205–217. [Google Scholar] [CrossRef]
- Zhang, J.; Singh, A.; Kulkarni, H.R.; Schuchardt, C.; Müller, D.; Wester, H.J.; Maina, T.; Rosch, F.; van der Meulen, N.P.; Müller, C.; et al. From bench to bedside-the Bad Berka experience with first-in-human studies. Semin. Nucl. Med. 2019, 49, 422–437. [Google Scholar] [CrossRef]
- Chatalic, K.L.; Kwekkeboom, D.J.; de Jong, M. Radiopeptides for imaging and therapy: A radiant future. J. Nucl. Med. 2015, 56, 1809–1812. [Google Scholar] [CrossRef] [Green Version]
- Moreno, P.; Ramos-Alvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging, targeting and treatment. Expert. Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef] [Green Version]
- Delle Fave, G.; Annibale, B.; de Magistris, L.; Severi, C.; Bruzzone, R.; Puoti, M.; Melchiorri, P.; Torsoli, A.; Erspamer, V. Bombesin effects on human gi functions. Peptides 1985, 6 (Suppl. 3), 113–116. [Google Scholar] [CrossRef]
- Bruzzone, R.; Tamburrano, G.; Lala, A.; Mauceri, M.; Annibale, B.; Severi, C.; de Magistris, L.; Leonetti, F.; Delle Fave, G. Effect of bombesin on plasma insulin, pancreatic glucagon, and gut glucagon in man. J. Clin. Endocrinol. Metab. 1983, 56, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Bitar, K.N.; Zhu, X.X. Expression of bombesin-receptor subtypes and their differential regulation of colonic smooth muscle contraction. Gastroenterology 1993, 105, 1672–1680. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.A.; Kulkarni, H.; Singh, A.; Baum, R.P. Theranostic prospects of gastrin-releasing peptide receptor-radioantagonists in oncology. PET Clin. 2017, 12, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Nicolas, G.P.; Wild, D. Somatostatin receptor antagonists for imaging and therapy. J. Nucl. Med. 2017, 58 (Suppl. 2), 61S–66S. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Coy, D.H.; Taylor, J.E.; Jiang, N.Y.; Moreau, J.P.; Huang, S.C.; Frucht, H.; Haffar, B.M.; Jensen, R.T. Des-Met carboxyl-terminally modified analogues of bombesin function as potent bombesin receptor antagonists, partial agonists, or agonists. J. Biol. Chem. 1990, 265, 15695–15703. [Google Scholar] [PubMed]
- Wang, L.H.; Coy, D.H.; Taylor, J.E.; Jiang, N.Y.; Kim, S.H.; Moreau, J.P.; Huang, S.C.; Mantey, S.A.; Frucht, H.; Jensen, R.T. Desmethionine alkylamide bombesin analogues: A new class of bombesin receptor antagonists with potent antisecretory activity in pancreatic acini and antimitotic activity in Swiss 3T3 cells. Biochemistry 1990, 29, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.; Nikolopoulou, A.; Chiotellis, E.; Loudos, G.; Maintas, D.; Reubi, J.C.; Maina, T. [99mTc]demobesin 1, a novel potent bombesin analogue for grp receptor-targeted tumour imaging. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 247–258. [Google Scholar] [CrossRef]
- Maina, T.; Bergsma, H.; Kulkarni, H.R.; Mueller, D.; Charalambidis, D.; Krenning, E.P.; Nock, B.A.; de Jong, M.; Baum, R.P. Preclinical and first clinical experience with the gastrin-releasing peptide receptor-antagonist [68Ga]SB3 and PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 964–973. [Google Scholar] [CrossRef]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, I.L.; van Tiel, S.T.; Haeck, J.; Doeswijk, G.N.; de Blois, E.; Segbers, M.; Maina, T.; Nock, B.A.; de Jong, M.; Dalm, S.U. In vivo stabilized SB3, an attractive GRPR antagonist, for pre- and intra-operative imaging for prostate cancer. Mol. Imaging Biol. 2018, 20, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nock, B.A.; Charalambidis, D.; Sallegger, W.; Waser, B.; Mansi, R.; Nicolas, G.P.; Ketani, E.; Nikolopoulou, A.; Fani, M.; Reubi, J.C.; et al. New gastrin releasing peptide receptor-directed [99mTc]demobesin 1 mimics: Synthesis and comparative evaluation. J. Med. Chem. 2018, 61, 3138–3150. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.; Maina, T. Tetraamine-coupled peptides and resulting 99mTc-radioligands: An effective route for receptor-targeted diagnostic imaging of human tumors. Curr. Top. Med. Chem. 2012, 12, 2655–2667. [Google Scholar] [CrossRef]
- Roques, B.P.; Noble, F.; Dauge, V.; Fournie-Zaluski, M.C.; Beaumont, A. Neutral endopeptidase 24.11: Structure, inhibition, and experimental and clinical pharmacology. Pharmacol. Rev. 1993, 45, 87–146. [Google Scholar]
- Roques, B.P. Zinc metallopeptidases: Active site structure and design of selective and mixed inhibitors: New approaches in the search for analgesics and anti-hypertensives. Biochem. Soc. Trans. 1993, 21 Pt 3, 678–685. [Google Scholar] [CrossRef]
- Suda, H.; Aoyagi, T.; Takeuchi, T.; Umezawa, H. Letter: A thermolysin inhibitor produced by actinomycetes: Phosphoramidon. J. Antibiot. (Tokyo) 1973, 26, 621–623. [Google Scholar] [CrossRef] [Green Version]
- Nock, B.A.; Maina, T.; Krenning, E.P.; de Jong, M. “To serve and protect”: Enzyme inhibitors as radiopeptide escorts promote tumor targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Kaloudi, A.; Lymperis, E.; Kanellopoulos, P.; Waser, B.; de Jong, M.; Krenning, E.P.; Reubi, J.C.; Nock, B.A.; Maina, T. Localization of 99mTc-GRP analogs in GRPR-expressing tumors: Effects of peptide length and neprilysin inhibition on biological responses. Pharmaceuticals 2019, 12, 42. [Google Scholar] [CrossRef] [Green Version]
- Lymperis, E.; Kaloudi, A.; Sallegger, W.; Bakker, I.L.; Krenning, E.P.; de Jong, M.; Maina, T.; Nock, B.A. Radiometal-dependent biological profile of the radiolabeled gastrin-releasing peptide receptor antagonist SB3 in cancer theranostics: Metabolic and biodistribution patterns defined by neprilysin. Bioconjug. Chem. 2018, 29, 1774–1784. [Google Scholar] [CrossRef]
- Lymperis, E.; Kaloudi, A.; Kanellopoulos, P.; Krenning, E.P.; de Jong, M.; Maina, T.; Nock, B.A. Comparative evaluation of the new GRPR-antagonist 111In-SB9 and 111In-AMBA in prostate cancer models: Implications of in vivo stability. J. Labelled Comp. Radiopharm. 2019, 62, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Lymperis, E.; Kaloudi, A.; Kanellopoulos, P.; de Jong, M.; Krenning, E.P.; Nock, B.A.; Maina, T. Comparing Gly11/dAla11-replacement vs. the in-situ neprilysin-inhibition approach on the tumor-targeting efficacy of the 111In-SB3/111In-SB4 radiotracer pair. Molecules 2019, 24, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatalic, K.L.; Konijnenberg, M.; Nonnekens, J.; de Blois, E.; Hoeben, S.; de Ridder, C.; Brunel, L.; Fehrentz, J.A.; Martinez, J.; van Gent, D.C.; et al. In vivo stabilization of a gastrin-releasing peptide receptor antagonist enhances PET imaging and radionuclide therapy of prostate cancer in preclinical studies. Theranostics 2016, 6, 104–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitran, B.; Rinne, S.S.; Konijnenberg, M.W.; Maina, T.; Nock, B.A.; Altai, M.; Vorobyeva, A.; Larhed, M.; Tolmachev, V.; de Jong, M.; et al. Trastuzumab cotreatment improves survival of mice with PC-3 prostate cancer xenografts treated with the GRPR antagonist 177Lu-DOTAGA-PEG2-RM26. Int. J. Cancer 2019, 145, 3347–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkema, R.; Froberg, A.; Maina, T.; Nock, B.A.; de Blois, E.; Melis, M.L.; Konijnenberg, M.W.; Koolen, S.L.W.; Peeters, R.P.; de Herder, W.W.; et al. Clinical translation of the pepprotect: A novel method to improve the detection of cancer and metastases by peptide scanning under the protection of enzyme inhibitors. Eur. J. Nucl. Med. Mol. Imaging 2019, 46 (Suppl. 1), S701–S702. [Google Scholar] [CrossRef]
- Shipp, M.A.; Tarr, G.E.; Chen, C.Y.; Switzer, S.N.; Hersh, L.B.; Stein, H.; Sunday, M.E.; Reinherz, E.L. CD10/neutral endopeptidase 24.11 hydrolyzes bombesin-like peptides and regulates the growth of small cell carcinomas of the lung. Proc. Natl. Acad. Sci. USA 1991, 88, 10662–10666. [Google Scholar] [CrossRef] [Green Version]
- Linder, K.E.; Metcalfe, E.; Arunachalam, T.; Chen, J.; Eaton, S.M.; Feng, W.; Fan, H.; Raju, N.; Cagnolini, A.; Lantry, L.E.; et al. In vitro and in vivo metabolism of Lu-AMBA, a GRP-receptor binding compound, and the synthesis and characterization of its metabolites. Bioconjug. Chem. 2009, 20, 1171–1178. [Google Scholar] [CrossRef]
- Fleischmann, A.; Laderach, U.; Friess, H.; Buechler, M.W.; Reubi, J.C. Bombesin receptors in distinct tissue compartments of human pancreatic diseases. Lab. Investig. 2000, 80, 1807–1817. [Google Scholar] [CrossRef] [Green Version]
- Varasteh, Z.; Rosenstrom, U.; Velikyan, I.; Mitran, B.; Altai, M.; Honarvar, H.; Rosestedt, M.; Lindeberg, G.; Sörensen, J.; Larhed, M.; et al. The effect of mini-PEG-based spacer length on binding and pharmacokinetic properties of a 68Ga-labeled NOTA-conjugated antagonistic analog of bombesin. Molecules 2014, 19, 10455–10472. [Google Scholar] [CrossRef] [Green Version]
- Jamous, M.; Tamma, M.L.; Gourni, E.; Waser, B.; Reubi, J.C.; Maecke, H.R.; Mansi, R. PEG spacers of different length influence the biological profile of bombesin-based radiolabeled antagonists. Nucl. Med. Biol. 2014, 41, 464–470. [Google Scholar] [CrossRef]
- Gourni, E.; Mansi, R.; Jamous, M.; Waser, B.; Smerling, C.; Burian, A.; Buchegger, F.; Reubi, J.C.; Maecke, H.R. N-terminal modifications improve the receptor affinity and pharmacokinetics of radiolabeled peptidic gastrin-releasing peptide receptor antagonists: Examples of 68Ga- and 64Cu-labeled peptides for PET imaging. J. Nucl. Med. 2014, 55, 1719–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abiraj, K.; Mansi, R.; Tamma, M.L.; Fani, M.; Forrer, F.; Nicolas, G.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J. Nucl. Med. 2011, 52, 1970–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reile, H.; Armatis, P.E.; Schally, A.V. Characterization of high-affinity receptors for bombesin/gastrin releasing peptide on the human prostate cancer cell lines PC-3 and DU-145: Internalization of receptor bound 125I-(Tyr4)bombesin by tumor cells. Prostate 1994, 25, 29–38. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
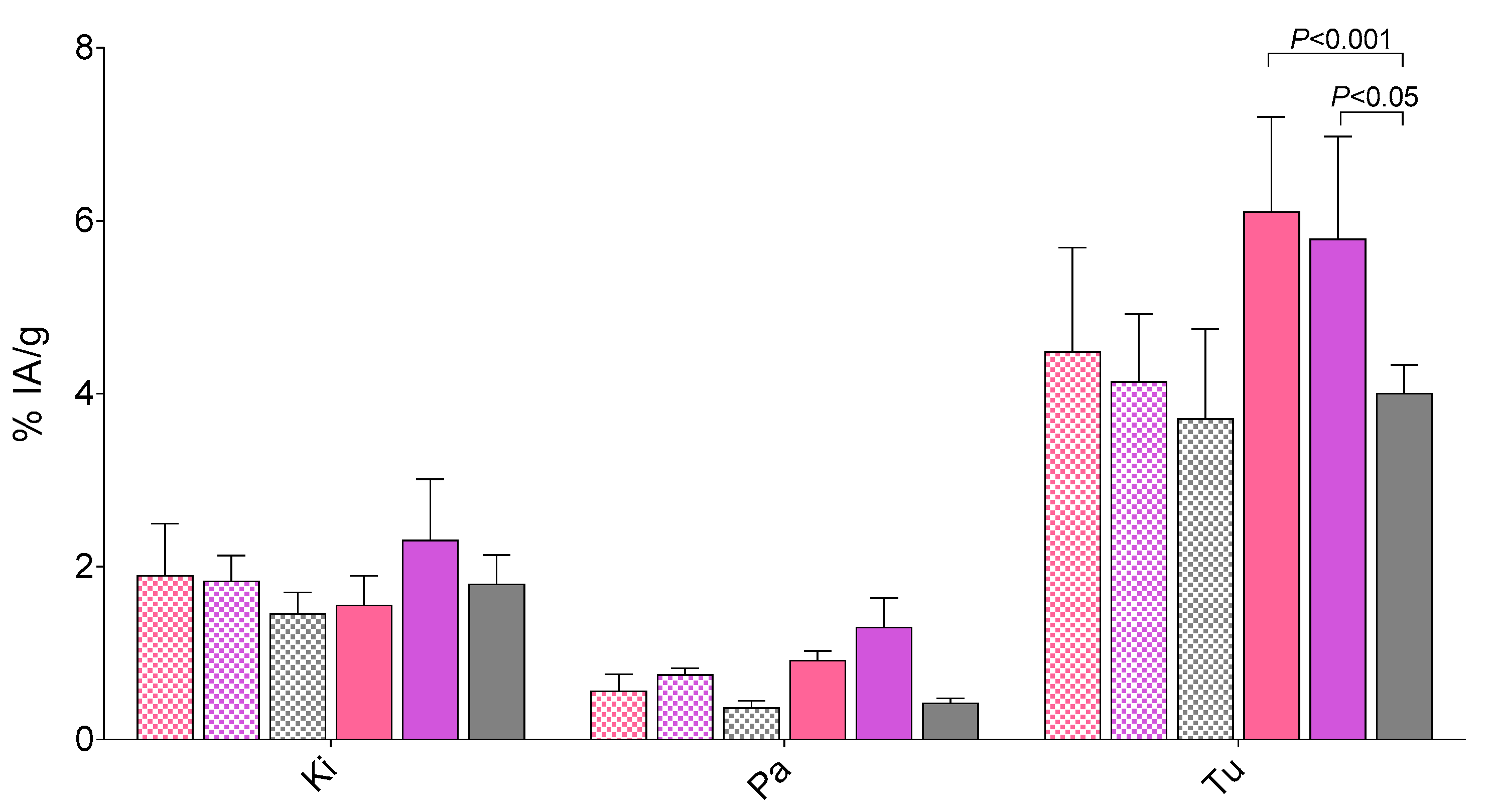{kind=link}
| Tissue | [99mTc]Tc-DB7 | ||||
|---|---|---|---|---|---|
| Block 1 | Controls | PA 2 | |||
| Blood | 0.08 ± 0.03 | 0.05 ± 0.01 | 0.06 ± 0.02 | ||
| Liver | 2.11 ± 0.43 | 0.96 ± 0.23 | 0.87 ± 0.12 | ||
| Heart | 0.09 ± 0.03 | 0.04 ± 0.01 | 0.07 ± 0.03 | ||
| Kidneys | 2.38 ± 0.56 | 1.89 ± 0.61 | 1.55 ± 0.34 | ||
| Stomach | 0.45 ± 0.34 | 0.27 ± 0.17 | 0.22 ± 0.05 | ||
| Intestines | 2.85 ± 0.39 | 1.38 ± 0.27 | 1.64 ± 0.72 | ||
| Spleen | 1.33 ± 0.37 | 0.24 ± 0.07 | 0.28 ± 0.1 | ||
| Muscle | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | ||
| Lungs | 0.55 ± 0.26 | 0.16 ± 0.12 | 0.14 ± 0.02 | ||
| Pancreas | 0.37 ± 0.09 | ⇤ p > 0.05 ⇥ | 0.56 ± 0.2 | ⇤ p > 0.05 ⇥ | 0.91 ± 0.12 |
| Tumor | 0.53 ± 0.20 | ⇤ p < 0.0001 ⇥ | 4.49 ± 1.20 | ⇤ p < 0.0001 ⇥ | 6.10 ± 1.1 |
| Tissue | [99mTc]Tc-DB13 | ||||
|---|---|---|---|---|---|
| Block 1 | Controls | PA 2 | |||
| Blood | 0.09 ± 0.01 | 0.06 ± 0.02 | 0.19 ± 0.19 | ||
| Liver | 1.8 ± 0.15 | 0.75 ± 0.24 | 0.88 ± 0.25 | ||
| Heart | 0.18 ± 0.07 | 0.07 ± 0.03 | 0.16 ± 0.08 | ||
| Kidneys | 3.5 ± 1.59 | 1.83 ± 0.3 | 2.66 ± 1.14 | ||
| Stomach | 1.25 ± 0.86 | 0.9 ± 0.18 | 1.09 ± 0.47 | ||
| Intestines | 5.37 ± 1.34 | 3.69 ± 0.72 | 4.23 ± 0.98 | ||
| Spleen | 1.14 ± 0.47 | 0.29 ± 0.07 | 0.36 ± 0.13 | ||
| Muscle | 0.04 ± 0.02 | 0.03 ± 0.01 | 0.16 ± 0.32 | ||
| Lungs | 0.67 ± 0.31 | 0.13 ± 0.01 | 0.24 ± 0.08 | ||
| Pancreas | 0.3 ± 0.09 | ⇤ p > 0.05 ⇥ | 0.75 ± 0.08 | ⇤ p > 0.05 ⇥ | 1.82 ± 1.04 |
| Tumor | 1.12 ± 0.23 | ⇤ p < 0.0001 ⇥ | 4.14 ± 0.78 | ⇤ p < 0.001 ⇥ | 5.79 ± 1.18 |
| Tissue | [99mTc]Tc-DB14 | ||||
|---|---|---|---|---|---|
| Block 1 | Controls | PA 2 | |||
| Blood | 0.21 ± 0.13 | 0.06 ± 0.03 | 0.07 ± 0.01 | ||
| Liver | 1.69 ± 0.15 | 0.79 ± 0.32 | 0.93 ± 0.13 | ||
| Heart | 0.23 ± 0.08 | 0.08 ± 0.05 | 0.15 ± 0.03 | ||
| Kidneys | 7.05 ± 4.24 | 1.46 ± 0.25 | 1.79 ± 0.34 | ||
| Stomach | 0.85 ± 0.10 | 0.75 ± 0.72 | 0.21 ± 0.06 | ||
| Intestines | 6.25 ± 2.51 | 2.03 ± 0.81 | 1.83 ± 0.77 | ||
| Spleen | 0.89 ± 0.15 | 0.19 ± 0.07 | 0.24 ± 0.05 | ||
| Muscle | 0.07 ± 0.04 | 0.04 ± 0.03 | 0.03 ± 0.01 | ||
| Lungs | 0.78 ± 0.25 | 0.15 ± 0.06 | 0.16 ± 0.05 | ||
| Pancreas | 0.32 ± 0.09 | ⇤ p > 0.05 ⇥ | 0.36 ± 0.09 | ⇤ p > 0.05 ⇥ | 0.42 ± 0.06 |
| Tumor | 2.16 ± 0.59 | ⇤ p < 0.01 ⇥ | 3.71 ± 1.04 | ⇤ p > 0.05 ⇥ | 4.00 ± 0.34 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanellopoulos, P.; Lymperis, E.; Kaloudi, A.; de Jong, M.; Krenning, E.P.; Nock, B.A.; Maina, T. [99mTc]Tc-DB1 Mimics with Different-Length PEG Spacers: Preclinical Comparison in GRPR-Positive Models. Molecules 2020, 25, 3418. https://doi.org/10.3390/molecules25153418
Kanellopoulos P, Lymperis E, Kaloudi A, de Jong M, Krenning EP, Nock BA, Maina T. [99mTc]Tc-DB1 Mimics with Different-Length PEG Spacers: Preclinical Comparison in GRPR-Positive Models. Molecules. 2020; 25(15):3418. https://doi.org/10.3390/molecules25153418
Chicago/Turabian StyleKanellopoulos, Panagiotis, Emmanouil Lymperis, Aikaterini Kaloudi, Marion de Jong, Eric P. Krenning, Berthold A. Nock, and Theodosia Maina. 2020. "[99mTc]Tc-DB1 Mimics with Different-Length PEG Spacers: Preclinical Comparison in GRPR-Positive Models" Molecules 25, no. 15: 3418. https://doi.org/10.3390/molecules25153418