
Properly Substituted Cyclic Bis-(2-bromobenzylidene) Compounds Behaved as Dual p300/CARM1 Inhibitors and Induced Apoptosis in Cancer Cells

 ,
,  ,
, 
 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
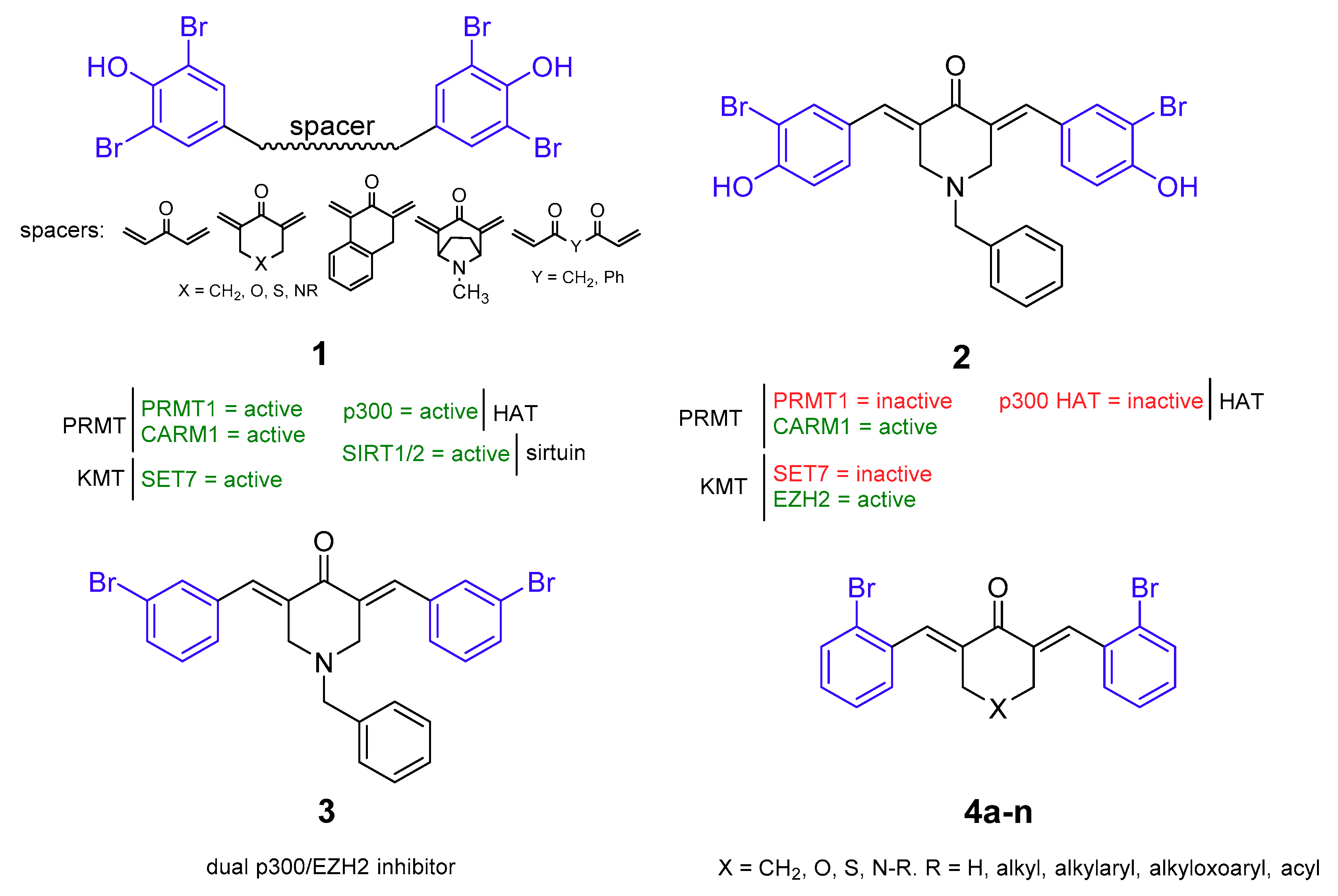
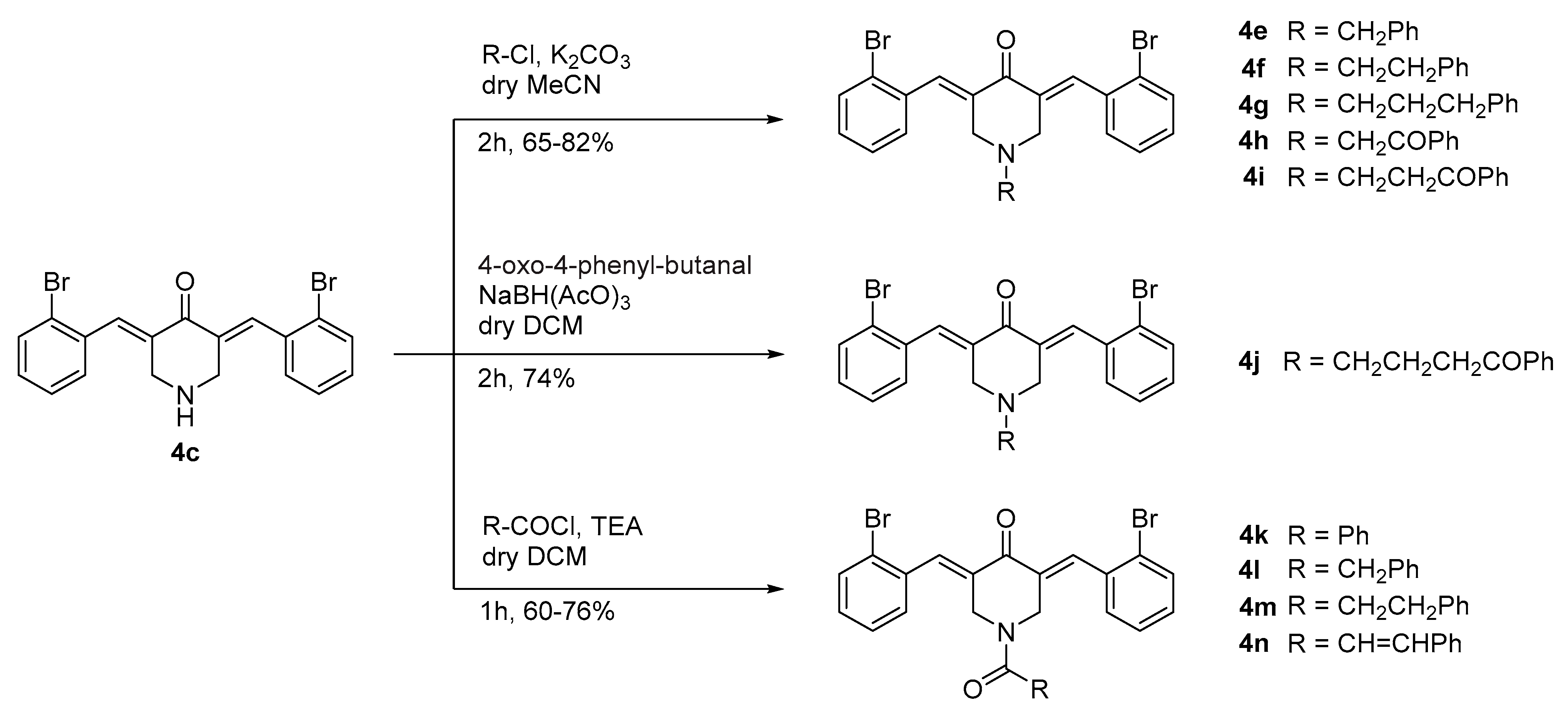
2.1. Chemistry
2.2. p300, PCAF, SIRT1/2, EZH2, and CARM1 Enzyme Assays
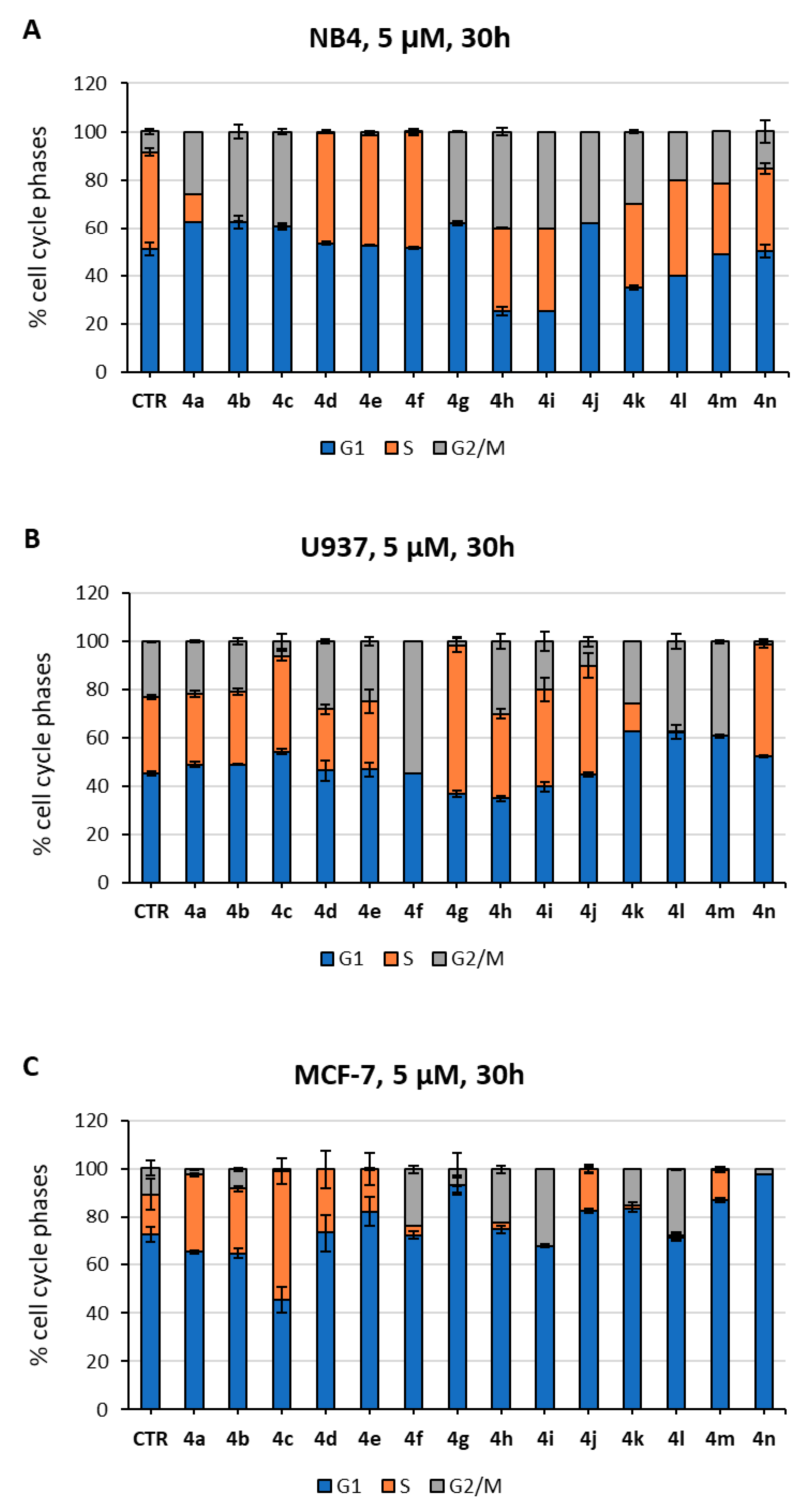
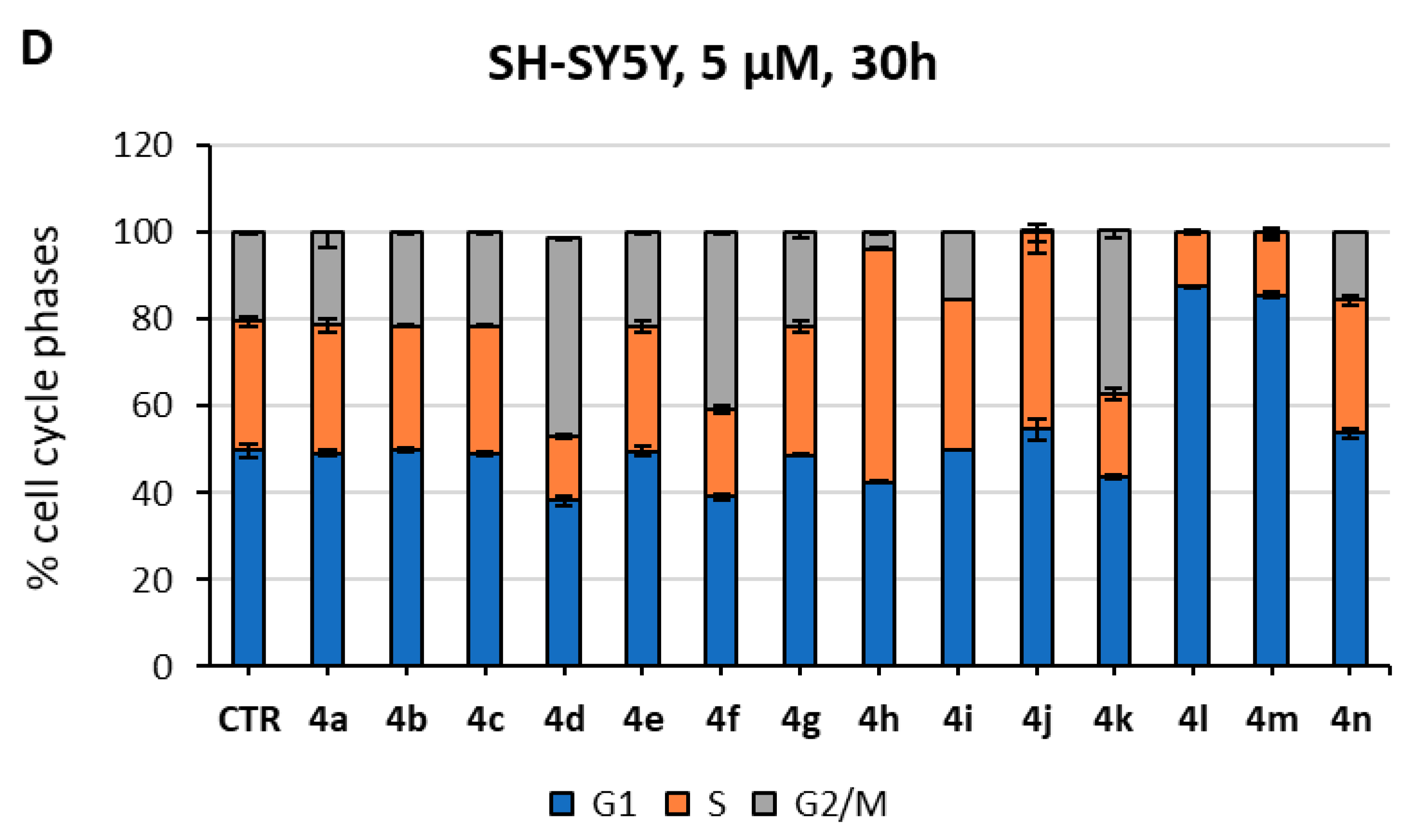
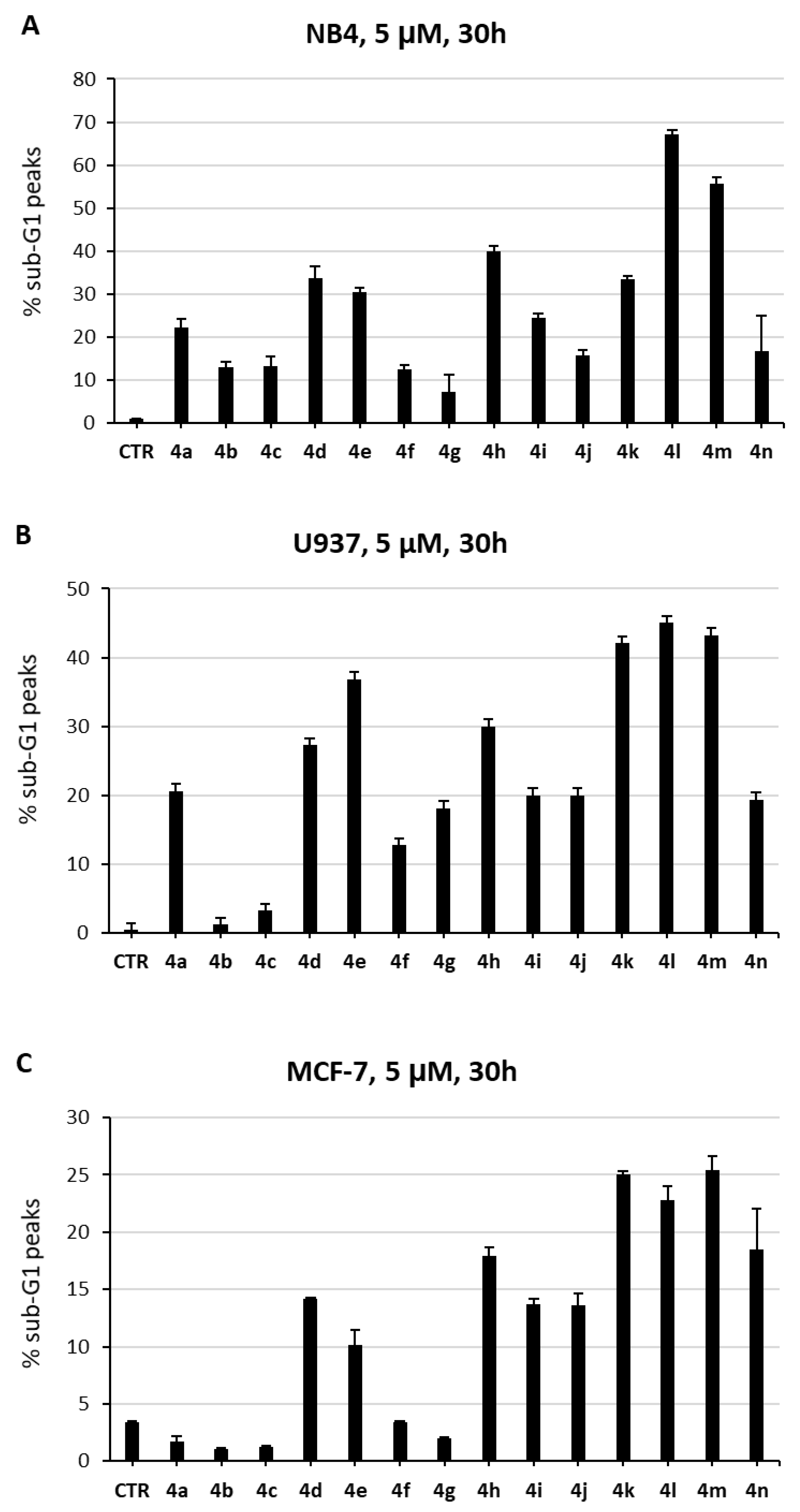
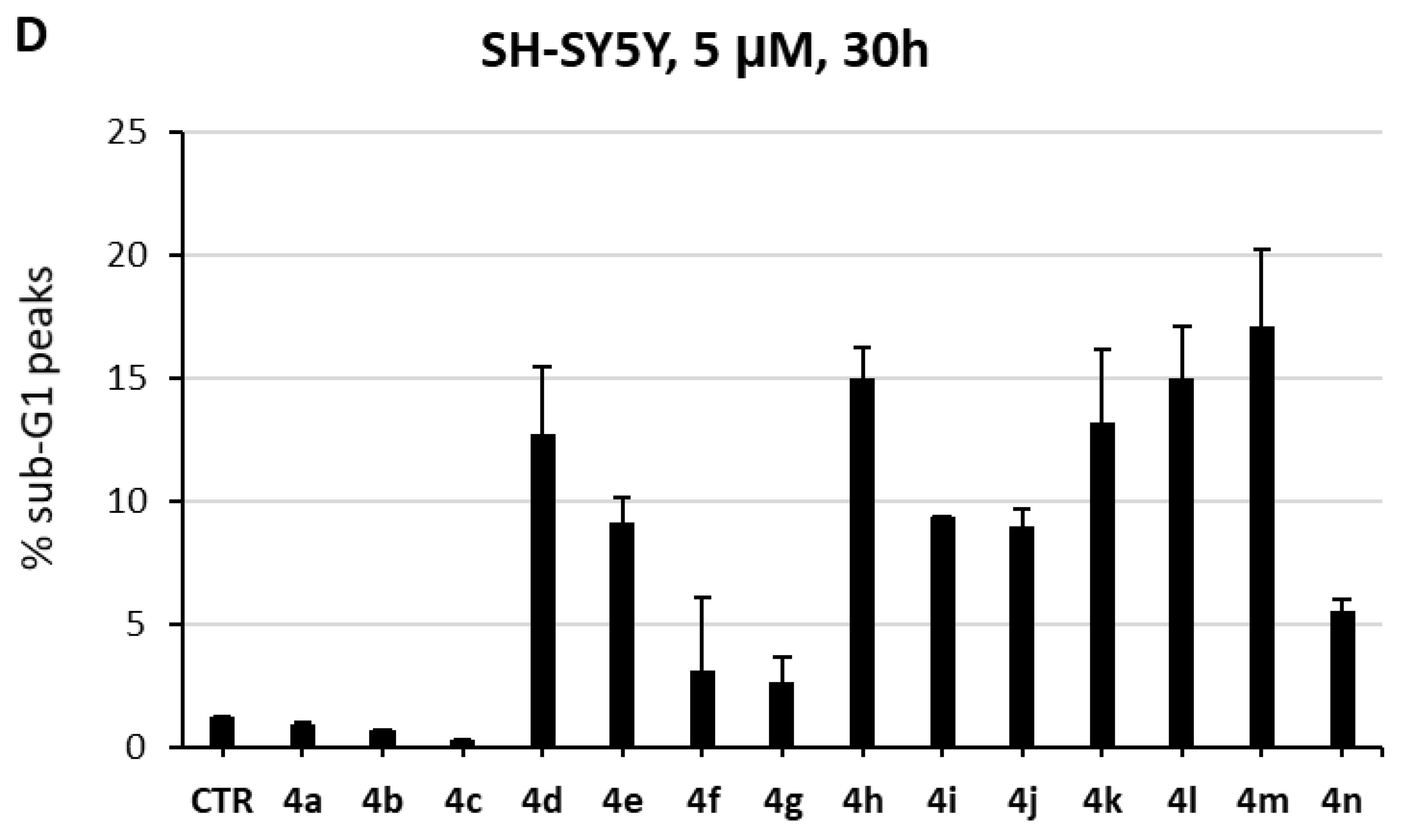
2.3. Biological Evaluation in Cancer Cells
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of the N-Alkylaryl 3,5-Bis((E)-2-bromobenzylidene)piperid-4-ones (4e–i). Example: 1-benzyl-3,5-bis((E)-2-bromobenzylidene)piperidin-4-one (4e)
3.1.2. Synthesis of 3,5-Bis((E)-2-bromobenzylidene)-1-(4-oxo-4-phenylbutyl)piperidin-4-one (4j)
3.1.3. General Procedure for the Synthesis of N-Acyl-3,5-Bis((E)-2-bromobenzylidene)piperidin-4-ones (4k–n). Example: 3,5-Bis((E)-2-bromobenzylidene)-1-cinnamoylpiperidin-4-one (4n)
3.2. Biochemistry
3.2.1. HAT Assays
3.2.2. SIRT1/2 Assay
3.2.3. EZH2/PRC2 Assay
3.2.4. CARM1 Assay
3.3. Cellular Studies
3.3.1. Cell Cultures
3.3.2. Cell Proliferation, Cell Cycle, and Cell Death Analyses
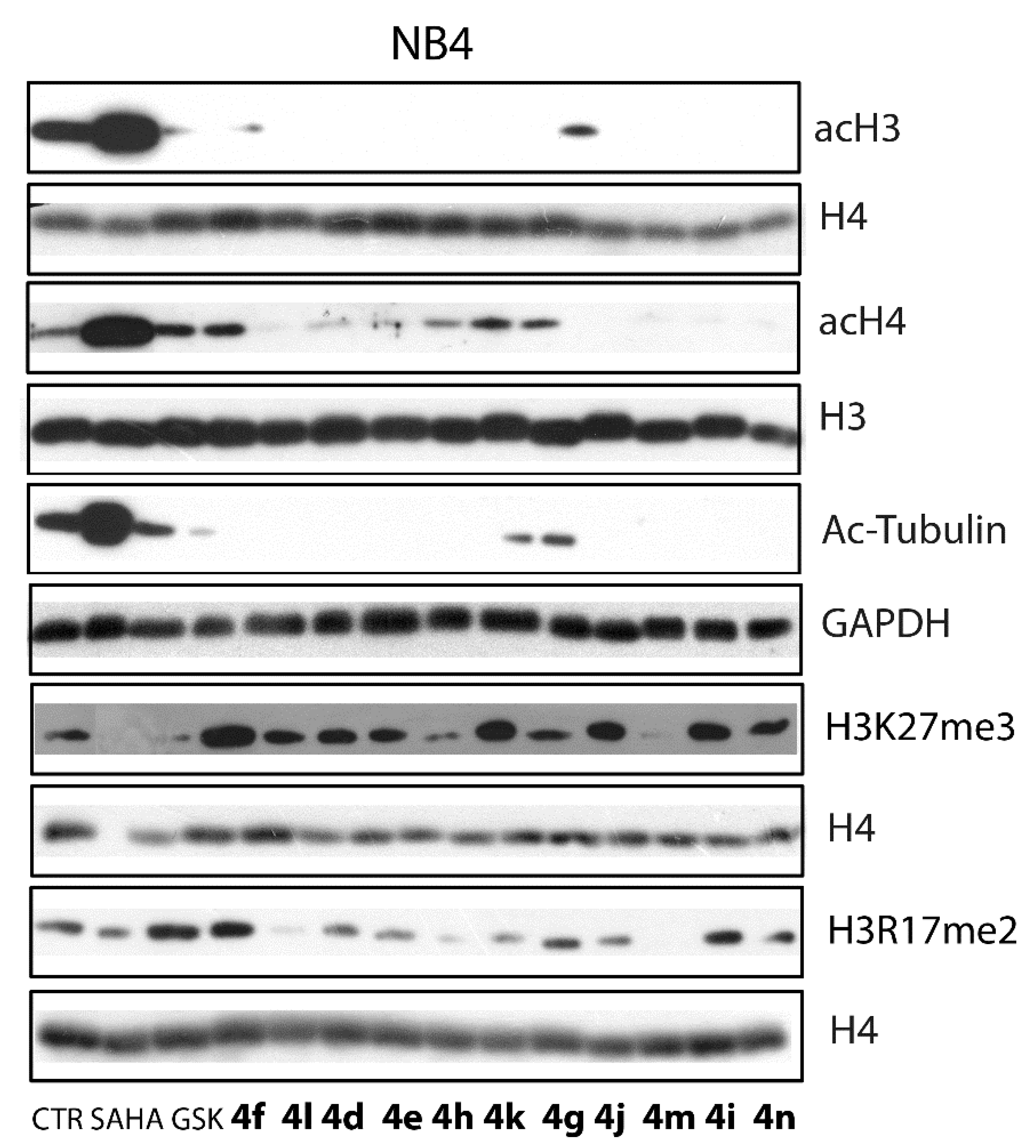
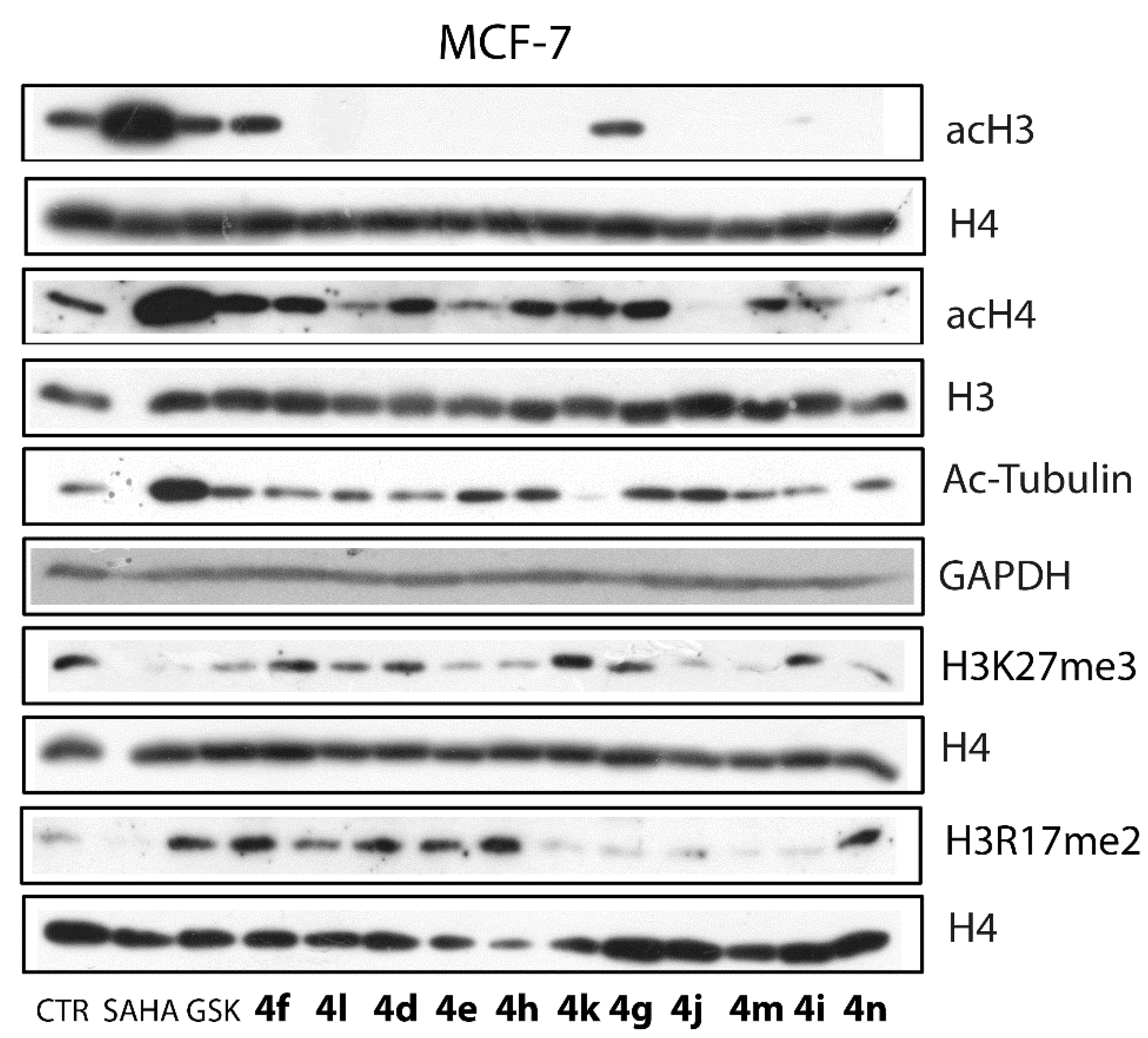
3.3.3. Western Blot Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Conte, M.; Iside, C.; Altucci, L. Epigenetic-based therapy: From single- to multi-target approaches. Int. J. Biochem. Cell Boil. 2015, 69, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2019, 40, 190–244. [Google Scholar] [CrossRef]
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef]
- Zwergel, C.; Valente, S.; Jacob, C.; Mai, A. Emerging approaches for histone deacetylase inhibitor drug discovery. Expert Opin. Drug Discov. 2015, 10, 599–613. [Google Scholar] [CrossRef]
- Kaniskan, H.U.; Konze, K.D.; Jin, J. Selective Inhibitors of Protein Methyltransferases. J. Med. Chem. 2014, 58, 1596–1629. [Google Scholar] [CrossRef]
- Fioravanti, R.; Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. Six Years (2012–2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. Chem. Rec. 2018, 18, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Mai, A. Targeting Histone Demethylases: A New Avenue for the Fight against Cancer. Genes Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Wimalasena, V.K.; Wang, T.; Sigua, L.H.; Durbin, A.D.; Qi, J. Using Chemical Epigenetics to Target Cancer. Mol. Cell 2020, 78, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Yadav, N.; King, R.W.; Swanson, M.S.; Weinstein, E.J.; Bedford, M.T.; Dihazi, H.; Kessler, R.; Eschrich, K. Small Molecule Regulators of Protein Arginine Methyltransferases. J. Boil. Chem. 2004, 279, 23892–23899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, A.; Valente, S.; Cheng, D.; Perrone, A.; Ragno, R.; Simeoni, S.; Sbardella, G.; Brosch, G.; Nebbioso, A.; Conte, M.; et al. Synthesis and Biological Validation of Novel Synthetic Histone/Protein Methyltransferase Inhibitors. ChemMedChem 2007, 2, 987–991. [Google Scholar] [CrossRef]
- Mai, A.; Cheng, N.; Bedford, M.T.; Valente, S.; Nebbioso, A.; Perrone, A.; Brosch, G.; Sbardella, G.; De Bellis, F.; Miceli, M.; et al. Epigenetic Multiple Ligands: Mixed Histone/Protein Methyltransferase, Acetyltransferase, and Class III Deacetylase (Sirtuin) Inhibitors. J. Med. Chem. 2008, 51, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Lepore, I.; Dell’Aversana, C.; Tardugno, M.; Castellano, S.; Sbardella, G.; Tomassi, S.; Di Maro, S.; Novellino, E.; Di Santo, R.; et al. Identification of PR-SET7 and EZH2 selective inhibitors inducing cell death in human leukemia U937 cells. Biochimie 2012, 94, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.; Di Santo, R.; Artico, M.; Miele, G.; Valentini, P.; Novellino, E.; Cereseto, A. Cinnamoyl Compounds as Simple Molecules that Inhibit p300 Histone Acetyltransferase. J. Med. Chem. 2007, 50, 1973–1977. [Google Scholar] [CrossRef]
- Madia, V.N.; Benedetti, R.; Barreca, M.L.; Ngo, L.; Pescatori, L.; Messore, A.; Pupo, G.; Saccoliti, F.; Valente, S.; Mai, A.; et al. Structure-Activity Relationships on Cinnamoyl Derivatives as Inhibitors of p300 Histone Acetyltransferase. ChemMedChem 2017, 12, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Valente, S.; Castellano, S.; Sbardella, G.; Di Santo, R.; Costi, R.; Bedford, M.T.; Mai, A. Novel 3,5-Bis(bromohydroxybenzylidene)piperidin-4-ones as Coactivator-Associated Arginine Methyltransferase 1 Inhibitors: Enzyme Selectivity and Cellular Activity. J. Med. Chem. 2011, 54, 4928–4932. [Google Scholar] [CrossRef] [Green Version]
- Ruta, V.; Longo, C.; Boccaccini, A.; Madia, V.N.; Saccoliti, F.; Tudino, V.; Di Santo, R.; Lorrai, R.; Ioio, R.D.; Sabatini, S.; et al. Inhibition of Polycomb Repressive Complex 2 activity reduces trimethylation of H3K27 and affects development in Arabidopsis seedlings. BMC Plant Boil. 2019, 19, 429. [Google Scholar] [CrossRef]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/p38α/Polycomb Signaling to Pax7 Locus in Satellite Cells Links Inflammation to the Epigenetic Control of Muscle Regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Ciarapica, R.; Carcarino, E.; Adesso, L.; De Salvo, M.; Bracaglia, G.; Leoncini, P.P.; Dall’Agnese, A.; Verginelli, F.; Milano, G.; Boldrini, R.; et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 2014, 14, 139. [Google Scholar] [CrossRef] [Green Version]
- Petraglia, F.; Singh, A.; Carafa, V.; Nebbioso, A.; Conte, M.; Scisciola, L.; Valente, S.; Baldi, A.; Mandoli, A.; Petrizzi, V.B.; et al. Combined HAT/EZH2 modulation leads to cancer-selective cell death. Oncotarget 2018, 9, 25630–25646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Petraglia, F.; Nebbioso, A.; Yi, G.; Conte, M.; Valente, S.; Mandoli, A.; Scisciola, L.; Lindeboom, R.; Kerstens, H.; et al. Multi-omics profiling reveals a distinctive epigenome signature for high-risk acute promyelocytic leukemia. Oncotarget 2018, 9, 25647–25660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Li, X.; Chen, L.; Yang, S.; Wu, X.; Studer, E.; Gurley, E.; Hylemon, P.B.; Ye, F.; Li, Y.; et al. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Bioorg. Med. Chem. Lett. 2008, 18, 1525–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, Z.; Wu, J.; Bai, B.; Chen, H.; Xiao, Z.; Chen, L.; Zhao, Y.; Lum, H.; Wang, Y.; et al. New MD2 inhibitors derived from curcumin with improved anti-inflammatory activity. Eur. J. Med. Chem. 2018, 148, 291–305. [Google Scholar] [CrossRef]
- Jin, R.; Chen, Q.; Yao, S.; Bai, E.; Fu, W.; Wang, L.; Wang, J.; Du, X.; Wei, T.; Xu, H.; et al. Synthesis and anti-tumor activity of EF24 analogues as IKKβ inhibitors. Eur. J. Med. Chem. 2018, 144, 218–228. [Google Scholar] [CrossRef]
- Miceli, M.; Franci, G.; Dell’Aversana, C.; Ricciardiello, F.; Petraglia, F.; Carissimo, A.; Perone, L.; Maruotti, G.; Savarese, M.; Martinelli, P.; et al. MePR: A Novel Human Mesenchymal Progenitor Model with Characteristics of Pluripotency. Stem Cells Dev. 2013, 22, 2368–2383. [Google Scholar] [CrossRef] [Green Version]
- Miceli, M.; Dell’Aversana, C.; Russo, R.; Rega, C.; Cupelli, L.; Ruvo, M.; Altucci, L.; Chambery, A. Secretome profiling of cytokines and growth factors reveals that neuro-glial differentiation is associated with the down-regulation of Chemokine Ligand 2 (MCP-1/CCL2) in amniotic fluid derived-mesenchymal progenitor cells. Proteomics 2016, 16, 674–688. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Iii, A.D.P.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Álvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2004, 11, 77–84. [Google Scholar] [CrossRef]
- Conte, M.; Dell’Aversana, C.; Benedetti, R.; Petraglia, F.; Carissimo, A.; Petrizzi, V.B.; D’Arco, A.M.; Abbondanza, C.; Nebbioso, A.; Altucci, L. HDAC2 deregulation in tumorigenesis is causally connected to repression of immune modulation and defense escape. Oncotarget 2014, 6, 886–901. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Hou, G.-G.; Zhao, F.; Cong, W.; Li, H.; Liu, W.; Wang, C.-H. Synthesis, antiproliferative and multidrug resistance reversal activities of heterocyclic α,β-unsaturated carbonyl compounds. Chem. Biol. Drug Des. 2016, 88, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Zha, G.-F.; Zhang, C.-P.; Qin, H.-L.; Jantan, I.; Sher, M.; Amjad, M.W.; Hussain, M.A.; Hussain, Z.; Bukhari, S.N.A. Biological evaluation of synthetic α,β-unsaturated carbonyl based cyclohexanone derivatives as neuroprotective novel inhibitors of acetylcholinesterase, butyrylcholinesterase and amyloid-β aggregation. Bioorg. Med. Chem. 2016, 24, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.-L.; Leng, J.; Zhang, C.-P.; Jantan, I.; Amjad, M.W.; Sher, M.; Naeem-Ul-Hassan, M.; Hussain, M.A.; Bukhari, S.N.A. Synthesis of α,β-Unsaturated Carbonyl-Based Compounds, Oxime and Oxime Ether Analogs as Potential Anticancer Agents for Overcoming Cancer Multidrug Resistance by Modulation of Efflux Pumps in Tumor Cells. J. Med. Chem. 2016, 59, 3549–3561. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, S.; Li, H.; Jiang, W.; Hou, G.-G.; Zhao, F.; Cong, W. Synthesis, antitumor activity evaluation of some new N-aroyl-α,β-unsaturated piperidones with fluorescence. J. Enzym. Inhib. Med. Chem. 2015, 31, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Li, H.; Bai, H.; Su, Z.; Xiang, Q.; Wang, C.; Zhao, B.; Zhang, Y.; Zhang, Q.; Chu, Y.; et al. Synthesis of novel curcumin analogues for inhibition of 11β-hydroxysteroid dehydrogenase type 1 with anti-diabetic properties. Eur. J. Med. Chem. 2014, 77, 223–230. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Cai, Y.; Wang, J.; Weng, B.; Tang, Q.; Chen, X.; Pan, Z.; Liang, G.; Yang, S. Discovery and evaluation of piperid-4-one-containing mono-carbonyl analogs of curcumin as anti-inflammatory agents. Bioorg. Med. Chem. 2013, 21, 3058–3065. [Google Scholar] [CrossRef]
- Lagisetty, P.; Vilekar, P.; Sahoo, K.; Anant, S.; Awasthi, V. CLEFMA—An anti-proliferative curcuminoid from structure–activity relationship studies on 3,5-bis(benzylidene)-4-piperidones. Bioorg. Med. Chem. 2010, 18, 6109–6120. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds 4a–n are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50, μM | |||||
|---|---|---|---|---|---|---|
| p300 | PCAF | SIRT1 | SIRT2 | PRC2/EZH2 | CARM1 | |
| 4a | 1.38 ± 0.24 | >200 | >200 | >200 | 117 ± 6 | 16.8 ± 3.4 |
| 4b | 2.56 ± 0.23 | >200 | >200 | >200 | >200 | 23.3 ± 0.5 |
| 4c | 23.2 ± 0.51 | >200 | >200 | >200 | >200 | 44.8 ± 6.0 |
| 4d | 2.66 ± 0.15 | >200 | >200 | >200 | 46.7 ± 1.1 | 3.24 ± 1.22 |
| 4e | 2.19 ± 0.77 | >200 | >200 | >200 | 44.2 ± 0.2 | 6.50 ± 0.86 |
| 4f | 30.17 ± 2.71 | >200 | >200 | >200 | >200 | 18.5 ± 0.9 |
| 4g | 40.7 ± 2.39 | >200 | >200 | >200 | >200 | 12.6 ± 2.6 |
| 4h | 1.03 ± 0.22 | >200 | >200 | >200 | 46.0 ± 8.1 | 5.52 ± 0.78 |
| 4i | 1.57 ± 0.72 | >200 | >200 | >200 | 60.6 ± 10.5 | 6.96 ± 0.90 |
| 4j | 2.13 ± 1.35 | >200 | >200 | >200 | 68.5 ± 6.2 | 4.80 ± 1.72 |
| 4k | 2.09 ± 0.79 | >200 | >200 | >200 | 40.7 ± 15.3 | 1.33 ± 0.14 |
| 4l | 0.45 ± 0.03 | >200 | >200 | >200 | 15.2 ± 2.2 | 0.43 ± 0.12 |
| 4m | 0.46 ± 0.10 | >200 | >200 | >200 | 11.3 ± 0.08 | 0.79 ± 0.06 |
| 4n | 1.23 ± 0.29 | >200 | >200 | >200 | 13.9 ± 1.8 | 7.14 ± 2.54 |
| C-646 | 0.12 ± 0.01 | |||||
| anacardic acid | 40.0 ± 10.9 | |||||
| selisistat | 0.16 ± 0.01 | 48.5 ± 1.9 | ||||
| SAH | 33.8 ± 1.4 | 0.30 ± 0.07 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fioravanti, R.; Tomassi, S.; Di Bello, E.; Romanelli, A.; Plateroti, A.M.; Benedetti, R.; Conte, M.; Novellino, E.; Altucci, L.; Valente, S.; et al. Properly Substituted Cyclic Bis-(2-bromobenzylidene) Compounds Behaved as Dual p300/CARM1 Inhibitors and Induced Apoptosis in Cancer Cells. Molecules 2020, 25, 3122. https://doi.org/10.3390/molecules25143122
Fioravanti R, Tomassi S, Di Bello E, Romanelli A, Plateroti AM, Benedetti R, Conte M, Novellino E, Altucci L, Valente S, et al. Properly Substituted Cyclic Bis-(2-bromobenzylidene) Compounds Behaved as Dual p300/CARM1 Inhibitors and Induced Apoptosis in Cancer Cells. Molecules. 2020; 25(14):3122. https://doi.org/10.3390/molecules25143122
Chicago/Turabian StyleFioravanti, Rossella, Stefano Tomassi, Elisabetta Di Bello, Annalisa Romanelli, Andrea Maria Plateroti, Rosaria Benedetti, Mariarosaria Conte, Ettore Novellino, Lucia Altucci, Sergio Valente, and et al. 2020. "Properly Substituted Cyclic Bis-(2-bromobenzylidene) Compounds Behaved as Dual p300/CARM1 Inhibitors and Induced Apoptosis in Cancer Cells" Molecules 25, no. 14: 3122. https://doi.org/10.3390/molecules25143122