
Phytocomplex Characterization and Biological Evaluation of Powdered Fruits and Leaves from Elaeagnus angustifolia

 , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
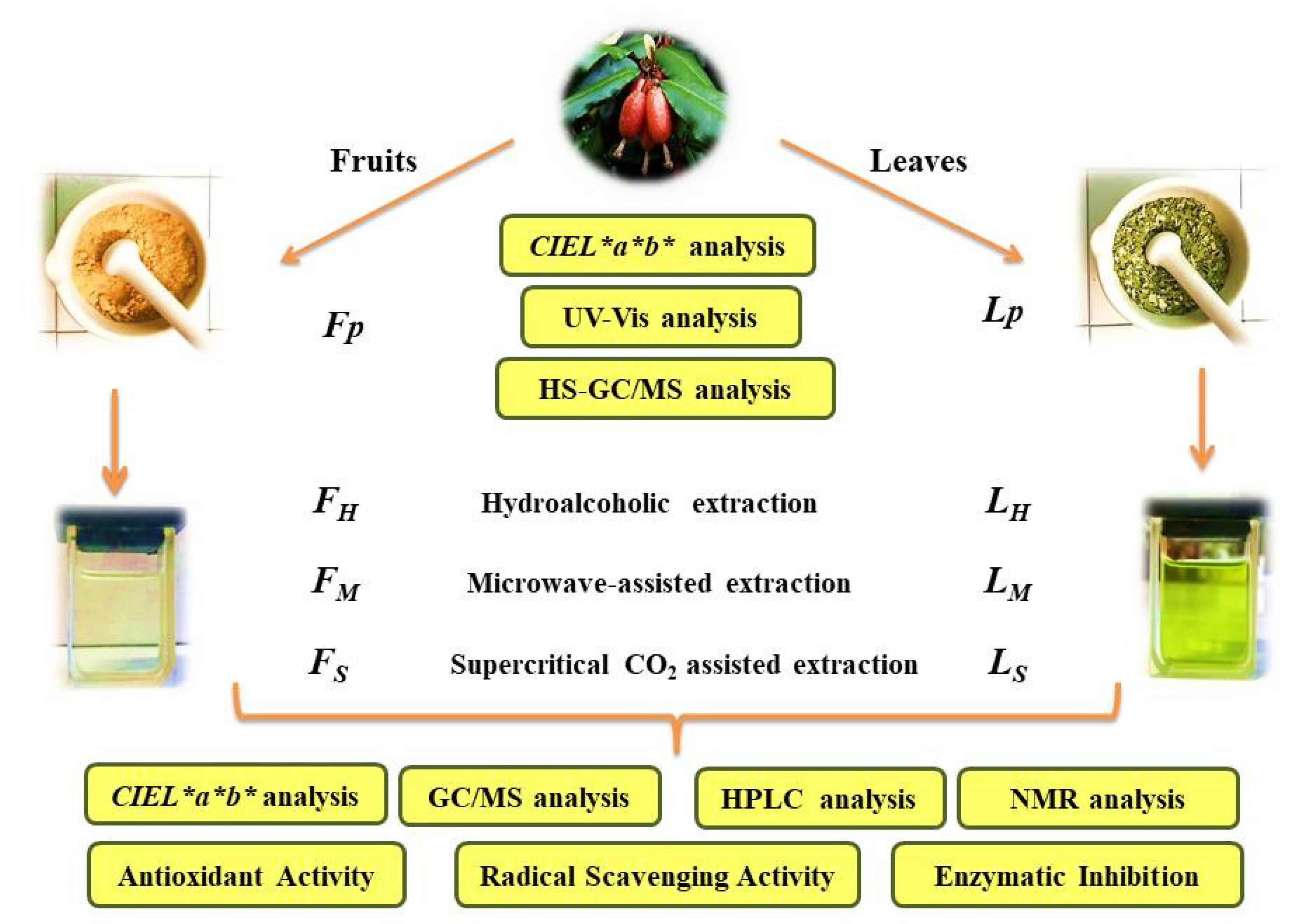
2.1. Extraction Methods
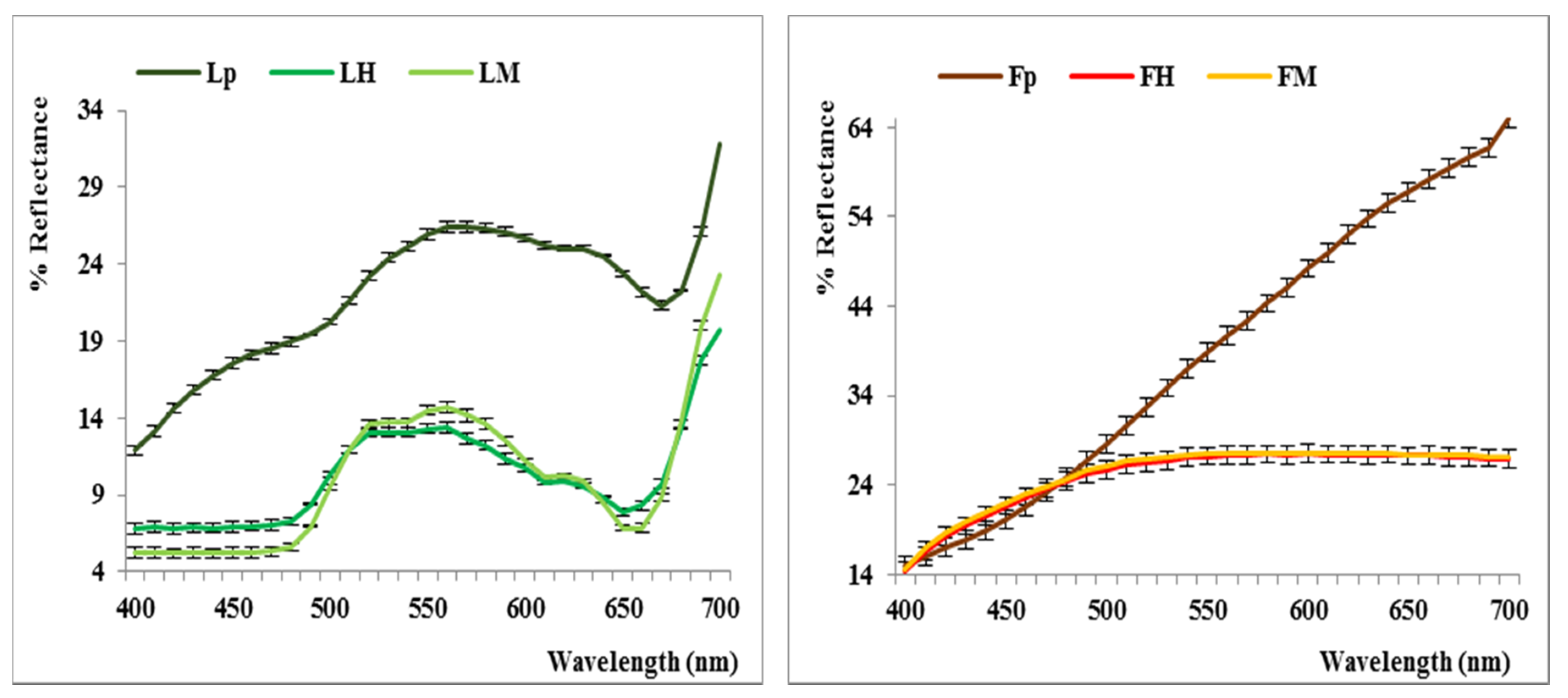
2.2. Color Analysis
2.3. Carotenoid and Chlorophyll Analysis
2.4. HS-GC/MS and GC/MS Results
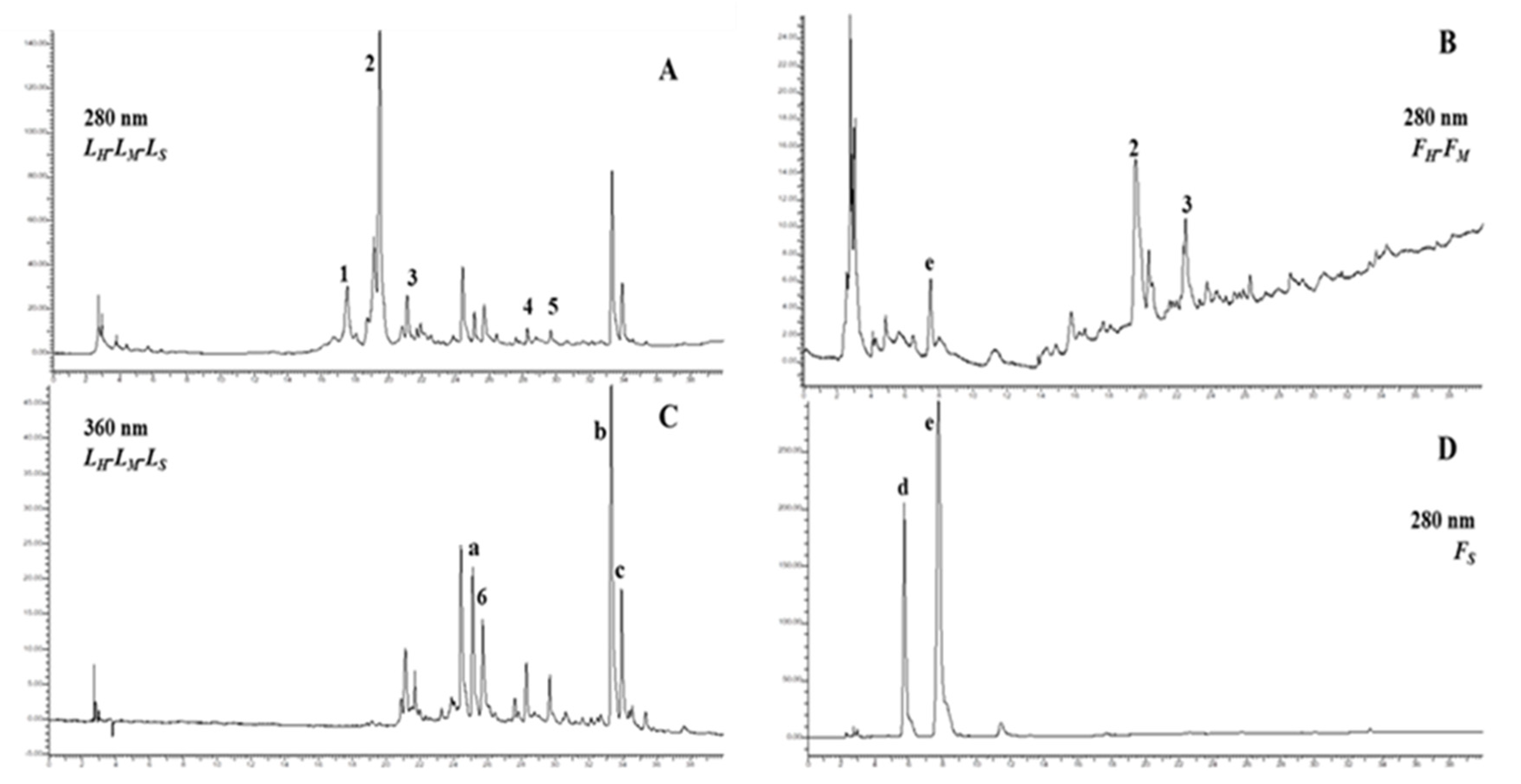
2.5. HPLC-DAD Analysis
2.6. H- and 13C-NMR Analysis
2.7. TPC, TFC, and Antioxidant In Vitro Assays
2.8. Enzyme Inhibition Assays In Vitro
3. Materials and Methods
3.1. Materials
3.2. Sample Preparation
3.3. Extraction Methods
3.3.1. Hydro-Alcoholic Extraction (HAE) by Maceration
3.3.2. Microwave-Assisted Extraction (MAE)
3.3.3. Supercritical CO2 Assisted Extraction (SCE)
3.4. Colorimetric Analysis
3.5. Carotenoid and Chlorophyll Analysis
3.6. HS-GC/MS Analysis
3.7. GC/MS Analyses
3.8. HPLC-DAD Analyses
3.9. Semipreparative HPLC-Refractive Index Detector
3.10. H- and 13C-MR Analysis
3.11. Determination of Total Bioactive Components: Total Phenolic Content (TPC) and Total Flavonoid Content (TFC)
3.12. Antioxidant and Metal Chelating Spectrophotometric Assays
3.13. Enzyme Inhibition Assays
3.14. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Katz, G.L.; Shafroth, P.B. Biology, ecology and management of Elaeagnus angustifolia L. (Russian olive) in western North America. Wetlands 2003, 23, 763–777. [Google Scholar] [CrossRef]
- Hamidpour, R.; Hamidpour, S.; Hamidpour, M.; Shahlari, M.; Sohraby, M.; Shahlari, N.; Hamidpour, R. Russian olive (Elaeagnus angustifolia L.): From a variety of traditional medicinal applications to its novel roles as active antioxidant, anti-inflammatory, anti-mutagenic and analgesic agent. J. Tradit. Complementary Med. 2017, 7, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehranizadeh, Z.A.; Baratian, A.; Hosseinzadeh, H. Russian olive (Elaeagnus angustifolia) as a herbal healer. BioImpacts 2016, 6, 155. [Google Scholar] [CrossRef] [PubMed]
- Farzaei, M.H.; Bahramsoltani, R.; Abbasabadi, Z.; Rahimi, R. A comprehensive review on phytochemical and pharmacological aspects of Elaeagnus angustifolia L. J. Pharm. 2015, 67, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, F.A.; Bertoft, E. Sugar and phenolic acid composition of stored commercial oleaster fruits. J. Food Compos. Anal. 2001, 14, 505–511. [Google Scholar] [CrossRef]
- Saboonchian, F.; Jamei, R.; Sarghein, S.H. Phenolic and flavonoid content of Elaeagnus angustifolia L. (leaf and flower). Avicenna J. Phytomedicine 2014, 4, 231. [Google Scholar]
- Carradori, S.; D’Ascenzio, M.; Chimenti, P.; Secci, D.; Bolasco, A. Selective MAO-B inhibitors: A lesson from natural products. Mol. Divers. 2014, 18, 219–243. [Google Scholar] [CrossRef]
- Torbati, M.; Asnaashari, S.; Afshar, F.H. Essential oil from flowers and leaves of Elaeagnus angustifolia (Elaeagnaceae): Composition, radical scavenging and general toxicity activities. Adv. Pharm. Bull. 2016, 6, 163. [Google Scholar] [CrossRef]
- Gürbüz, I.; Üstün, O.; Yesilada, E.; Sezik, E.; Kutsal, O. Anti-ulcerogenic activity of some plants used as folk remedy in Turkey. J. Ethnopharmacol. 2003, 88, 93–97. [Google Scholar] [CrossRef]
- Farahbakhsh, S.; Arbabian, S.; Emami, F.; Rastegar Moghadam, B.; Ghoshooni, H.; Noroozzadeh, A.; Sahraei, H.; Golmanesh, L.; Jalili, C.; Zrdooz, H. Inhibition of cyclooxygenase type 1 and 2 enzymes by aqueous extract of Elaeagnus angustifolia in mice. Basic Clin. Neurosci. 2011, 2, 31–37. [Google Scholar]
- Hosseinzadeh, H.; Ramezani, M.; Namjo, N. Muscle relaxant activity of Elaeagnus angustifolia L. fruit seeds in mice. J. Ethnopharmacol. 2003, 84, 275–278. [Google Scholar] [CrossRef]
- Faramarz, S.; Dehghan, G.; Jahanban-Esfahlan, A. Antioxidants in different parts of oleaster as a function of genotype. BioImpacts 2015, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Mahboubi, M. Elaeagnus angustifolia and its therapeutic applications in osteoarthritis. Ind. Crop. Prod. 2018, 121, 36–45. [Google Scholar] [CrossRef]
- Koohi-Hosseinabadi, O.; Ranjbar, Z.; Sepehrimanesh, M.; AndisheTadbir, A.; Poorbaghi, S.L.; Bahranifard, H.; Tanideh, N.; Koohi-Hosseinabadi, M.; Iraji, A. Biochemical, hematological, and pathological related healing effects of Elaeagnus angustifolia hydroalcoholic extract in 5-fluorouracil-induced oral mucositis in male golden hamster. Env. Sci. Pollut. Res. 2017, 24, 24447–24453. [Google Scholar] [CrossRef] [PubMed]
- Dabbaghmanesh, M.H.; Noorafshan, A.; Talezadeh, P.; Tanideh, N.; Koohpeyma, F.; Iraji, A.; Bakhshayeshkaram, M.; Montazeri-Najafabady, N. Stereological investigation of the effect of Elaeagnus angustifolia fruit hydroalcoholic extract on osteoporosis in ovariectomized rats. Avicenna J. Phytomedicine 2017, 7, 261. [Google Scholar]
- Amereh, Z.; Hatami, N.; Shirazi, F.H.; Gholami, S.; Hosseini, S.H.; Noubarani, M.; Kamalinejad, M.; Andalib, S.; Keyhanfar, F.; Reza Eskandari, M. Cancer chemoprevention by oleaster (Elaeagnus angustifolia L.) fruit extract in a model of hepatocellular carcinoma induced by diethylnitrosamine in rats. Exp. Clin. Sci. J. 2017, 16, 1046. [Google Scholar]
- Dehghan, M.H.; Soltani, J.; Farnad, M.; Kalantar, E.; Kamalinejad, M.; Khodaii, Z.; Hatami, S.; Natanzi, M.M. Characterization of an antimicrobial extract from Elaeagnus angustifolia. Int. J. Enteric Pathog. 2014, 2, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Okmen, G.; Turkcan, O. A study on antimicrobial, antioxidant and antimutagenic activities of Elaeagnus angustifolia L. leaves. Afr. J. Tradit. Complementary Altern. Med. 2014, 11, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Sharifian-Nejad, M.S.; Shekarchizadeh, H. Physicochemical and functional properties of oleaster (Elaeagnus angustifolia L.) polysaccharides extracted under optimal conditions. Int. J. Biol. Macromol. 2019, 124, 946–954. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, J.; Du, H.; Li, Q.; Chen, J.; Zhang, G.; Liu, H.; Wang, J. Structural characterization and antioxidant activities of polysaccharides extracted from the pulp of Elaeagnus angustifolia L. Int. J. Mol. Sci. 2014, 15, 11446–11455. [Google Scholar] [CrossRef]
- Du, H.; Chen, J.; Tian, S.; Gu, H.; Li, N.; Sun, Y.; Ru, J.; Wang, J.-R. Extraction optimization, preliminary characterization and immunological activities in vitro of polysaccharides from Elaeagnus angustifolia L. pulp. Carbohydr. Polym. 2016, 151, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Rawson, A.; Tiwari, B.K.; Brunton, N.; Brennan, C.; Cullen, P.J.; O’donnell, C.P. Application of supercritical carbon dioxide to fruit and vegetables: Extraction, processing, and preservation. Food Rev. Int. 2012, 28, 253–276. [Google Scholar] [CrossRef]
- Lucien, F.P.; Foster, N.R. Solubilities of solid mixtures in supercritical carbon dioxide: A review. J. Supercrit. Fluids 2000, 17, 111–134. [Google Scholar] [CrossRef]
- Witowski, J.; Jorres, A. Glucose degradation products: Relationship with cell damage. Perit. Dial. Int. 2000, 20, S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Beejmohun, V.; Fliniaux, O.; Grand, É.; Lamblin, F.; Bensaddek, L.; Christen, P.; Kovensky, J.; Fliniaux, M.-A.; Mesnard, F. Microwave-assisted extraction of the main phenolic compounds in flaxseed. Phytochem. Anal.: Int. J. Plant. Chem. Biochem. Tech. 2007, 18, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Cairone, F.; Carradori, S.; Locatelli, M.; Casadei, M.A.; Cesa, S. Reflectance colorimetry: A mirror for food quality-a mini review. Eur. Food Res. Technol. 2020, 246, 259–272. [Google Scholar] [CrossRef]
- Patsilinakos, A.; Ragno, R.; Carradori, S.; Petralito, S.; Cesa, S. Carotenoid content of Goji berries: CIELAB, HPLC-DAD analyses and quantitative correlation. Food Chem. 2018, 268, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Boutaoui, N.; Zaiter, L.; Benayache, F.; Benayache, S.; Carradori, S.; Cesa, S.; Giusti, A.M.; Campestre, C.; Menghini, L.; Innosa, D.; et al. Qualitative and quantitative phytochemical analysis of different extracts from Thymus algeriensis aerial parts. Molecules 2018, 23, 463. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Chung, H.Y. GC-MS analysis of the volatile components in dried boxthorn (Lycium chinensis) fruit. J. Korean Soc. Appl. Biol. Chem. 2009, 52, 516–524. [Google Scholar] [CrossRef]
- Bucur, L.; Vlase, L.; Istudor, V.; Popescu, A. HPLC-MS analysis of the polyphenols in two soft extracts of Elaeagnus angustifolia L. Note 2. Soft extract of young branches analysis. Farmacia 2009, 57, 736–742. [Google Scholar]
- Ge, Y.; Zhang, F.; Qin, Q.; Shang, Y.; Wan, D. In vivo evaluation of the antiasthmatic, antitussive, and expectorant activities and chemical components of three Elaeagnus leaves. Evid.-Based Complementary Altern. Med. 2015, 2015, 428208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, S.M.; Kabir, T.M.; Niaz, K.; Jeandet, P.; Clément, C.; Mathew, B.; Rauf, A.; Rengasamy, K.R.R.; Sobarzo-Sánchez, E.; Ashraf, G.M.; et al. Molecular insight into the therapeutic promise of flavonoids against Alzheimer’s disease. Molecules 2020, 25, 1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balli, D.; Cecchi, L.; Khatib, M.; Bellumori, M.; Cairone, F.; Carradori, S.; Zengin, G.; Cesa, S.; Innocenti, M.; Mulinacci, N. Characterization of arils juice and peel decoction of fifteen varieties of Punica granatum L.: A focus on anthocyanins, ellagitannins and polysaccharides. Antioxidants 2020, 9, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.X.; Wen, L.; Zhong, W.J.; Xiong, L.; Liang, J.; Wang, H.L. Quercetin, kaempferol and isorhamnetin in Elaeagnus pungens Thunb. leaf: Pharmacological activities and quantitative determination studies. Chem. Biodivers. 2018, 15, e1800129. [Google Scholar] [CrossRef]
- Murai, N.; Yonaga, M.; Tanaka, K. Palladium-catalyzed direct hydroxymethylation of aryl halides and triflates with potassium acetoxymethyltrifluoroborate. Org. Lett. 2012, 14, 1278–1281. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; González-Paramás, A.M.; Buzati Pereira, B.L.; Hammami, R.; Power, K.A.; Bordenave, N. Plant phenolics as fuctional food ingredients. Adv. Food Nutr. Res. 2019, 90, 183–257. [Google Scholar]
- Kwon, S.H.; Lee, H.K.; Kim, J.A.; Hong, S.I.; Kim, H.C.; Jo, T.H.; Park, Y.I.; Lee, C.K.; Kim, Y.B.; Lee, S.Y.; et al. Neuroprotective effects of chlorogenic acid on scopolamine-induced amnesia via anti-acetylcholinesterase and anti-oxidative activities in mice. Eur. J. Pharm. 2010, 649, 210–217. [Google Scholar] [CrossRef]
- de Freitas, M.M.; Fontes, P.R.; Souza, P.M.; William Fagg, C.; Neves Silva Guerra, E.; de Medeiros Nóbrega, Y.K.; Silveira, D.; Fonseca-Bazzo, Y.; Simeoni, L.A.; Homem-de-Mello, M.; et al. Extracts of Morus nigra L. leaves standardized in chlorogenic acid, rutin and isoquercitrin: Tyrosinase inhibition and cytotoxicity. PLoS ONE 2016, 11, e0163130. [Google Scholar] [CrossRef]
- Ko, R.K.; Kim, G.O.; Hyun, C.G.; Jung, D.S.; Lee, N.H. Compounds with tyrosinase inhibition, elastase inhibition and DPPH radical scavenging activities from the branches of Distylium racemosum Sieb. et Zucc. Phytother. Res. 2011, 25, 1451–1456. [Google Scholar] [CrossRef]
- Maruyama, H.; Kawakami, F.; Lwin, T.T.; Imai, M.; Shamsa, F. Biochemical characterization of ferulic acid and caffeic acid which effectively inhibit melanin synthesis via different mechanisms in B16 melanoma cells. Biol. Pharm. Bull. 2018, 41, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Caro-Ordieres, T.; Marín-Royo, G.; Opazo-Ríos, L.; Jiménez-Castilla, L.; Moreno, J.A.; Gómez-Guerrero, C.; Egido, J. The coming age of flavonoids in the treatment of diabetic complications. J. Clin. Med. 2020, 9, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assefa, S.T.; Yang, E.Y.; Chae, S.Y.; Song, M.; Lee, J.; Cho, M.C.; Jang, S. Alpha glucosidase inhibitory activities of plants with focus on common vegetables. Plants 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesa, S.; Carradori, S.; Bellagamba, G.; Locatelli, M.; Casadei, M.A.; Masci, A.; Paolicelli, P. Evaluation of processing effects on anthocyanin content and colour modifications of blueberry (Vaccinium spp.) extracts: Comparison between HPLC-DAD and CIELAB analyses. Food Chem. 2017, 232, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, A.P.; Mannina, L.; Capitani, D.; Sanzò, G.; Ingallina, C.; Botta, B.; Fornarini, S.; Crestoni, M.E.; Chiavarino, B.; Carradori, S.; et al. A multi-methodological approach in the study of Italian PDO “Cornetto di Pontecorvo” red sweet pepper. Food Chem. 2018, 255, 120–131. [Google Scholar] [CrossRef]
- Oliva, A.; Costantini, S.; De Angelis, M.; Garzoli, S.; Božović, M.; Mascellino, M.; Vullo, V.; Ragno, R. High potency of Melaleuca alternifolia essential oil against multi-drug resistant Gram-negative bacteria and methicillin-resistant Staphylococcus aureus. Molecules 2018, 23, 2584. [Google Scholar] [CrossRef] [Green Version]
- Oliva, A.; Garzoli, S.; De Angelis, M.; Marzuillo, C.; Vullo, V.; Mastroianni, C.M.; Ragno, R. In-vitro evaluation of different antimicrobial combinations with and without colistin against carbapenem-resistant Acinetobacter baumannii. Molecules 2019, 24, 886. [Google Scholar] [CrossRef] [Green Version]
- Garzoli, S.; Turchetti, G.; Giacomello, P.; Tiezzi, A.; Laghezza Masci, V.; Ovidi, E. Liquid and vapour phase of Lavandin (Lavandula × intermedia) essential oil: Chemical composition and antimicrobial activity. Molecules 2019, 24, 2701. [Google Scholar] [CrossRef] [Green Version]
- Garzoli, S.; Laghezza Masci, V.; Turchetti, G.; Pesci, L.; Tiezzi, A.; Ovidi, E. Chemical investigations of male and female leaf extracts from Schinus molle L. Nat. Prod. Res. 2019, 33, 1980–1983. [Google Scholar] [CrossRef]
- Zengin, G.; Menghini, L.; Di Sotto, A.; Mancinelli, R.; Sisto, F.; Carradori, S.; Cesa, S.; Fraschetti, C.; Filippi, A.; Angiolella, L.; et al. Chromatographic analyses, in vitro biological activities and cytotoxicity of Cannabis sativa L. essential oil: A multidisciplinary study. Molecules 2018, 23, 3266. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the material are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Lp | LH | LM | Fp | FH | FM | |
|---|---|---|---|---|---|---|
| L* | 56.29 ± 1.52 | 40.52 ± 0.33 | 41.22 ± 2.44 | 68.61 ± 0.64 | 58.66 ± 2.46 | 58.90 ± 1.46 |
| a* | −1.47 ± 0.49 | −7.80 ± 0.49 | −8.98 ± 3.26 | 7.73 ± 0.02 | −2.15 ± 0.20 | −2.30 ± 0.32 |
| b* | 13.14 ± 1.68 | 14.41 ± 1.72 | 22.41 ± 4.58 | 27.23 ± 0.31 | 8.62 ± 0.16 | 8.46 ± 3.07 |
| C* | 13.24 ± 1.61 | 16.39 ± 1.73 | 24.18 ± 5.45 | 28.31 ± 0.29 | 8.88 ± 0.12 | 8.78 ± 3.04 |
| hab | 96.62 ± 2.96 | 118.57 ± 1.58 | 111.22 ± 3.36 | 74.14 ± 0.18 | 104.01 ± 1.46 | 106.29 ± 3.70 |
| Lp * | Fp * | |
|---|---|---|
| Chlorophyll a | 43.8 ± 8.6 | 1.0 ± 0.4 |
| Chlorophyll b | 24.0 ± 4.0 | 1.9 ± 0.2 |
| Total carotenoids | 18.3 ± 2.5 | 3.2 ± 0.6 |
| N° | Compound 1 | LRI 2 | LRIlit 3 | FP (%) | LP (%) |
|---|---|---|---|---|---|
| 1 | dihydro-2-methyl-furanone | 773 | 775 | 5.8 | - |
| 2 | pyrimidine, 2-methyl | 790 | + | - | 7.2 |
| 3 | furfural | 798 | 802 | 36.7 | 18.8 |
| 4 | 2-furanmethanol | 833 | 835 | 7.0 | 5.8 |
| 5 | acetol acetate | 858 | 862 | 0.2 | 3.2 |
| 6 | butyrolactone | 861 | 863 | - | - |
| 7 | p-xylene | 867 | 869 | - | 5.3 |
| 8 | 2(5H)-furanone | 868 | 871 | 1.2 | - |
| 9 | 4-cyclopentene-1,3-dione | 880 | 884 | 4.5 | - |
| 10 | acetyl furan | 910 | 914 | 2.5 | 1.8 |
| 11 | pyrrolidine, 2-(cyanomethylene) | 922 | + | - | 8.1 |
| 12 | 5-methylfurfural, | 930 | 933 | 25.3 | 9.6 |
| 13 | 5-hepten-2-one, 6-methyl- | 956 | 962 | - | 2.0 |
| 14 | 2-furanmethanol, acetate | 960 | 966 | 0.4 | - |
| 15 | 2-ethyl-6-metylpyrazine | 982 | 981 | - | 2.1 |
| 16 | 2,4-dihydroxy-2,5-dimethyl-3-(2H)-furanone | 980 | 989° | 1.0 | 3.2 |
| 17 | 2-cyclopenten-1-one, 2-hydroxy-3-methyl- | 997 | 1000 | 0.7 | - |
| 18 | 1H-pyrrole-2-carboxaaldehyde | 1004 | 1009 | - | 6.4 |
| 19 | 2,5-furandione, 3,4-dimethyl- | 1032 | 1038 | 1.4 | - |
| 20 | 2,5-dimethyl-4-hydroxy-3(2H)-furanone | 1062 | 1064 | 4.7 | 3.2 |
| 21 | 2-acetylpyrrole | 1066 | 1065 | 0.9 | 4.3 |
| 22 | nicotinyl acetate | 1103 | 1100 | - | - |
| 23 | pyrimidine-4,6-diol,5-methyl | 1105 | - | 1.1 | - |
| 24 | pyranone | 1110 | 1107 | 1.8 | - |
| 25 | 5-hydroxymethylfurfural | 1202 | 1208 | 2.7 | - |
| 26 | p-vinylguiacol | 1287 | 1282 | - | 9.3 |
| 27 | 5-acetoxymethyl-2-furaldheyde | 1305 | * | 0.6 | - |
| 28 | naphthalene,1,2-dihydro-1,1,6-trimethyl- | 1328 | 1332° | - | 3.7 |
| 29 | geranylacetone | 1429 | 1426 | - | 1.7 |
| 30 | dehydro β-ionone | 1435 | 1433 | - | 1.2 |
| 31 | dihydroactinidiolid | 1461 | 1458 | - | 1.4 |
| 32 | hexahydrofarnesyl acetone | 1850 | 1846 | - | 1.2 |
| Total (%) | 98.5 | 99.5 |
| N° | Compound 1 | LRI 2 | LRIlit 3 | FM (%) | LM (%) | FH (%) | LH (%) |
|---|---|---|---|---|---|---|---|
| 1 | Acetol | 1319 | 1317 | 6.4 | 20.4 | 18.6 | 24.0 |
| 2 | methyl pyruvate | 1322 | * | - | 7.5 | - | 11.4 |
| 3 | acetic acid | 1440 | 1442 | - | 14.1 | - | 16.0 |
| 4 | furfural | 1458 | 1465 | 28.7 | - | 2.3 | - |
| 5 | acetylfuran | 1495 | 1497 | 2.1 | - | 2.9 | - |
| 6 | 5-methyl furfural | 1600 | 1604 | 11.3 | - | 8.9 | - |
| 7 | 2-furanmethanol | 1655 | 1659 | 7.7 | - | 18.2 | - |
| 8 | ionone | 1840 | 1846 | - | 10.0 | - | 8.9 |
| 9 | furaneol | 2056 | 2060 | 16.9 | - | 5.4 | - |
| 10 | p-vinylguaiacol | 2160 | 2166 | - | 48.0 | - | 39.5 |
| 11 | 5-acetoxymethyl-2-furaldheyde | 2195 | 2199 | 4.7 | - | 4.7 | - |
| 12 | pyranone | 2271 | 2274 | 10.9 | - | 17.3 | - |
| 13 | 5-hydroxymethylfurfural | 2529 | 2532 | 11.3 | - | 21.6 | - |
| Total (%) | 100.0 | 100.0 | 99.9 | 99.8 |
| LH | LM | LS | FH | FM | FS | |
|---|---|---|---|---|---|---|
| Epicatechin | 13.30 ± 2.60 | 43.10 ± 1.70 | nd | nd | nd | nd |
| Chlorogenic acid | 40.8 ± 1.52 | 41.9 ± 1.32 | 4.85 ± 0.10 | 1.10 ± 0.20 | 3.50 ± 0.80 | nd |
| Caffeic acid | 2.40 ± 0.12 | 3.18 ± 0.50 | nd | 0.19 ± 0.06 | 0.40 ± 0.10 | nd |
| p-Coumaric acid | 0.18 ± 0.06 | 0.20 ± 0.04 | nd | nd | nd | nd |
| Ferulic acid | 0.50 ± 0.09 | 0.70 ± 0.08 | nd | nd | nd | nd |
| Flavonoids | 5.40 ± 0.35 | 6.79 ± 0.10 | 0.80 ± 0.08 | nd | nd | nd |
| Samples |
TPC (mg GAE/g) |
TFC (mg RE/g) |
DPPH (mg TE/g) |
ABTS (mg TE/g) |
CUPRAC (mg TE/g) |
FRAP (mg TE/g) |
Metal Chelating Ability (mg EDTAE/g) | Phosphomolybdenum Assay (mmol TE/g) |
|---|---|---|---|---|---|---|---|---|
| FH | 5.15 ± 0.03 d | 0.22 ± 0.02 d | na | 5.06 ± 0.05 d | 11.92 ± 0.09 c | 6.47 ± 0.03 c | 2.64 ± 0.30 c | 0.64 ± 0.02 d |
| FM | 4.84 ± 0.05 d | 0.21 ± 0.08 d | na | 3.62 ± 0.39 e | 12.34 ± 0.51 c | 6.51 ± 0.10 c | 2.43 ± 0.25 c | 0.56 ± 0.03 d |
| FS | 3.32 ± 0.04 e | 0.17 ± 0.02 d | na | 2.22 ± 0.21 f | 11.59 ± 0.07 c | 6.03 ± 0.04 c | 2.97 ± 0.16 c | 0.50 ± 0.02 d |
| LH | 65.35 ± 0.52 a | 32.91 ± 0.12 b | 48.93 ± 0.06a | 72.44 ± 0.40 a | 86.67 ± 2.51 a | 45.66 ± 0.67 a | 11.97 ± 1.36 b | 2.46 ± 0.14 b |
| LM | 57.67 ± 0.09 b | 36.58 ± 0.35 a | 48.85 ± 0.01 a | 69.64 ± 0.38 b | 86.24 ± 1.85 a | 45.40 ± 0.86 a | 11.13 ± 1.14 b | 2.20 ± 0.08 c |
| LS | 17.86 ± 0.70 c | 24.34 ± 0.24 c | 2.57 ± 0.29 b | 13.18 ± 0.40 c | 67.03 ± 1.29 b | 20.33 ± 0.19 b | 18.25 ± 0.44 a | 2.73 ± 0.14 a |
| Samples | AChE Inhibition (mg GALAE/g) | BChE Inhibition (mg GALAE/g) | Tyrosinase Inhibition (mg KAE/g) | α-Amylase Inhibition (mmol ACAE/g) | α-Glucosidase Inhibition (mmol ACAE/g) |
|---|---|---|---|---|---|
| FH | 1.72 ± 0.08 c | 3.37 ± 0.31 b | 35.42 ± 0.75 c | 0.15 ± 0.01 e | na |
| FM | 2.02 ± 0.10 b | 3.51 ± 0.19 b | 37.53 ± 2.62 b,c | 0.22 ± 0.01 d | na |
| FS | 2.54 ± 0.01 a | 4.53 ± 0.10 a | 37.21 ± 2.35 b,c | 0.38 ± 0.01 c | 0.69 ± 0.02 b |
| LH | 2.08 ± 0.07 b | 1.11 ± 0.06 c | 61.20 ± 3.65 a | 0.39 ± 0.01 c | 0.83 ± 0.01 a |
| LM | 2.21 ± 0.04 b | 1.48 ± 0.13 c | 58.84 ± 1.02 a | 0.45 ± 0.01 b | 0.85 ± 0.01 a |
| LS | 0.88 ± 0.09 d | 4.81 ± 0.30 a | 43.21 ± 3.56 b | 0.60 ± 0.02 a | 0.65 ± 0.01 c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carradori, S.; Cairone, F.; Garzoli, S.; Fabrizi, G.; Iazzetti, A.; Giusti, A.M.; Menghini, L.; Uysal, S.; Ak, G.; Zengin, G.; et al. Phytocomplex Characterization and Biological Evaluation of Powdered Fruits and Leaves from Elaeagnus angustifolia. Molecules 2020, 25, 2021. https://doi.org/10.3390/molecules25092021
Carradori S, Cairone F, Garzoli S, Fabrizi G, Iazzetti A, Giusti AM, Menghini L, Uysal S, Ak G, Zengin G, et al. Phytocomplex Characterization and Biological Evaluation of Powdered Fruits and Leaves from Elaeagnus angustifolia. Molecules. 2020; 25(9):2021. https://doi.org/10.3390/molecules25092021
Chicago/Turabian StyleCarradori, Simone, Francesco Cairone, Stefania Garzoli, Giancarlo Fabrizi, Antonia Iazzetti, Anna Maria Giusti, Luigi Menghini, Sengul Uysal, Gunes Ak, Gokhan Zengin, and et al. 2020. "Phytocomplex Characterization and Biological Evaluation of Powdered Fruits and Leaves from Elaeagnus angustifolia" Molecules 25, no. 9: 2021. https://doi.org/10.3390/molecules25092021