All melting points were taken on a Büchi apparatus and were uncorrected. NMR spectra were recorded on a Bruker Avance 400 spectrometer (400 MHz for 1H NMR, 100 MHz for 13C NMR) using residual solvent such as chloroform (δ = 7.26) as the internal standard. 1H and 13C chemical shifts are expressed in ppm (δ) referring to TMS. Spectral data are reported using the following abbreviations: s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, t = triplet and m = multiplet and coupling constants are reported in Hz, followed by integration. Chromatographic separations were performed on a silica gel column by flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck). Yields were given after purification, unless otherwise stated.
Compounds were named following IUPAC rules as applied by ChemBioDraw Ultra 14.0 software (Cambridgesoft, Perkin Elmer, Milan, Italy). When reactions were performed in anhydrous conditions, the mixtures were maintained under nitrogen. Free bases 15–28 were transformed into the hydrochloride by treatment with a solution of acetyl chloride (1.1 equivalents) in anhydrous CH3OH. The salts were crystallized from abs. ethanol/petroleum ether.
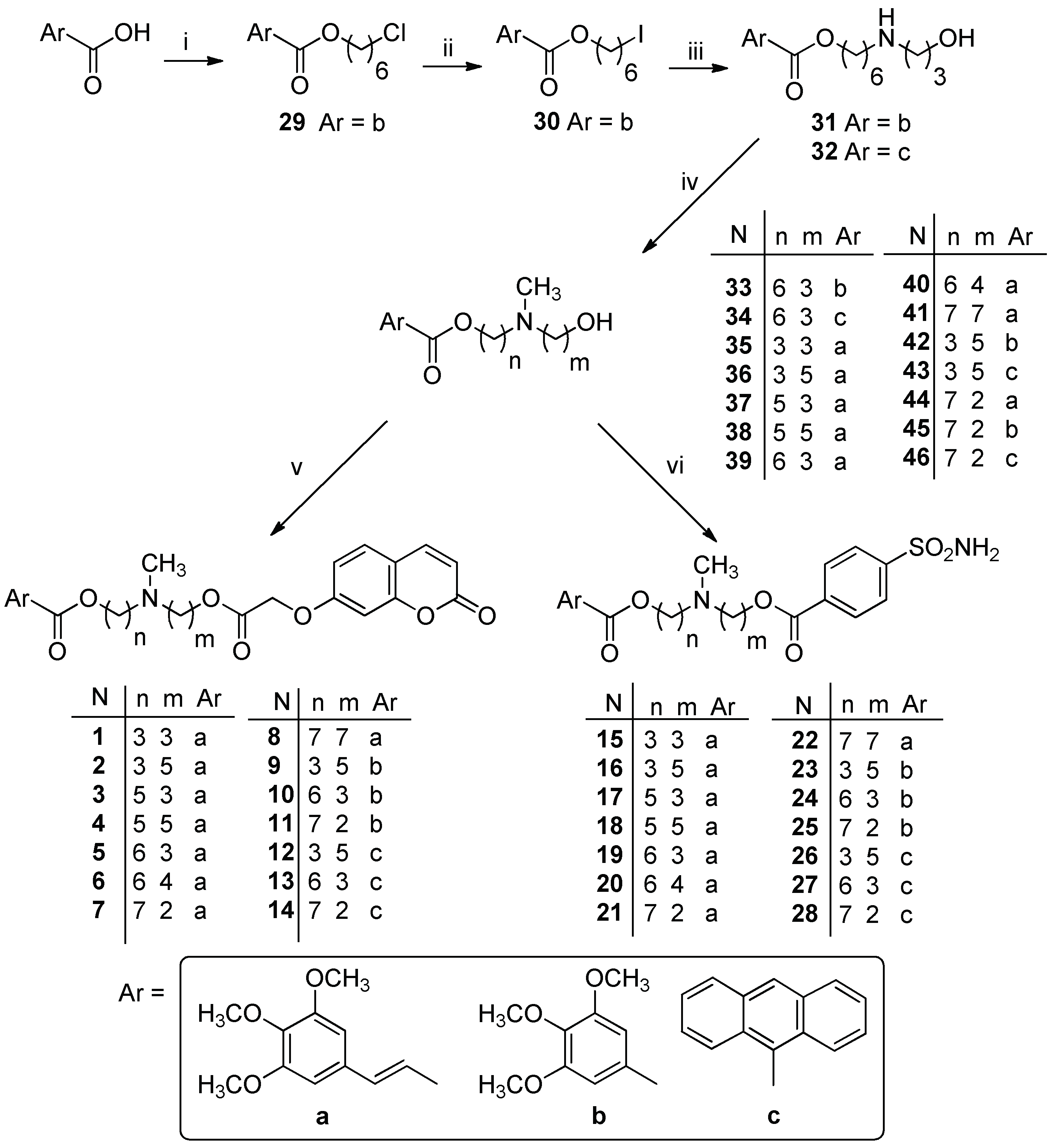
3.1.1. General Procedure for the Synthesis of Diester Compounds 1–14
To a solution of 48 (0.26 mmol) in 25 mL of anhydrous CH3CN, 0.33 mmol of EDC hydrochloride and 0.33 mmol of HOBt were added. The mixture was stirred at room temperature for 1 h, and then the suitable (hydroxyalkyl) methylaminoester 33–46 (0.22 mmol) dissolved in 5 mL of anhydrous CH3CN was added. The reaction mixture was stirred for 4 h at room temperature and the solvent was removed under reduce pressure. Then CH2Cl2 was added and the organic layer was washed twice with a saturated solution of NaHCO3. After drying with Na2SO4, the solvent was removed under reduced pressure. The crude product was then purified by flash chromatography, using the proper eluting system, yielding the desired compound as an oil.
(E)-3-(methyl(3-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)propyl)amino)propyl 3-(3,4,5-trimethoxyphenyl)acrylate1. From (hydroxyalkyl)methylaminoester
35 [35]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 95:5. Yield: 37%.
1H-NMR (400 MHz, CDCl
3) δ: 7.63 (d,
J = 9.4 Hz, 1H, CH); 7.58 (d,
J = 16.0 Hz, 1H, C
H=CH); 7.38 (d,
J = 8.4 Hz, 1H, CH); 6.87 (dd,
J = 8.4, 2.2 Hz, 1H, CH); 6.77 (d,
J = 2.2 Hz, 1H, CH); 6.74 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, C
H=CH); 6.26 (d,
J = 9.4 Hz, 1H, CH); 4.68 (s, 2H, OCH
2); 4.30–4.22 (m, 4H, OCH
2); 3.88 (s, 6H, OCH
3); 3.87 (s, 3H, OCH
3); 2.47–2.35 (m, 4H, NCH
2); 2.22 (s, 3H, NCH
3); 1.87–1.81 (m, 4H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.97 (C); 166.95 (C); 160.84 (C); 155.62 (C); 153.44 (C); 144.73 (CH); 143.16 (CH); 129.88 (C); 128.97 (CH); 117.31 (CH); 113.78 (CH); 133.33 (C); 112.80 (CH); 105.27 (CH); 101.70 (CH); 65.34 (CH
2); 64.02 (CH
2); 62.85 (CH
2); 60.95 (OCH
3); 56.17 (OCH
3); 54.20 (CH
2); 53.85 (CH
2)
; 41.95 (NCH
3); 26.67 (CH
2); 26.47 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
30H
36NO
10 = 570.2334, found 570.2340.
(E)-3-(methyl(5-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)pentyl)amino)propyl 3-(3,4,5-trimethoxyphenyl)acrylate2. From
36 [
37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 93:7. Yield: 29%.
1H-NMR (400 MHz, CDCl
3) δ: 7.60 (d,
J = 9.6 Hz, 1H, CH); 7.56 (d,
J = 16.0 Hz, 1H, C
H=CH); 7.36 (d,
J = 8.4 Hz, 1H, CH); 6.85 (dd,
J = 8.4, 2.2 Hz, 1H, CH); 6.74 (d,
J = 2.2 Hz, 1H, CH); 6.72 (s, 2H, CH); 6.31 (d,
J = 16.0 Hz, 1H, C
H=CH); 6.23 (d,
J = 9.6 Hz, 1H, CH); 4.66 (s, 2H, OCH
2); 4.23–4.16 (m, 4H, OCH
2); 3.85 (s, 6H, OCH
3); 3.84 (s, 3H, OCH
3); 2.42 (t,
J = 6.8 Hz, 2H, NCH
2); 2.30 (t,
J = 6.8 Hz, 2H, NCH
2); 2.19 (s, 3H, NCH
3); 1.88–1.83 (m, 2H, CH
2); 1.71–1.60 (m, 2H, CH
2); 1.50–1.40 (m, 2H, CH
2); 1.38–1.28 (m, 2H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.98 (C); 166.92 (C); 160.81 (C); 155.63 (C); 153.41 (C); 144.65 (CH); 143.18 (CH); 129.87 (C); 128.95 (CH); 117.33 (CH); 113.70 (CH); 113.29 (C); 112.79 (CH); 105.26 (CH); 101.71 (CH); 65.63 (CH
2); 65.32 (CH
2); 62.98 (CH
2); 60.92 (OCH
3); 57.45 (CH
2); 56.15 (OCH
3); 54.19 (CH
2); 42.08 (NCH
3); 28.42 (CH
2); 26.87 (CH
2); 26.67 (CH
2); 23.67 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
32H
40NO
10 = 598.2647, found 598.2651.
(E)-5-(methyl(3-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)propyl)amino)pentyl 3-(3,4,5-trimethoxyphenyl)acrylate3. From
37 [37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 95:5. Yield: 33%.
1H-NMR (400 MHz, CDCl
3) δ: 7.59 (d,
J = 9.6 Hz, 1H, CH); 7.53 (d,
J = 15.6 Hz, 1H, CH=CH); 7.34 (d,
J = 8.4 Hz, 1H, CH); 6.82 (dd,
J = 8.4, 2.2 Hz, 1H, CH); 6.72 (d,
J = 2.2 Hz, 1H, CH); 6.70 (s, 2H, CH); 6.30 (d,
J = 15.6 Hz, 1H, CH=CH); 6.21 (d,
J = 9.6 Hz, 1H, CH); 4.64 (s, 2H, OCH
2); 4.22 (t,
J = 6.4 Hz, 2H, OCH
2); 4.15 (t,
J = 6.4 Hz, 2H, OCH
2); 3.83 (s, 6H, OCH
3); 3.82 (s, 3H, OCH
3); 2.34 (t,
J = 6.8 Hz, 2H, NCH
2); 2.29 (t,
J = 7.2 Hz, 2H, NCH
2); 2.15 (s, 3H, NCH
3); 1.82–1.75 (m, 2H, CH
2); 1.69–1.64 (m, 2H, CH
2); 1.52–1.32 (m, 4H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.95 (C); 166.97 (C); 160.77 (C); 155.61 (C); 153.38 (C); 144.58 (CH); 143.20 (CH); 140.04 (C); 129.90 (C); 128.98 (CH); 117.41 (CH); 113.68 (CH); 113.29 (C); 112.73 (CH); 105.21 (CH); 101.68 (CH); 65.29 (CH
2); 64.47 (CH
2); 64.06 (CH
2); 60.90 (OCH
3); 57.51 (CH
2); 56.12 (OCH
3); 53.88 (CH
2); 41.97 (NCH
3); 28.64 (CH
2); 26.81 (CH
2); 26.37 (CH
2); 23.81 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
32H
40NO
10 = 598.2647, found 598.2642.
(E)-5-(methyl(5-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)pentyl)amino)pentyl 3-(3,4,5-trimethoxyphenyl)acrylate4. From
38 [35]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 95:5. Yield: 46%.
1H-NMR (400 MHz, CDCl
3) δ: 7.62 (d,
J = 9.6 Hz, 1H, CH); 7.57 (d,
J = 16.0 Hz, 1H, CH=CH); 7.38 (d,
J = 8.8 Hz, 1H, CH); 6.87 (dd,
J = 8.8, 2.2 Hz, 1H, CH); 6.76 (d,
J = 2.2 Hz, 1H, CH); 6.74 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, CH=CH); 6.25 (d,
J = 9.6 Hz, 1H, CH); 4.67 (s, 2H, OCH
2); 4.22–4.15 (m, 4H, OCH
2); 3.87 (s, 6H, OCH
3); 3.86 (s, 3H, OCH
3); 2.35–2.29 (m, 4H, NCH
2); 2.20 (s, 3H, NCH
3); 1.75–1.63 (m, 4H, CH
2); 1.55–1.30 (m, 8H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.99 (C); 167.01 (C); 160.86 (C); 160.82 (C); 155.66 (C); 153.43 (C); 144.61 (CH); 143.20 (CH); 140.21 (C); 129.93 (C); 128.97 (CH); 117.44 (CH); 113.76 (CH); 113.32 (C); 112.85 (CH); 105.24 (CH); 101.70 (CH); 65.66 (CH
2); 65.34 (CH
2); 64.54 (CH
2); 60.95 (OCH
3); 57.64 (CH
2); 57.54 (CH
2); 56.16 (OCH
3); 42.15 (NCH
3); 28.70 (CH
2); 28.45 (CH
2); 26.88 (CH
2); 26.80 (CH
2); 23.97 (CH
2); 23.77 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
34H
44NO
10 = 626.2960, found 626.2951.
(E)-6-(methyl(3-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)propyl)amino)hexyl 3-(3,4,5-trimethoxyphenyl)acrylate5. From
39 [
38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 96:4. Yield: 80%.
1H-NMR (400 MHz, CDCl
3) δ: 7.61 (d,
J = 9.6 Hz, 1H, CH); 7.56 (d,
J = 16.0 Hz, 1H, CH=CH); 7.36 (d,
J = 8.4 Hz, 1H, CH); 6.85 (dd,
J = 8.4, 2.2 Hz, 1H, CH); 6.74 (d,
J = 2.2 Hz, 1H, CH); 6.72 (s, 2H, CH); 6.32 (d,
J = 16.0 Hz, 1H, CH=CH); 6.24 (d,
J = 9.6 Hz, 1H, CH,); 4.66 (s, 2H, OCH
2); 4.24 (t,
J = 6.4 Hz, 2H, OCH
2); 4.16 (t,
J = 6.4 Hz, 2H, OCH
2); 3.85 (s, 6H, OCH
3); 3.84 (s, 3H, OCH
3); 2.35 (t,
J = 7.2 Hz, 2H, NCH
2); 2.28 (t,
J = 7.6 Hz, 2H, NCH
2); 2.16 (s, 3H, NCH
3); 1.84–1.77 (m, 2H, CH
2); 1.70–1.64 (m, 2H, CH
2); 1.47–1.30 (m, 6H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.96 (C); 167.01 (C); 160.84 (C); 160.79 (C); 155.64 (C); 153.41 (C); 144.57 (CH); 143.20 (CH); 140.06 (C); 129.93 (C); 128.98 (CH); 117.45 (CH); 113.73 (CH); 113.31 (C); 112.77 (CH); 105.21 (CH); 101.70 (CH); 65.31 (CH
2); 64.55 (CH
2); 64.15 (CH
2); 60.93 (OCH
3); 57.62 (CH
2); 56.14 (OCH
3); 53.91 (CH
2); 42.01 (NCH
3); 28.71 (CH
2); 27.09 (CH
2); 26.40 (CH
2); 25.90 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
33H
42NO
10 = 612.2803, found 612.2794.
(E)-6-(methyl(4-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)butyl)amino)hexyl 3-(3,4,5-trimethoxyphenyl)acrylate6. From
40 [
38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 97:3:0.3. Yield: 100%.
1H-NMR (400 MHz, CDCl
3) δ: 7.61 (d,
J = 9.6 Hz, 1H, CH); 7.55 (d,
J = 16.0 Hz, 1H, CH=CH); 7.36 (d,
J = 8.4 Hz, 1H, CH); 6.84 (dd,
J = 8.4, 2.2 Hz, 1H, CH); 6.73 (d,
J = 2.2 Hz, 1H, CH); 6.71 (s, 2H, CH); 6.31 (d,
J = 16.0 Hz, 1H, CH=CH); 6.23 (d,
J = 9.6 Hz, 1H, CH); 4.65 (s, 2H, OCH
2); 4.21–4.14 (m, 4H, OCH
2); 3.85 (s, 6H, OCH
3); 3.84 (s, 3H, OCH
3) 2.32–2.27 (m, 4H, NCH
2); 2.16 (s, 3H, NCH
3); 1.69–1.62 (m, 4H, CH
2); 1.51–1.29 (m, 8H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.98 (C); 166.98 (C); 160.85 (C); 160.77 (C); 155.59 (C); 153.38 (C); 144.54 (CH); 143.26 (CH); 139.99 (C); 129.91 (C); 129.00 (CH); 117.44 (CH); 113.66 (CH); 113.29 (C); 112.73 (CH); 105.16 (CH); 101.69 (CH); 65.50 (CH
2); 65.28 (CH
2); 64.53 (CH
2); 60.92 (OCH
3); 57.57 (CH
2); 56.96 (CH
2); 56.12 (OCH
3); 41.88 (NCH
3); 28.68 (CH
2); 27.11 (CH
2); 26.44 (CH
2); 25.89 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
34H
44NO
10 = 626.2960, found 626.2966.
(E)-7-(methyl(2-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)ethyl)amino)heptyl 3-(3,4,5-trimethoxyphenyl)acrylate7. From
44 [
38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 95:5. Yield: 10%.
1H-NMR (400 MHz, CDCl
3) δ: 7.63 (d,
J = 9.6 Hz, 1H, CH); 7.58 (d,
J = 16.0 Hz, 1H, CH=CH); 7.38 (d,
J = 8.8 Hz, 1H, CH); 6.88 (dd,
J = 8.8, 2.2 Hz, 1H, CH); 6.78 (d,
J = 2.2 Hz, 1H, CH); 6.74 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, CH=CH); 6.27 (d,
J = 9.6 Hz, 1H, CH); 4.70 (s, 2H, OCH
2); 4.31 (t,
J = 5.6 Hz, 2H, OCH
2); 4.18 (t,
J = 5.6 Hz, 2H, OCH
2); 3.88 (s, 6H, OCH
3); 3.87 (s, 3H, OCH
3); 2.64 (t,
J = 5.6 Hz 2H, NCH
2); 2.38 (t,
J = 7.2 Hz, 2H, NCH
2); 2.26 (s, 3H, NCH
3); 1.71–1.65 (m, 4H, CH
2); 1.47–1.30 (m, 6H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.93 (C); 167.05 (C); 160.89 (C); 160.74 (C); 153.43 (C); 144.64 (CH); 143.21 (CH); 129.93 (C); 129.03 (CH); 117.43 (CH); 113.80 (CH); 113.38 (C); 112.79 (CH); 105.22 (CH); 101.84 (CH); 65.35 (CH
2); 65.26 (CH
2); 64.52 (CH
2); 60.97 (OCH
3); 57.53 (CH
2); 56.17 (OCH
3); 54.99 (CH
2); 52.50 (NCH
3); 29.02 (CH
2); 28.66 (CH
2); 27.07 (CH
2); 25.86 (CH
2) ppm. ESI-HRMS (
m/z) calculated for [M + H]
+ ion species C
33H
42NO
10 = 612.2803, found 612.2794.
(E)-7-(methyl(7-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)heptyl)amino)heptyl 3-(3,4,5-trimethoxyphenyl)acrylate8. From
41 [35]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 95:5. Yield: 31%.
1H-NMR (400 MHz, CDCl
3) δ: 7.61 (d,
J = 9.6 Hz, 1H, CH); 7.56 (d,
J = 16.0 Hz, 1H, CH=CH); 7.37 (d,
J = 8.8 Hz, 1H, CH); 6.84 (dd,
J = 8.8, 2.2 Hz, 1H, CH); 6.75 (d,
J = 2.2 Hz, 1H, CH); 6.73 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, CH=CH); 6.24 (d,
J = 9.6 Hz, 1H, CH); 4.66 (s, 2H, OCH
2); 4.20–4.15 (m, 4H, OCH
2); 3.86 (s, 6H, OCH
3); 3.85 (s, 3H, OCH
3); 2.34–2.28 (m, 4H, NCH
2); 2.20 (s, 3H, NCH
3); 1.70–1.62 (m, 4H, CH
2); 1.50–1.20 (m, 16H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 168.00 (C); 167.03 (C); 160.87 (C); 160.82 (C); 155.65 (C); 153.41 (C); 144.54 (CH); 143.22 (CH); 140.06 (C); 129.94 (C); 128.97 (CH); 117.48 (CH); 113.72 (CH); 113.30 (C); 112.82 (CH); 105.21 (CH); 101.70 (CH); 65.73 (CH
2); 65.33 (CH
2); 64.63 (CH
2); 60.94 (OCH
3); 57.77 (CH
2); 57.74 (CH
2); 56.14 (OCH
3); 42.15 (NCH
3); 29.20 (CH
2); 29.08 (CH
2); 28.69 (CH
2); 28.43 (CH
2); 27.44 (CH
2); 27.38 (CH
2); 27.04 (CH
2); 27.02 (CH
2); 25.92 (CH
2); 25.72 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
38H
52NO
10 = 682.3586, found 682.3573.
3-(methyl(5-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)pentyl)amino)propyl 3,4,5-trimethoxy benzoate9. From
42 [37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 90:10:1. Yield: 86%.
1H-NMR (400 MHz, CDCl
3) δ: 7.61 (d,
J = 9.2 Hz, 1H, CH); 7.36 (d,
J =8.4 Hz, 1H, CH); 7.25 (s, 2H, CH); 6.85 (dd,
J = 8.4, 2.2 Hz, 1H, CH); 6.73 (d,
J = 2.2 Hz, 1H, CH); 6.23 (d,
J = 9.2 Hz, 1H, CH); 4.66 (s, 2H, OCH
2); 4.33 (t,
J = 6.4 Hz, 2H, OCH
2); 4.16 (t,
J = 6.4 Hz, 2H, OCH
2); 3.86 (s, 9H, OCH
3); 2.56 (t,
J = 6.8 Hz, 2H, NCH
2); 2.41 (t,
J =7.2 Hz, 2H, NCH
2); 2.29 (s, 3H, NCH
3); 2.00–1.95 (m, 2H, CH
2); 1.67–1.60 (m, 2H, CH
2); 1.52–1.46 (m, 2H, CH
2); 1.35–1.27 (m, 2H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 168.01 (C); 166.15 (C); 160.88 (C); 160.80 (C); 155.61 (C); 152.91 (C); 143.27 (CH); 142.19 (C); 129.00 (CH); 125.24 (C); 113.69 (CH); 113.28 (C); 112.86 (CH); 106.78 (CH); 101.63 (CH); 65.50 (CH
2); 65.30 (CH
2); 63.30 (CH
2); 60.90 (OCH
3); 57.19 (CH
2); 56.24 (OCH
3); 53.96 (CH
2); 41.70 (NCH
3); 28.36 (CH
2); 26.37 (CH
2); 26.18 (CH
2); 23.62 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
30H
38NO
10 = 572.2490, found 572.2479.
6-(methyl(3-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)propyl)amino)hexyl 3,4,5-trimethoxy benzoate10. From 33. Oil. Chromatographic eluent: CH2Cl2/CH3OH/NH4OH 90:10:1. Yield: 83%. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.6 Hz, 1H, CH); 7.37 (d, J = 8.4 Hz, 1H, CH); 7.27 (s, 2H, CH); 6.85 (d, J = 8.4 Hz, 1H, CH); 6.75 (s, 1H, CH); 6.24 (d, J = 9.6 Hz, 1H, CH); 4.67 (s, 2H, OCH2); 4.29–4.23 (m, 4H, OCH2); 3.88 (s, 9H, OCH3); 2.40 (t, J = 7.2 Hz, 2H, NCH2); 2.33 (t, J = 7.2 Hz, 2H, NCH2); 2.20 (s, 3H, NCH3); 1.83–1.73 (m, 4H, CH2); 1.47–1.34 (m, 6H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 167.92 (C); 166.18 (C); 160.77 (C); 155.60 (C); 152.87 (C); 143.18 (CH); 142.14 (C); 128.97 (CH); 125.45 (C); 113.69 (CH); 113.29 (C); 112.72 (CH); 106.80 (CH); 101.68 (CH); 65.29 (CH2); 65.09 (CH2); 63.99 (CH2); 60.85 (OCH3); 57.49 (CH2); 56.22 (OCH3); 53.85 (CH2); 41.81 (NCH3); 28.68 (CH2); 27.02 (CH2); 26.89 (CH2); 26.22 (CH2); 25.89 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C31H40NO10 = 586.2647, found 586.2643.
7-(methyl(2-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)ethyl)amino)heptyl 3,4,5-trimethoxy benzoate11. From
45 [38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 89%.
1H-NMR (400 MHz, CDCl
3) δ: 7.58 (d,
J = 9.6 Hz, 1H, CH); 7.33 (d,
J = 8.4 Hz, 1H, CH); 7.24 (s, 2H, CH); 6.83 (d,
J = 8.4 Hz, 1H, CH); 6.73 (s, 1H, CH); 6.20 (d,
J = 9.6 Hz, 1H, CH); 4.66 (s, 2H, OCH
2); 4.29 (t,
J = 5.6 Hz, 2H, OCH
2); 4.24 (t,
J = 6.4 Hz, 2H, OCH
2); 3.85 (s, 9H, OCH
3); 2.67 (t,
J = 5.2 Hz, 2H, NCH
2); 2.40 (t,
J =7.6 Hz, 2H, NCH
2); 2.27 (s, 3H, NCH
3); 1.73–1.69 (m, 2H, CH
2); 1.45–1.26 (m, 8H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.86 (C); 166.08 (C); 160.70 (C); 155.52 (C); 152.82 (C); 143.21 (CH); 142.07 (C); 128.96 (CH); 125.43 (C); 113.54 (CH); 113.23 (C); 112.60 (CH); 106.74 (CH); 101.72 (CH); 65.21 (CH
2); 65.06 (CH
2); 62.40 (CH
2); 60.76 (OCH
3); 57.53 (CH
2); 56.15 (OCH
3); 55.10 (CH
2); 42.00 (NCH
3); 29.01 (CH
2); 28.58 (CH
2); 27.08 (CH
2); 26.46 (CH
2); 25.84 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
31H
40NO
10 = 586.2647, found 586.2638.
3-(methyl(5-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)pentyl)amino)propyl anthracene-9-carboxylate12. From
43 [37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH 95:5. Yield: 81%.
1H-NMR (400 MHz, CDCl
3) δ: 8.51 (s, 1H, CH); 8.05–8.00 (m, 4H, CH); 7.56–7.46 (m, 5H, CH); 7.30 (d,
J = 8.8 Hz, 1H, CH); 6.82 (dd,
J = 8.8, 2.2 Hz, 1H, CH); 6.73 (d,
J = 2.2 Hz, 1H, CH); 6.23 (d,
J = 9.2 Hz, 1H, CH); 4.68 (t,
J = 6.4 Hz, 2H, OCH
2); 4.64 (s, 2H, OCH
2); 4.19 (t,
J = 6.4 Hz, 2H, OCH
2); 2.54 (t,
J =7.2 Hz, 2H, NCH
2); 2.35 (t,
J = 7.2 Hz, 2H, NCH
2); 2.25 (s, 3H, NCH
3); 2.11–2.02 (m, 2H, CH
2); 1.68–1.61 (m, 2H, CH
2); 1.53–1.45 (m, 2H, CH
2); 1.39–1.30 (m, 2H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 169.60 (C); 167.99 (C); 160.87 (C); 160.77 (C); 155.59 (C); 143.19 (CH); 130.97 (C); 129.29 (CH); 128.93 (CH); 128.64 (CH); 128.38 (C); 127.98 (C); 126.96 (CH); 125.48 (CH); 124.97 (CH); 113.66 (CH); 113.24 (C); 112.74 (CH); 101.68 (CH); 65.55 (CH
2); 65.31 (CH
2); 64.04 (CH
2); 57.40 (CH
2); 54.13 (CH
2); 41.89 (NCH
3); 28.38 (CH
2); 26.59 (CH
2); 26.39 (CH
2); 23.67 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
35H
36NO
7 = 582.2486, found 582.2489.
6-(methyl(3-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)propyl)amino)hexyl anthracene-9-carboxylate13. From 34. Oil. Chromatographic eluent: CH2Cl2/CH3OH/NH4OH 90:10:1. Yield: 98%. 1H-NMR (400 MHz, CDCl3) δ: 8.38 (s, 1H, CH); 7.96 (d, J = 8.4 Hz, 2H, CH); 7.90 (d, J = 8.4 Hz, 2H, CH); 7.47–7.37 (m, 5H, CH); 7.16 (d, J = 8.8 Hz, 1H, CH); 6.70 (dd, J = 8.8, 2.2 Hz, 1H, CH); 6.64 (d, J = 2.2 Hz, 1H, CH); 6.10 (d, J = 9.6 Hz, 1H, CH); 4.57–4.52 (m, 4H, OCH2); 4.17 (t, J = 6.4 Hz, 2H, OCH2); 2.27 (t, J = 6.8 Hz, 2H, NCH2); 2.21 (t, J = 6.8 Hz, 2H, NCH2); 2.09 (s, 3H, NCH3); 1.85–1.69 (m, 4H, CH2); 1.46–1.35 (m, 4H, CH2); 1.33–1.26 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 169.71 (C); 167.94 (C); 160.85 (C); 160.75 (C); 155.60 (C); 143.16 (CH); 142.69 (C); 141.90 (C); 130.98 (C); 129.22 (CH); 128.95 (CH); 128.63 (CH); 128.36 (C); 128.16 (C); 126.93 (CH); 125.47 (CH); 124.99 (CH); 113.70 (CH); 113.28 (C); 112.69 (CH); 101.70 (CH); 65.84 (CH2); 65.30 (CH2); 64.06 (CH2); 57.52 (CH2); 53.87 (CH2); 41.91 (NCH3); 28.75 (CH2); 27.04 (CH2); 26.95 (CH2); 26.30 (CH2); 26.04 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C36H38NO7 = 596.2643, found 596.2652.
7-(methyl(2-(2-((2-oxo-2H-chromen-7-yl)oxy)acetoxy)ethyl)amino)heptyl anthracene-9-carboxylate14. From
46 [38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 96:4:0.4. Yield: 68%.
1H-NMR (400 MHz, CDCl
3) δ: 8.48 (s, 1H, CH); 8.03–7.98 (m, 4H, CH); 7.54–7.44 (m, 5H, CH); 7.28 (d,
J = 8.4 Hz, 1H, CH); 6.80 (dd,
J = 8.4, 2.2 Hz, 1H, CH); 6.73 (d,
J = 2.2 Hz, 1H, CH); 6.20 (d,
J = 9.6 Hz, 1H, CH); 4.64 (s, 2H, OCH
2); 4.60 (t,
J = 6.8 Hz, 2H, OCH
2); 4.29 (t,
J = 5.6 Hz, 2H, OCH
2); 2.62 (t,
J = 5.6 Hz, 2H, NCH
2); 2.36 (t,
J = 7.2 Hz, 2H, NCH
2); 2.24 (s, 3H, NCH
3); 1.90–1.83 (m, 2H, CH
2); 1.50–1.25 (m, 8H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 169.71 (C); 167.94 (C); 160.85 (C); 160.73 (C); 155.59 (C); 143.15 (CH); 130.98 (C); 129.19 (CH); 128.91 (CH); 128.61 (CH); 128.35 (C); 126.90 (CH); 125.45 (CH); 124.99 (CH); 113.68 (CH); 113.26 (C); 112.71 (CH); 101.75 (CH); 65.87 (CH
2); 65.29 (CH
2); 63.11 (CH
2); 57.91 (CH
2); 55.48 (CH
2); 42.49 (NCH
3); 29.14 (CH
2); 28.72 (CH
2); 27.24 (CH
2); 27.07 (CH
2); 26.07 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
36H
38NO
7 = 596.2643, found 596.2631.
3.1.2. General Procedure for the Synthesis of Diester Compounds 15–28
A 1 mmol portion of 4-sulfamoylbenzoic acid was transformed into the acyl chloride by reaction with SOCl2 (2 mmol) in 5 mL of CHCl3 (free of ethanol) at 60 °C for 5 h. The reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure; the mixture was then treated twice with cyclohexane and the solvent removed under reduced the pressure. The acyl chloride obtained was dissolved in CHCl3 (free of ethanol), and the suitable (hydroxyalkyl)methylaminoester 33–46 (1 eq) was added. The mixture was stirred for 17 h at room temperature and the solvent was removed under reduce pressure. Then CH2Cl2 was added and the organic layer was washed twice with a saturated solution of NaHCO3. After drying with Na2SO4, the solvent was removed under reduced pressure. The crude product was then purified by flash chromatography using the proper eluting system, yielding the desired compound as an oil.
All the compounds were transformed into the corresponding hydrochloride as a white solid. The salts were crystallized from abs. ethanol/petroleum ether.
(E)-3-(methyl(3-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)propyl)amino)propyl 4-sulfamoyl benzoate15. From (hydroxyalkyl)methylaminoester
35 [35]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 63%.
1H-NMR (400 MHz, CDCl
3) δ: 8.02 (d,
J = 8.4 Hz, 2H, CH); 7.88 (d,
J = 8.4 Hz, 2H, CH); 7.54 (d,
J = 16.0 Hz, 1H, CH=CH); 6.69 (s, 2H, CH); 6.30 (d,
J = 16.0 Hz, 1H, CH=CH); 4.35 (t,
J = 6.4 Hz, 2H, OCH
2); 4.22 (t,
J = 6.0 Hz, 2H, OCH
2); 3.83 (s, 6H, OCH
3); 3.82 (s, 3H, OCH
3); 2.51–2.46 (m, 4H, NCH
2); 2.23 (s, 3H, NCH
3); 1.95–1.81 (m, 4H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.04 (C); 165.14 (C); 153.31 (C); 146.14 (C); 144.75 (CH); 139.89 (C); 133.72 (C); 130.10 (CH); 129.94 (C); 126.29 (CH); 117.29 (CH); 105.28 (CH); 64.04 (CH
2); 62.70 (CH
2); 60.95 (OCH
3); 56.16 (OCH
3); 54.00 (CH
2); 53.69 (CH
2); 41.99 (NCH
3); 26.47 (CH
2); 26.43 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
26H
35N
2O
9S = 551.2058, found 551.2050. Hydrochloride: low melting solid.
(E)-5-(methyl(3-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)propyl)amino)pentyl 4-sulfamoyl benzoate16. From
36 [37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 21%.
1H-NMR (400 MHz, CDCl
3) δ: 8.09 (d,
J = 8.4 Hz, 2H, CH); 7.95 (d,
J = 8.4 Hz, 2H, CH); 7.58 (d,
J = 16.0 Hz, 1H, CH=CH); 6.73 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, CH=CH); 4.33 (t,
J = 6.8 Hz, 2H, OCH
2); 4.22 (t,
J = 6.4 Hz, 2H, OCH
2); 3.86 (s, 6H, OCH
3); 3.85 (s, 3H, OCH
3); 2.48 (t,
J = 7.2 Hz, 2H, NCH
2); 2.39 (t,
J = 7.2 Hz, 2H, NCH
2); 2.24 (s, 3H, NCH
3); 1.89–1.74 (m, 4H, CH
2); 1.59–1.41 (m, 4H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.06 (C); 165.15 (C); 153.39 (C); 146.05 (C); 144.83 (CH); 140.09 (C); 133.97 (C); 130.17 (CH); 129.90 (C); 126.37 (CH); 117.25 (CH); 105.35 (CH); 65.67 (CH
2); 62.88 (CH
2); 60.94 (OCH
3); 57.43 (CH
2); 56.18 (OCH
3); 53.93 (CH
2); 41.99 (NCH
3); 28.48 (CH
2); 26.76 (CH
2); 26.48 (CH
2); 23.84 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
28H
39N
2O
9S = 579.2371, found 579.2380. Hydrochloride: mp 98–100 °C.
(E)-3-(methyl(5-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)pentyl)amino)propyl 4-sulfamoyl benzoate17. From
37 [37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 100%.
1H-NMR (400 MHz, CDCl
3) δ: 8.10 (d,
J = 8.4 Hz, 2H, CH); 7.96 (d,
J = 8.4 Hz, 2H, CH); 7.58 (d,
J = 16.0 Hz, 1H, CH=CH); 6.73 (s, 2H, CH); 6.34 (d,
J = 16.0 Hz, 1H, CH=CH); 4.40 (t,
J = 6.4 Hz, 2H, OCH
2); 4.15 (t,
J = 6.8 Hz, 2H, OCH
2); 3.87 (s, 6H, OCH
3); 3.86 (s, 3H, OCH
3); 2.48 (t,
J = 6.8 Hz, 2H, NCH
2); 2.35 (t,
J = 7.2 Hz, 2H, NCH
2); 2.22 (s, 3H, NCH
3); 1.96–1.90 (m, 2H, CH
2); 1.71–1.64 (m, 2H, CH
2); 1.51–1.40 (m, 4H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.23 (C); 165.14 (C); 153.35 (C); 146.22 (C); 144.81 (CH); 139.97 (C); 133.79 (C); 130.14 (CH); 129.93 (C); 126.35 (CH); 117.33 (CH); 105.27 (CH); 64.53 (CH
2); 64.06 (CH
2); 60.94 (OCH
3); 57.50 (CH
2); 56.15 (OCH
3); 53.75 (CH
2); 42.14 (NCH
3); 28.66 (CH
2); 26.87 (CH
2); 26.39 (CH
2); 23.81 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
28H
39N
2O
9S = 579.2371, found 579.2364. Hydrochloride: mp 83–86 °C.
(E)-5-(methyl(5-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)pentyl)amino)pentyl 4-sulfamoyl benzoate18. From
38 [35]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 97:3:0.3. Yield: 19%.
1H-NMR (400 MHz, CDCl
3) δ: 8.11 (d,
J = 8.4 Hz, 2H, CH); 7.96 (d,
J = 8.4 Hz, 2H, CH); 7.58 (d,
J = 16.0 Hz, 1H, CH=CH); 6.74 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, CH=CH); 4.33 (t,
J = 6.4 Hz, 2H, OCH
2); 4.17 (t,
J = 6.8 Hz, 2H, OCH
2); 3.87 (s, 6H, OCH
3); 3.85 (s, 3H, OCH
3); 2.40–2.32 (m, 4H, NCH
2); 2.22 (s, 3H, NCH
3); 1.83–1.66 (m, 4H, CH
2); 1.58–1.37 (m, 8H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.15 (C); 165.17 (C); 153.41 (C); 146.25 (C); 144.76 (CH); 140.14 (C); 133.94 (C); 130.17 (CH); 129.92 (C); 126.36 (CH); 117.36 (CH); 105.36 (CH); 65.79 (CH
2); 65.63 (CH
2); 64.53 (CH
2); 60.92 (OCH
3); 57.41 (CH
2); 57.38 (CH
2); 56.17 (OCH
3); 42.05 (NCH
3); 28.65 (CH
2); 28.53 (CH
2); 26.64 (CH
2); 23.96 (CH
2); 23.90 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
30H
43N
2O
9S = 607.2684, found 607.2672. Hydrochloride: mp 83–85 °C.
(E)-3-(methyl(6-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)hexyl)amino)propyl 4-sulfamoyl benzoate19. From
39 [38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 90:10:1. Yield: 40%.
1H-NMR (400 MHz, CDCl
3) δ: 8.07 (d,
J = 8.4 Hz, 2H, CH); 7.94 (d,
J = 8.4 Hz, 2H, CH); 7.57 (d,
J = 16.0 Hz, 1H, CH=CH); 6.73 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, CH=CH); 4.99 (bs, 2H, NH
2); 4.35 (t,
J = 6.4 Hz, 2H, OCH
2); 4.12 (t,
J = 6.8 Hz, 2H, OCH
2); 3.85 (s, 6H, OCH
3); 3.83 (s, 3H, OCH
3); 2.49 (t,
J = 7.2 Hz, 2H, NCH
2); 2.35 (t,
J = 7.2 Hz, 2H, NCH
2); 2.22 (s, 3H, NCH
3); 1.95–1.88 (m, 2H, CH
2); 1.69–1.60 (m, 2H, CH
2); 1.49–1.32 (m, 6H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.28 (C); 165.16 (C); 153.35 (C); 146.38 (C); 144. 83 (CH); 139.98 (C); 133.70 (C); 130.15 (CH); 129.91 (C); 126.34 (CH); 117.31 (CH); 105.26 (CH); 64.63 (CH
2); 63.96 (CH
2); 60.93 (OCH
3); 57.51 (CH
2); 56.15 (OCH
3); 53.69 (CH
2); 42.00 (NCH
3); 28.63 (CH
2); 27.01 (CH
2); 26.93 (CH
2); 26.22 (CH
2); 25.87 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
29H
41N
2O
9S = 593.2527, found 593.2522. Hydrochloride: mp 73–76 °C.
(E)-4-(methyl(6-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)hexyl)amino)butyl 4-sulfamoyl benzoate20. From
40 [38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 49%.
1H-NMR (400 MHz, CDCl
3) δ: 8.06 (d,
J = 8.8 Hz, 2H, CH); 7.92 (d,
J = 8.8 Hz, 2H, CH); 7.57 (d,
J = 16.0 Hz, 1H, CH=CH); 6.73 (s, 2H, CH); 6.33 (d,
J = 16.0 Hz, 1H, CH=CH); 5.50 (bs, 2H, NH
2); 4.32 (t,
J = 6.4 Hz, 2H, OCH
2); 4.15 (t,
J = 6.8 Hz, 2H, OCH
2); 3.85 (s, 6H, OCH
3); 3.84 (s, 3H, OCH
3) 2.40–2.31 (m, 4H, NCH
2); 2.20 (s, 3H, NCH
3); 1.79–1.72 (m, 2H, CH
2); 1.70–1.55 (m, 4H, CH
2); 1.50–1.30 (m, 6H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.22 (C); 165.21 (C); 153.35 (C); 146.39 (C); 144.75 (CH); 139.95 (C); 133.72 (C); 130.15 (CH); 129.92 (C); 126.31 (CH); 117.36 (CH); 105.21 (CH); 65.53 (CH
2); 64.61 (CH
2); 60.94 (OCH
3); 57.45 (CH
2); 57.03 (CH
2); 56.14 (OCH
3); 42.05 (NCH
3); 28.66 (CH
2); 27.14 (CH
2); 26.84 (CH
2); 26.60 (CH
2); 25.86 (CH
2); 23.62 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
30H
43N
2O
9S = 607.2684, found 607.2683. Hydrochloride: mp 89–91 °C.
(E)-2-(methyl(7-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)heptyl)amino)ethyl 4-sulfamoyl benzoate21. From
44 [38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 45%.
1H-NMR (400 MHz, CDCl
3) δ: 8.11 (d,
J = 8.8 Hz, 2H, CH); 7.95 (d, J = 8.8 Hz, 2H, CH); 7.59 (d,
J = 15.6 Hz, 1H, CH=CH); 6.75 (s, 2H, CH); 6.34 (d,
J = 15.6 Hz, 1H, CH=CH); 4.43 (t,
J = 5.6 Hz, 2H, OCH
2); 4.14 (t,
J = 6.4 Hz, 2H, OCH
2); 3.87 (s, 6H, OCH
3); 3.86 (s, 3H, OCH
3); 2.76 (t,
J = 5.6 Hz, 2H, NCH
2); 2.43 (t,
J = 7.2 Hz, 2H, NCH
2); 2.31 (s, 3H, NCH
3); 1.66–1.62 (m, 2H, CH
2); 1.49–1.45 (m, 2H, CH
2); 1.33–1.25 (m, 6H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.37 (C); 165.14 (C); 153.40 (C); 146.19 (C); 144.92 (CH); 140.07 (C); 133.83 (C); 130.29 (CH); 129.90 (C); 126.36 (CH); 117.29 (CH); 105.29 (CH); 64.73 (CH
2); 63.46 (CH
2); 60.96 (OCH
3); 57.80 (CH
2); 56.17 (OCH
3); 55.47 (CH
2); 42.73 (NCH
3); 29.13 (CH
2); 28.66 (CH
2); 27.20 (CH
2); 27.17 (CH
2); 25.92 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
29H
41N
2O
9S = 593.2527, found 593.2524. Hydrochloride: mp 70–73 °C.
(E)-7-(methyl(7-((3-(3,4,5-trimethoxyphenyl)acryloyl)oxy)heptyl)amino)heptyl 4-sulfamoyl benzoate22. From
41 [35]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 22%.
1H-NMR (400 MHz, CDCl
3) δ: 8.15 (d,
J = 8.4 Hz, 2H, CH); 8.00 (d,
J = 8.4 Hz, 2H, CH); 7.61 (d,
J = 15.6 Hz, 1H, CH=CH); 6.77 (s, 2H, CH); 6.37 (d, J = 15.6 Hz, 1H, CH=CH); 4.36 (t,
J = 6.8 Hz, 2H, OCH
2); 4.20 (t,
J = 6.4 Hz, 2H, OCH
2); 3.90 (s, 6H, OCH
3); 3.89 (s, 3H, OCH
3); 2.40–2.35 (m, 4H, NCH
2); 2.26 (s, 3H, NCH
3); 1.82–1.65 (m, 4H, CH
2); 1.52–1.30 (m, 16H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 167.13 (C); 165.18 (C); 153.42 (C); 146.07 (C); 144.64 (CH); 139.96 (C); 134.09 (C); 130.22 (CH); 129.96 (C); 126.42 (CH); 117.46 (CH); 105.29 (CH); 65.78 (CH
2); 64.66 (CH
2); 60.95 (OCH
3); 57.57 (CH
2); 56.17 (OCH
3); 42.02 (NCH
3); 29.14 (CH
2); 28.67 (CH
2); 28.53 (CH
2); 27.37 (CH
2); 27.36 (CH
2); 26.77 (CH
2); 25.95 (CH
2); 25.89 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
34H
51N
2O
9S = 663.3310, found 663.3298. Hydrochloride: mp 68–70 °C.
3-(methyl(5-((4-sulfamoylbenzoyl)oxy)pentyl)amino)propyl 3,4,5-trimethoxybenzoate23. From
42 [37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 22%.
1H-NMR (400 MHz, CDCl
3) δ: 8.11 (d,
J = 8.8 Hz, 2H, CH); 7.97 (d,
J = 8.8 Hz, 2H, CH); 7.28 (s, 2H, CH); 4.37–4.32 (m, 4H, CH
2); 3.90 (s, 9H, OCH
3); 2.62 (t,
J = 6.8 Hz, 2H, NCH
2); 2.51 (t,
J = 6.8 Hz, 2H, NCH
2); 2.35 (s, 3H, NCH
3); 2.05–1.98 (m, 2H, CH
2); 1.83–1.76 (m, 2H, CH
2); 1.65–1.58 (m, 2H, CH
2); 1.51–1.43 (m, 2H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 166.25 (C); 165.15 (C); 152.91 (C); 146.07 (C); 133.93 (C); 130.21 (CH); 126.41 (CH); 125.20 (C); 106.87 (CH); 65.56 (CH
2); 63.28 (CH
2); 60.92 (OCH
3); 57.29 (CH
2); 56.28 (OCH
3); 54.00 (CH
2); 41.74 (NCH
3); 28.42 (CH
2); 26.33 (CH
2); 26.16 (CH
2); 23.82 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
26H
37N
2O
9S = 553.2214, found 553.2218. Hydrochloride: mp 52–55 °C.
6-(methyl(3-((4-sulfamoylbenzoyl)oxy)propyl)amino)hexyl 3,4,5-trimethoxybenzoate24. From 33. Oil. Chromatographic eluent: CH2Cl2/CH3OH/NH4OH 93:7:0.3. Yield: 55%. 1H-NMR (400 MHz, CDCl3) δ: 8.01 (d, J = 8.4 Hz, 2H, CH); 7.88 (d, J = 8.4 Hz, 2H, CH); 7.21 (s, 2H, CH); 5.56 (bs, 2H, NH2); 4.31 (t, J = 6.0 Hz, 2H, OCH2); 4.19 (t, J = 6.8 Hz, 2H, OCH2); 3.82 (s, 6H, OCH3); 3.81 (s, 3H, OCH3); 2.45 (t, J = 7.2 Hz, 2H, NCH2); 2.31 (t, J = 7.2 Hz, 2H, NCH2); 2.18 (s, 3H, NCH3); 1.90–1.85 (m, 2H, CH2); 1.69–1.65 (m, 2H, CH2); 1.42–1.28 (m, 6H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 166.39 (C); 165.13 (C); 152.86 (C); 146.37 (C); 142.11 (C); 133.70 (C); 130.13 (CH); 126.33 (CH); 125.41 (C); 106.85 (CH); 65.20 (CH2); 63.98 (CH2); 60.87 (OCH3); 57.50 (CH2); 56.25 (OCH3); 53.77 (CH2); 41.94 (NCH3); 28.64 (CH2); 27.03 (CH2); 26.89 (CH2); 26.23 (CH2); 25.90 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C27H39N2O9S = 567.2371, found 567.2380. Hydrochloride: mp 53–56 °C.
7-(methyl(2-((4-sulfamoylbenzoyl)oxy)ethyl)amino)heptyl 3,4,5-trimethoxybenzoate25. From
45 [38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 40%.
1H-NMR (400 MHz, CDCl
3) δ: 8.08 (d,
J = 8.4 Hz, 2H, CH); 7.93 (d,
J = 8.4 Hz, 2H, CH); 7.27 (s, 2H, CH); 4.43 (t,
J = 5.6 Hz, 2H, OCH
2); 4.26 (t,
J = 6.8 Hz, 2H, OCH
2); 3.89 (s, 6H, OCH
3); 3.87 (s, 3H, OCH
3); 2.78 (t,
J = 5.6 Hz, 2H, NCH
2); 2.44 (t,
J = 7.2 Hz, 2H, NCH
2); 2.32 (s, 3H, NCH
3); 1.74–1.71 (m, 2H, CH
2); 1.50–1.46 (m, 2H, CH
2); 1.40–1.30 (m, 6H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 166.48 (C); 165.14 (C); 152.88 (C); 146.32 (C); 142.14 (C); 133.65 (C); 130.24 (CH); 126.31 (CH); 125.40 (C); 106.86 (CH); 65.29 (CH
2); 63.35 (CH
2); 60.88 (OCH
3); 57.81 (CH
2); 56.26 (OCH
3); 55.40 (CH
2); 42.63 (NCH
3); 29.12 (CH
2); 28.63 (CH
2); 27.19 (CH
2); 27.02 (CH
2); 25.93 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
27H
39N
2O
9S = 567.2371, found 567.2368. Hydrochloride: mp 60–63 °C.
3-(methyl(5-((4-sulfamoylbenzoyl)oxy)pentyl)amino)propyl anthracene-9-carboxylate26. From
43 [37]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 41%.
1H-NMR (400 MHz, CDCl
3) δ: 8.52 (s, 1H, CH); 8.10 (d,
J = 8.4 Hz, 2H, CH); 8.02 (d,
J = 8.4 Hz, 4H, CH); 7.93 (d,
J = 8.4 Hz, 2H, CH); 7.55–7.47 (m, 4H, CH); 4.66 (t,
J = 6.4 Hz, 2H, OCH
2); 4.32 (t,
J = 6.4 Hz, 2H, OCH
2); 2.60 (t,
J = 7.2 Hz, 2H, NCH
2); 2.44 (t,
J = 7.2 Hz, 2H, NCH
2); 2.29 (s, 3H, NCH
3); 2.12–2.05 (m, 2H, CH
2); 1.79–1.72 (m, 2H, CH
2); 1.60–1.40 (m, 4H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 169.68 (C); 165.17 (C); 146.00 (C); 133.88 (C); 130.94 (C); 130.20 (CH); 129.39 (CH); 128.67 (CH); 128.35 (C); 127.79 (C); 127.05 (CH); 126.37 (CH); 125.52 (CH); 124.89 (CH); 65.56 (CH
2); 63.95 (CH
2); 57.32 (CH
2); 53.98 (CH
2); 41.73 (NCH
3); 28.42 (CH
2); 26.06 (CH
2); 23.82 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
31H
35N
2O
6S = 563.2210, found 563.2211. Hydrochloride: mp 82–84 °C.
6-(methyl(3-((4-sulfamoylbenzoyl)oxy)propyl)amino)hexyl anthracene-9-carboxylate27. From 34. Oil. Chromatographic eluent: CH2Cl2/CH3OH/NH4OH 93:7:0.3. Yield: 29%. 1H-NMR (400 MHz, CDCl3) δ: 8.51 (s, 1H, CH); 8.08 (d, J = 8.4 Hz, 2H, CH); 8.01 (d, J = 8.4 Hz, 4H, CH); 7.93 (d, J = 8.4 Hz, 2H, CH); 7.55–7.46 (m, 4H, CH); 5.08 (bs, 2H, NH2); 4.59 (t, J = 6.8 Hz, 2H, OCH2); 4.37 (t, J = 6.4 Hz, 2H, OCH2); 2.51 (t, J = 7.2 Hz, 2H, NCH2); 2.38 (t, J = 7.2 Hz, 2H, NCH2); 2.24 (s, 3H, NCH3); 1.97–1.82 (m, 4H, CH2); 1.51–1.35 (m, 6H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 169.85 (C); 165.09 (C); 146.22 (C); 142.61 (C); 141.80 (C); 133.63 (C); 130.96 (C); 130.18 (CH); 129.28 (CH); 128.64 (CH); 128.32 (C); 128.01 (C); 127.00 (CH); 126.37 (CH); 125.50 (CH); 124.91 (CH); 65.85 (CH2); 63.71 (CH2); 57.23 (CH2); 53.71 (CH2); 41.60 (NCH3); 28.62 (CH2); 26.86 (CH2); 26.39 (CH2); 25.92 (CH2); 25.84 (CH2) ppm. ESI-HRMS (m/z) calculated for [M + H]+ ion species C32H37N2O6S = 577.2367, found 577.2361. Hydrochloride: mp 81–84 °C.
7-(methyl(2-((4-sulfamoylbenzoyl)oxy)ethyl)amino)heptyl anthracene-9-carboxylate28. From
46 [38]. Oil. Chromatographic eluent: CH
2Cl
2/CH
3OH/NH
4OH 93:7:0.3. Yield: 33%.
1H-NMR (400 MHz, CDCl
3) δ: 8.52 (s, 1H, CH); 8.11 (d,
J = 8.0 Hz, 2H, CH); 8.01 (d,
J = 8.0 Hz, 4H, CH); 7.93 (d,
J = 8.0 Hz, 2H, CH); 7.56–7.47 (m, 4H, CH); 5.31 (bs, 2H, NH
2); 4.60 (t,
J = 6.8 Hz, 2H, OCH
2); 4.46 (t,
J = 5.6 Hz, 2H, OCH
2); 2.80 (t,
J = 5.6 Hz, 2H, NCH
2); 2.47 (t,
J = 7.2 Hz, 2H, NCH
2); 2.35 (s, 3H, NCH
3); 1.88–1.82 (m, 2H, CH
2); 1.54–1.30 (m, 8H, CH
2) ppm.
13C-NMR (100 MHz, CDCl
3) δ: 169.93 (C); 165.08 (C); 146.27 (C); 133.47 (C); 130.96 (C); 130.28 (CH); 129.28 (CH); 128.64 (CH); 128.32 (C); 128.01 (C); 127.00 (CH); 126.31 (CH); 125.50 (CH); 124.92 (CH); 65.97 (CH
2); 62.86 (CH
2); 57.65 (CH
2); 55.18 (CH
2); 42.34 (NCH
3); 29.03 (CH
2); 28.64 (CH
2); 27.10 (CH
2); 26.53 (CH
2); 26.00 (CH
2) ppm. ESI-HRMS (
m/
z) calculated for [M + H]
+ ion species C
32H
37N
2O
6S = 577.2367, found 577.2374. Hydrochloride: mp 71–73 °C.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
