Constituents from the Seeds of Sophora Alopecuroides L.
by
 ,
,
Zi-Jian Rong
1, 
Gao-Sheng Hu
1,
Shi-Yi Lin
2,
Ting Yan
1,
Na Li
1,
Yue Zhao
1,
Jing-Ming Jia
1,* and
An-Hua Wang
1,* 1
School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang 110016, China
2
College of pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(2), 411; https://doi.org/10.3390/molecules25020411
Submission received: 4 December 2019
/
Revised: 13 January 2020
/
Accepted: 15 January 2020
/
Published: 19 January 2020
(This article belongs to the Section Natural Products Chemistry)
Abstract
:Three new isoflavone glucosides, kudonol A−C (1–3), two new ester derivatives of phenylpropanoid, kudolignan A and B (4–5) and five known compounds, (−)-maackiain (6), neoliquiritin (7), methyl 4-coumarate (8), methyl ferulate (9) and (+)-wikstromol (10), were isolated from an extract of dried seeds of the traditional Chinese medicinal plant Sophora alopecuroides L. Their structures were established by NMR and HRESIMS data analyses. The monosaccharide part’s configuration of isoflavone glucosides was confirmed by acid hydrolysis and analyzed by a JAsco OR-4090 chiral detector, comparing it to standard substance D-glucose. The cytotoxicity effects against HeLa, Hep3B, MCF-7 and H1299 cells were tested by CCK-8 assay.
1. Introduction
Sophora alopecuroides L. is a traditional Chinese herbal plant, known as “Ku-Dou-Zi” in China, and is a perennial herbaceous plant in the Leguminosae family, which is widely distributed in the deserts of northern China, especially in the Xinjiang and Ningxia provinces as well as the Inner Mongolia Autonomous Region. The seeds of S. alopecuroides have a long history as a traditional Chinese medicine, utilized for the treatment of eczema, acute pharyngolaryngeal infection, sore throat, acute dysentery, and gastrointestinal hemorrhage [1].
Previous phytochemical investigations on S. alopecuroides have led to the isolation of alkaloids, flavonoids, isoflavonoids, and rotenoids [2]. Compounds isolated from the seeds of this plant have been used to treat leukemia [3], trophoblastic tumors and inflammation in traditional Chinese medicine. For example, sophocarpine was found to be effective against tumors, both in cell proliferation and metastasis in liver cancer [4].
During our present exploration for the diterpenoids of S. alopecurioides, five previously undescribed compounds were isolated and identified successfully for the first time (Figure 1). The structures of the isolated isoflavone glucosides were determined from various spectroscopic data. Finally, the inhibitory effects of some of the isolates against four cancer cell lines were evaluated in vitro using a CCK-8 bioassay.
2. Results and Discussion
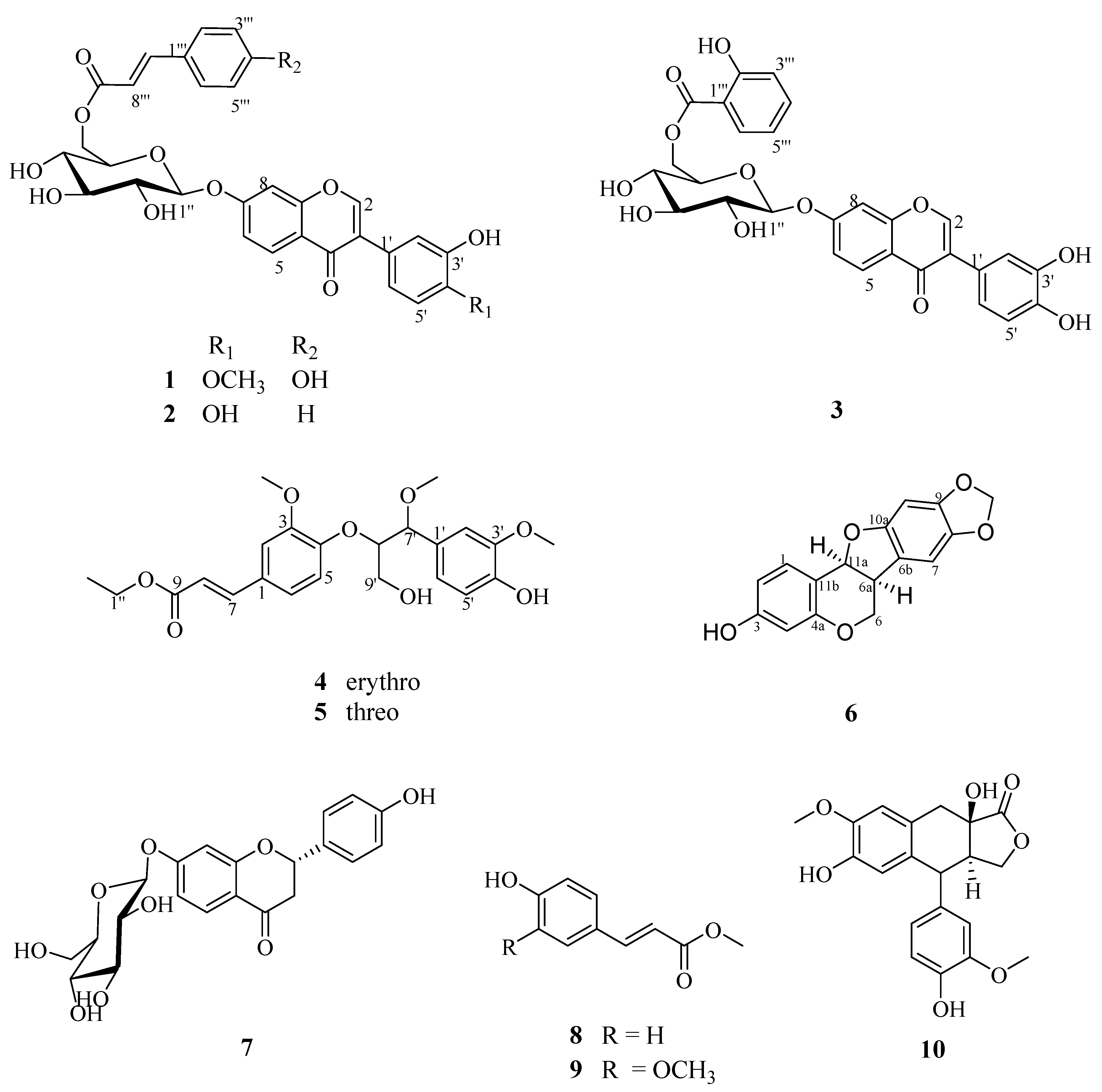
Compound 1 was obtained as a yellow powder and exhibited an ion peak at m/z 591.1512 ([M − H]−, calcd 591.1581) in its HRESIMS data, which indicated a molecular formula of C31H28O12. The indices of hydrogen deficiency (IHDs) of compound 1 was 18. The 13C NMR data of compound 1 comprised the parent nucleus of an isoflavonoid and a monosaccharide. On the basis of the 1H-NMR spectroscopic data (Table 1), six aromatic proton signals at δH 8.03 (1H, d, J = 8.4 Hz), δH 7.20 (1H, d, J = 2.4 Hz), δH 7.14 (1H, dd, J = 8.4, 2.4 Hz), δH 6.95 (1H, d, J = 8.4 Hz), δH 7.04 (1H, d, J = 1.8 Hz) and δH 6.81 (1H, dd, J = 8.4, 1.8 Hz) were observed, which were deduced to be two ABX coupled system in a sub-structure. Meanwhile, fifteen carbon signals at δC 153.2, 123.5, 174.5, 127.0, 115.6, 161.2, 103.4, 156.9, 118.5, 124.4, 116.4, 146.0, 147.6, 112.0 and 119.5 in the 13C NMR spectrum (Supplementary Materials) indicated the presence of structural fragments of isoflavones. A spin-spin system at δH 5.18(H-1″)/3.36(H-2″)/3.37(H-3″)/3.27(H-4″)/3.84(H-5″)/4.45(H-6″) was established on the basis of an 1H-1H COSY experiment, which indicated that there may be a six-carbon sugar in the structure. In its NOESY spectrum, the correlations δH 5.18(H-1″)/3.37(H-3″)/3.84(H-5″) and 3.36(H-2″)/3.27(H-4″)/4.45(H-6″) established the 1″a-H, 3″a-H, 5″a-H in the six-carbon sugar ring. Six carbon signals at δC 99.7, 73.0,76.3, 70.0, 73.9 and 63.3 suggested the existence of glucose as well as an anomeric proton at δH 5.18 (d, J = 7.2 Hz). Additionally, an acid hydrolysis experiment performed on compound 1 afforded D-glucose, which was determined using a JAsco OR-4090 detector with D-glucose standard sample. Then, a parahydroxy cinnamic acid was deduced in the structure from the signals at δH 7.52 (2H, d, J = 8.4 Hz) and 6.79 (2H, d, J = 8.4 Hz) and a pair of trans-ene hydrogen signals at δH 7.56 (1H, d, J = 15.6 Hz, 7‴-H) and 6.39 (1H, d, J = 15.6 Hz, 8‴-H) in the 1H NMR spectrum as well as a carbonyl carbon signal at δC 166.3 in the 13C NMR spectrum. The analysis of the spectroscopic data indicated that compound 1 was similar to the known compound calycosin-7-O-D-glucopyranoside [5], and the difference mainly lay in the parahydroxy cinnamic acid group. The exact structure of compound 1 was established by 2D-NMR. In the HMBC spectrum, a methoxide signal at δH 3.80 had a long-range correlation with δC 147.6(C-4′), which indicated a methoxy group connected on the C ring of this isoflavone. A long-range correlation was observed between δH 5.18 (H-1′) and δC 161.2 (C-7), which located the D-glucose at C-7 of the isoflavone, and the anomeric proton at δH 5.18 (d, J = 7.2 Hz) also indicated the presence of a β glycosidic bond. The parahydroxy cinnamic acid group was posited to the C-6 of D-glucose by the HMBC correlations of δH 4.45 (H-6″)/δC 166.3 (C-9‴). Based on the abovementioned data, the structure of compound 1 was elucidated as Calycosin-7-O-β-D-(6″-hydroxy cinnamate)-glucopyranoside and named as kudonol A.
Compound 2, a yellow powder, gave the molecular formula C30H26O11 through HRESIMS of its quasi-molecular ion peak at m/z 561.1405 ([M − H]−, calcd 561.1475). The spectroscopic data indicated that compound 2 was similar to compound 1, except for an absence of a methoxy group and an extra aromatic proton signal. Based on a long-range correlation of H-1″ (δH 5.12, J = 7.2 Hz)/C-7(δC 163.1) observed in the HMBC data, the glucose moiety was located at C-7 of the isoflavone fragments. Meanwhile, a structural fragment of cinnamic acid was evidenced by signals at δH 7.55 (1H, d, J = 8.4 Hz), 7.35 (1H, d, J = 8.4 Hz) and 7.34 (1H, m) in the 1H NMR spectrum and a carbonyl carbon signal at δC 168.1 in the 13C NMR spectrum. Additionally, a long-range correlation was observed from δH 4.46 (H-6″) to δC 168.1 (C-9‴), which located the cinnamic acid at C-6 of the glucose. Compared with compound 1, the structure of compound 2 has a hydroxy group instead of the methoxy group present in the B ring of the isoflavone parent nucleus. Consequently, the structure of compound 2 was elucidated to be 7,3′,4′-trihydroxyisoflavone-7-O-β-D-(6″-cinnamic acid)-glucopyranoside and named as kudonol B.
Compound 3 had the molecular formula C28H24O12 according to the HRESIMS data from its quasi-molecular ion peak at m/z 553.1336 ([M + H]+, calcd 553.1268). On the basis of spectroscopic data, compound 3 was deduced to be an isoflavone glycoside, which was similar to compound 1; the main difference was that cinnamic acid was replaced by a 2-hydroxy benzoyl group, as well as a hydroxy group instead of the methoxy at C-4′. The hydrolysis experiment on compound 3 gave a monosaccharide, which was identified to be of the D-glucopyranosyl group by the corresponding standard substances. Through the analysis of spectral data, a salicylic acid group was deduced from δC 113.5, 162.8, 118.4, 137.0, 120.4, 131.2 and 171.0 in the 13C NMR spectrum, and that was attached to the C-6 position of the glucose with the carboxyl of itself, which was indicated from the long-correlation between δH 4.81 (H-6″) and δC 171.0 (C-7‴). Consequently, the structure of compound 3 was elucidated to be 7,3′,4′-trihydroxyisoflavone-7-O-β-d-(6″-salicylic acid)-glucopyranoside and was named as kudonol C.
Compound 4 was obtained as a pale yellow powder and exhibited an ion peak at m/z 455.1673 ([M + Na]+, calcd 455.1682) in its HRESIMS data, which indicated a molecular formula of C23H28O8. The 1H NMR spectrum of compound 4 (see Table 2) showed six aromatic proton signals at δH 7.16 (1H, d, J = 1.8 Hz, H-2), 6.93 (1H, d, J = 8.4 Hz, H-5), 7.08 (1H, dd, J = 8.4, 1.8 Hz, H-6), 6.98 (1H, d, J = 1.8 Hz, H-2′), 6.76 (1H, d, J = 7.8 Hz, H-5′) and 6.84 (1H, dd, J = 7.8, 1.8 Hz, H-6′), revealing the presence of two ABX system aromatic rings. Two olefin proton signals were observed at δH 7.16 (1H, d, J = 16.2 Hz, H-7) and 6.40 (1H, d, J = 16.2 Hz, H-8) in the lower field region in the 1H NMR spectrum. Two oxymethine protons at δH 4.40 (1H, d, J = 6.6 Hz, H-7′) and 4.54 (1H, m, H-8′), and two oxymethylene protons at δH 3.86 (2H, d, J = 4.8 Hz, H-9′), established the presence of a 1,2,3-propanetriol moiety. Additionally, one ethoxy group at δH 4.26 (2H, q, J = 7.2 Hz, H-1″) and 1.35 (3H, t, J = 7.2 Hz, H-2″) and three methoxy groups at δH 3.83 (s, 3, OCH3), 3.82 (s, 3, OCH3) and 3.26 (s, 3, OCH3) were observed in the 1H NMR spectrum. The 13C NMR spectrum of compound 4 (see Table 2) showed 23 carbon signals. Aside from the carbon signal of the ethoxy unit and the three methoxy groups, the remaining 18 carbon signals included a carbonyl signal at δC 169.1 (C-9), double ring olefin carbon signals at δC 116.8 (C-8) and 146.1 (C-9), 12 aromatic carbons, and three aliphatic carbons at 83.7 (C-7′), 84.7 (C-8′) and 62.3 (C-9′). The HMBC correlations of H-7 at δH 7.61 with C-2, of H-8 at δH 6.40 with C-6 and C-9, and of H-7′ at δH 4.40 with C-2′, C-5′, C-6′ and C-8′ confirmed the presence of two phenyl propanoid units. In the HMBC spectrum, the correlation of H-8′ at δH 4.54 with C-4 at δC 151.8 suggested that compound 4 was an 8′-O-4 system neolignan. The methoxy group was determined to be at C-3, C-3′ and C-7′, based on the HMBC correlation of the methoxy groups at δH 3.83 with C-3 at δC 151.8 and at δH 3.82, with C-3′ at δC 148.9 and δH 3.26, and with C-7′ at δC 83.7. Compound 5, a pale yellow powder, gave the same molecular formula through HRESIMS, the spectroscopic data indicated that compound 5 was similar to compound 4, except for the difference of their absolute configuration. Regarding the configurations of compounds 4 and 5, the difference in chemical shifts of both H-9′ protons is the parameter that must be used to establish the relative configuration around the chiral centers H-7′ and H-8′ [6,7], in compound 4 both H-9′ protons seem to be isochronous (δH 3.86), whereas there is a difference of 0.20 ppm (δH 3.71 and 3.51) in the chemical shift of these protons in compound 5, thus confirming that 4 and 5 have erythro and threo relative configurations, respectively, namely these are an erythro/threo pair. Thus, the structure of compound 4 was determined to be erythro-4′,9′-dihydroxy-3,3′,7′-trimethoxy-9-ethyoxyl-8−4-oxyneolignan and was named as kudolignan A (4), and 5 was determined to be threo-4′,9′-dihydroxy-3,3′,7′-trimethoxy-9-ethyoxyl-8−4-oxyneolignan named as kudolignan B (5).
The other known compounds were identified as (−)-maackiain (6) [8], neoliquiritin (7) [9], methyl 4-coumarate (8) [10], methyl ferulate (9) [11] and (+)-Wikstromol (10) [12] from the comparison of their NMR data with that reported in the literature.
Compounds 1–6 were tested for their cytotoxicity against four human cancer cell lines (HeLa, Hep3B, MCF-7 and H1299) and one normal human liver cell line (LO2) (see Table 3). Experimental results show that all of the compounds showed no cytotoxicity to the LO2 cell line, compound 2 showed moderate inhibition of cell proliferation against several cancer cell lines, and compound 1 showed moderate inhibition of cell proliferation against Hep3B cells. On the basis of structural analysis, the activity of inhibiting the compounds’ cytotoxicity against tumor cells was probably due to the trans-cinnamic acid moiety.
3. Experimental Section
3.1. General Experimental Procedures
NMR spectra were run in DMSO-d6 and CD3OD on a Varian Mercury NMR spectrometer. Analytical HPLC data were collected on an Agilent 1260 infinity II instrument (Thermo Scientific dionex). The UV spectra were measured by an Agilent 1260 infinity II UV–vis spectrophotometer in methanol. Preparative HPLC was performed by a Saipuruisi MH-LC 52 instrument with an Elite UV2300 detector and an YMC C18 column (250 × 20 mm, 5 µm). The HRESIMS data were obtained using an Agilent 1290 series 6540 UHD accurate mass Q-TOF mass spectrometer using direction injection. The sugar configurations were determined by a JAsco OR-4090 chiral detector. Column chromatographic separations were carried out on silica gel H-60 (Qingdao Marine Chemical Group Corporation, Qingdao, China), Polyamide (Shanghai Yiyan biology technology Co. Ltd., Zhengzhou, China) and Sephadex LH-20 (Shanghai Yi-He biological technology Co. Ltd., Shanghai, China). The Acetonitrile and Methanol used were of chromatographic grade, and were purchased from Fisher in America. All other solvents used of chemical grade (Da-Mao Chemical Co. Ltd., Tianjin, China).
3.2. Plant Materials
The dry seeds of S. alopecuroides L. were collected from Alxa League in Inner Mongolia Autonomous Region and identified by Prof. Liu Yong, Jiang-Xi University of Chinese Medicine. The specimens were deposited in the specimen room of traditional Chinese Medicine in Shenyang Pharmaceutical University (SPU-2014-0714-06).
3.3. Extraction and Isolation
The dry seeds of S. alopecuroides L. (50 kg) were extracted by 70% ethanol aqueous solution, refluxed (1.5 h × 3) and adjusted to a pH of 2 by 1%HCl, and then partitioned with EtOAc. The EtOAc portion (336 g) was partitioned by polyamide chromatography (EtOH/H2O) and yielded four fractions (100% water, 30% EtOH, 60% EtOH, 90% EtOH). Afterwards, the fraction of 60% EtOH (51 g) was dealt with by a silica gel column by elution with CH2Cl2:CH3OH (100:1–0:100) in sequence to give fractions 1–19. Sub-fraction 12 was separated by medium pressure ODS CC (0.03% TFA, 45–100% CH3OH aqueous) to obtain 22 fractions, 1′–22′. Fraction 6′ was purified by preparative HPLC with CH3CN-H2O (30%, 0.03% TFA) to get compound 1 (12.7 mg, tR = 16.06 min), compound 2 (18.9 mg, tR = 21.50 min) and compound 3 (20.4 mg, tR = 17.45 min). Fraction 2′ was purified by preparative HPLC with CH3CN-H2O (15%, 0.03% TFA) to get compound 7 (2.2 mg, tR = 16.06 min). Sub-fraction 4 was separated by Sephadex LH-20 (90% CH3OH aqueous) to obtained 16 fractions 1″–16″. Fraction 9″ was purified by preparative HPLC with CH3OH–H2O (60%, 0.03% TFA) to get compound 4 (3.1 mg, tR = 21.44 min) and compound 5 (3.2 mg, tR = 23.01 min). Sub-fraction 3 was separated by medium pressure ODS CC (0.03% TFA, 10–100% CH3OH aqueous) to obtain 14 fractions 1‴–14‴. Fraction 10‴ was purified by preparative HPLC with CH3OH–H2O (40%, 0.03% TFA) to get compound 8 (2.1 mg, tR = 40.02 min) and compound 9 (2.5 mg, tR = 42.33 min). Fraction 11‴ was purified by preparative HPLC with CH3OH–H2O (50%, 0.03% TFA) to get compound 6 (42.4 mg, tR = 24.21 min). Fraction 9‴ was purified by preparative HPLC with CH3OH–H2O 35%, 0.03% TFA) to get compound 10 (2.3 mg, tR = 39.15 min).
3.3.1. Kurosol A (1)
A yellow powder; [α]D20 −39.12(c 0.3, MeOH); UV λmax 196, 264 and 292; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 1; HRESIMS m/z 591.1512 ([M − H]−, calcd 591.1581).
3.3.2. Kurosol B (2)
A yellow powder; [α]D20 −71.97(c 0.8, MeOH); UV λmax 200, 218 and 264; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz), see Table 1; HRESIMS m/z 561.1405 ([M − H]−, calcd 561.1475).
3.3.3. Kurosol C (3)
A yellow powder; [α]D20 −28.00(c 0.8, MeOH); UV λmax 204 and 290; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz), see Table 1; HRESIMS m/z 553.1336 ([M + H]+, calcd 553.1268).
3.3.4. Kudolignan A (4)
A pale yellow powder; [α]D20 −14.60(c 0.2, MeOH); UV λmax 198, 230 and 324; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz), see Table 2; HRESIMS m/z 455.1673 ([M + Na]+, calcd 455.1682).
3.3.5. Kudolignan B (5)
A pale yellow powder; [α]D20 −21.53(c 0.1, MeOH); UV λmax 200, 230 and 322; 1H NMR (CD3OD, 600 MHz) and 13C NMR (CD3OD, 150 MHz), see Table 2; HRESIMS m/z 455.1676 ([M + Na]+, calcd 455.1682).
3.4. Acid Hydrolysis
Five milligrams of powder of compounds 1, 2 and 3 were dissolved in CH3OH (the least amount) and participated in an acid hydrolysis reaction with 2 M of HCl in a 90 °C water bath over 5 hours [13]. The reaction mixture was cooled to room temperature and extracted by EtOAc three times. Then, the water layer was evaporated in vacuo by a rotatory evaporator to remove the remaining EtOAc. The residue was dissolved in water again and next analyzed by HPLC coupled to a JAsco OR-4090 chiral detector, comparing it to standard substance glucose (chromatography column: Shodex Asahipak NH2P-504E, CH3CN: H2O (3:1), 0.8 mL/min).
3.5. Cytotoxicity Assay
4. Conclusions
Three new isoflavone glucosides, termed kudonol A−C (1–3), two new ester derivatives of phenylpropanoid, kudolignan A and B (4–5) together with five known compounds (6–10), were isolated from the seeds of S. alopecuroides. The absolute configurations of compounds 4–5 were determined by the extensive analysis of spectroscopic data and quantum chemical ECD calculations. Unfortunately, the cytotoxicity of compounds 2 and 6 that were evaluated against the tumor cell lines showed only weak activity. In addition, further biological assays of these compounds and the structural diversity of S. alopecuroides are worth exploring in future research.
Supplementary Materials
The following are available online. HREIMS, 1H-NMR, 13C-NMR, HMBC, and HSQC spectra of new compounds are available as supporting information.
Author Contributions
Z.-J.R., G.-S.H. and S.-Y.L. performed the experiments; T.Y., N.L. and Y.Z. contributed to manuscript preparation; J.-M.J. dealt with the structure analysis of compounds; A.-H.W. designed the whole experiments, analyzed the data and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key R&D Program of China (2017YFC1701200) and the National Natural Science Foundation of China (No. 81374061, 81903789).
Acknowledgments
We appreciate the help of Ma Guang-Rui at the forestry bureau of Alxa league.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gao, H.-Y.; Li, G.-Y.; Wang, J.-H. A New Alkaloid from the Seeds of Sophora alopecuroides L. Helvetica Chim. Acta 2012, 95, 1108–1113. [Google Scholar] [CrossRef]
- Raksat, A.; Maneerat, W.; Andersen, R.J.; Pyne, S.G.; Laphookhieo, S. Antibacterial Prenylated Isoflavonoids from the Stems of Millettia extensa. J. Nat. Prod. 2018, 81, 1835–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-X.; Wang, G.; Zhu, J.-S.; Zhang, R.; Zhang, J. Traditional uses, phytochemistry, and pharmacological properties of Sophora alopecuroides L. Eur. J. Inflamm. 2016, 14, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-P.; Wang, P.-Q.; Qiao, C.-P.; Zhang, Q.; Zhang, J.-P.; Chen, F.; Zhang, X.; Xie, W.-F.; Yuan, Z.-L.; Li, Z.-S.; et al. Differentiation therapy of hepatocellular carcinoma by inhibiting the activity of AKT/GSK-3β/β-catenin axis and TGF-β induced EMT with sophocarpine. Cancer Lett. 2016, 376, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Du, X.G.; Bai, Y.J.; Liang, H.; Wang, Z.Y.; Zhao, Y.Y.; Zhang, Q.Y.; Huang, L.Q. Solvent effect in 1H NMR spectra of 3″-hydroxy-4″-methoxy isoflavonoids from Astragalus membranaceus var. Mongholicus. Magn. Reson. Chem. 2006, 44, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.-B.; Chai, D.-W.; Jin, X.-J.; Bi, Y.-R.; Yao, X.-J.; Wu, W.-S.; Zhu, Y. Triterpenes and neolignans from the roots of Nannoglottis carpesioides. Phytochem. 2011, 72, 1804–1813. [Google Scholar] [CrossRef]
- Huo, C.; Liang, H.; Zhao, Y.; Wang, B.; Zhang, Q. Neolignan glycosides from Symplocos caudata. Phytochem. 2008, 69, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Hillerns, P.I.; Wink, M. Binding of Flavonoids from Sophora flavescens to the Rat Uterine Estrogen Receptor. Planta Medica 2005, 71, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-X.; Lin, W.-H.; Wang, X.-L.; Yang, J.-S. Flavonoids from preparation of traditional Chinese medicines named Sini-Tang. J. Asian Nat. Prod. Res. 2005, 7, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, S.; Qin, N.; Zhai, H.; Duan, H. Antioxidant constituents from Smilax riparia. China J. Chin. Mater. Medica 2012, 37, 806–810. [Google Scholar]
- Lima, T.C.; Ferreira, A.R.; Silva, D.F.; Lima, E.O.; de Sousa, D.P. Antifungal activity of cinnamic acid and benzoic acid esters against Candida albicans strains. Nat. Prod. Res. 2018, 32, 572–575. [Google Scholar]
- Hu, K.; Kobayashi, H.; Dong, A.; Iwasaki, S.; Yao, X. Antifungal, Antimitotic and Anti-HIV-1 Agents from the Roots of Wikstroemia indica. Planta Med. 2000, 66, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yu, B.; Chen, C.; Duan, J.; Di, D.; Xiong, X.; Yang, Y.; Gao, H. New natural barrigenol-like triterpenoid isolated from the husks of Xanthoceras sorbifolia Bunge. Nat. Prod. Res. 2018, 32, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.J.; Guo, Y.J.; Yang, X.L.; Ou, Z.L. Anti-Cervical Cancer Role of Matrine, Oxymatrine and Sophora Flavescens Alkaloid Gels and its Mechanism. J. Cancer 2018, 9, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.H.; Gao, X.X.; Huo, X.K.; Huang, S.S.; Feng, L.; Sun, C.P.; Zhang, B.J.; Ma, X.C.; Jia, J.M.; Wang, C. Antioxidant acetophenone glycosides from the roots of Euphorbia ebracteolata Hayata. Nat. Prod. Res. 2018, 32, 2187–2192. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1, 2, 3 and 6 are available from the authors. Especially, compound 6, as a major component, can be got a lot. |
Figure 1.
Structures of compounds 1–10.


{kind=link}
Table 1.
1H NMR data (600 MHz) and 13C NMR data (150 MHz) of compounds 1, 2 and 3.
| Position | 1(DMSO-d6) | 2(CD3OD) | Position | 3(CD3OD) | |||
|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 2 | 8.03, s | 153.2 | 7.50, s | 154.7 | 2 | 8.04, s | 154.9 |
| 3 | - | 123.5 | - | 124.5 | 3 | - | 124.6 |
| 4 | - | 174.5 | - | 177.8 | 4 | - | 178.0 |
| 5 | 8.03, d (8.4) | 127.0 | 8.09, d (9.0) | 128.2 | 5 | 8.08, d (8.4) | 128.3 |
| 6 | 7.14, dd (8.4, 2.4) | 115.6 | 7.16, dd (9.0, 1.8) | 117.5 | 6 | 7.19, dd (8.4, 2.4) | 117.0 |
| 7 | - | 161.2 | - | 163.1 | 7 | - | 163.0 |
| 8 | 7.20, d (2.4) | 103.4 | 7.15, d (1.8) | 104.5 | 8 | 7.18, d (2.4) | 104.8 |
| 9 | - | 156.9 | - | 158.9 | 9 | - | 159.0 |
| 10 | - | 118.5 | - | 120.2 | 10 | - | 120.2 |
| 1′ | - | 124.4 | - | 126.0 | 1′ | - | 126.2 |
| 2′ | 7.04, d (1.8) | 116.4 | 6.93, d (1.8) | 117.4 | 2′ | 7.06, s | 117.5 |
| 3′ | - | 146.0 | - | 146.1 | 3′ | - | 146.2 |
| 4′ | - | 147.6 | - | 146.7 | 4′ | - | 146.8 |
| 5′ | 6.95, d (8.4) | 112.0 | 6.82, d (7.8) | 116.2 | 5′ | 6.94, m | 116.4 |
| 6′ | 6.81, dd (8.4, 1.8) | 119.5 | 6.67, dd (7.8, 1.8) | 121.6 | 6′ | 6.94, m | 121.7 |
| 1″ | 5.18, d (7.2) | 99.7 | 5.12, d (7.2) | 101.5 | 1″ | 5.21, d (7.8) | 101.2 |
| 2″ | 3.36, m | 73.0 | 3.60, m | 74.7 | 2″ | 3.62, m | 74.6 |
| 3″ | 3.37, m | 76.3 | 3.60, m | 77.8 | 3″ | 3.62, m | 77.8 |
| 4″ | 3.27, m | 70.0 | 3.47, m | 72.0 | 4″ | 3.51, m | 72.0 |
| 5″ | 3.84, m | 73.9 | 3.92, m | 75.7 | 5″ | 4.00, m | 75.5 |
| 6″ | 4.45, dd (12.0, 1.8) 4.21, m | 63.3 | 4.64, dd (12.0, 1.8) 4.45, m | 64.9 | 6″ | 4.81, dd (12.0, 1.8) 4.56, m | 65.6 |
| 1‴ | - | 125.0 | - | 135.7 | 1‴ | - | 113.5 |
| 2‴/6‴ | 7.52, d (8.4) | 130.3 | 7.55, d (8.4) | 129.2 | 2‴ | - | 162.8 |
| 3‴/5‴ | 6.79, d (8.4) | 115.8 | 7.35, d (8.4) | 130.2 | 3‴ | 6.94, m | 118.4 |
| 4‴ | - | 159.9 | 7.34, m | 131.7 | 4‴ | 7.52, m | 137.0 |
| 7‴ | 7.56, d (15.6) | 144.9 | 7.70, d (15.6) | 146.6 | 5‴ | 6.94, m | 120.4 |
| 8‴ | 6.39, d (15.6) | 113.9 | 6.56, d (15.6) | 118.7 | 6‴ | 7.95, dd (7.8, 1.8) | 131.2 |
| 9‴ | - | 166.3 | - | 168.1 | 7‴ | - | 171.0 |
| -OCH3 | 3.80, s | 55.6 | - | - | - | - | - |
Table 2.
1H NMR data (600 MHz) and 13C NMR data (150 MHz) of compounds 4–5 (CD3OD).
| Position | 4 | 5 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | - | 129.5 | - | 129.4 |
| 2 | 7.16, d (1.8) | 112.3 | 7.24, d (2.4) | 112.3 |
| 3 | - | 151.8 | - | 152.4 |
| 4 | - | 151.7 | - | 151.7 |
| 5 | 6.93, d (8.4) | 117.4 | 7.04, d (8.4) | 117.2 |
| 6 | 7.08, dd (8.4, 1.8) | 123.3 | 7.13, dd (8.4, 2.4) | 123.5 |
| 7 | 7.61, d (16.2) | 146.1 | 7.64, d (16.2) | 146.1 |
| 8 | 6.40, d (16.2) | 116.8 | 6.42, d (16.2) | 116.8 |
| 9 | - | 169.1 | - | 169.1 |
| 1′ | - | 130.6 | - | 130.8 |
| 2′ | 6.98, d (1.8) | 112.5 | 7.00, d (1.8) | 112.1 |
| 3′ | - | 148.9 | - | 149.1 |
| 4′ | - | 147.5 | - | 147.6 |
| 5′ | 6.76, d (7.8) | 115.6 | 6.81, d (7.8) | 116.0 |
| 6′ | 6.84, dd (7.8, 1.8) | 122.3 | 6.85, dd (7.8, 1.8) | 121.6 |
| 7′ | 4.40, d (6.6) | 83.7 | 4.47, d (6.0) | 84.2 |
| 8′ | 4.54, m | 84.7 | 4.50, m | 85.1 |
| 9′ | 3.86, d (4.8) | 62.3 | 3.71, dd (12.0, 4.2) 3.51, q (6.0) | 62.2 |
| 1″ | 4.26, q (7.2) | 61.5 | 4.26, q (7.2) | 61.5 |
| 2″ | 1.35, t (7.2) | 14.6 | 1.35, t (7.2) | 14.6 |
| 3-OCH3 | 3.83, s | 56.5 | 3.92, s | 56.6 |
| 3′-OCH3 | 3.82, s | 56.3 | 3.85, s | 56.3 |
| 7′-OCH3 | 3.26, s | 57.0 | 3.26, s | 57.2 |
Table 3.
Inhibition effects on the growth of tumor cells in vitro (IC50, μM).
| Compounds | IC50 ± SEM (μM) | ||||
|---|---|---|---|---|---|
| HeLa | Hep3B | MCF-7 | H1299 | LO2 | |
| 1 | 118.237 ± 15.524 | > 150 | > 150 | > 150 | > 150 |
| 2 | 68.033 ± 8.321 | 85.366 ± 27.313 | > 150 | 77.366 ± 17.309 | > 150 |
| 3 | > 150 | > 150 | > 150 | > 150 | > 150 |
| 4 | > 150 | > 150 | > 150 | > 150 | > 150 |
| 5 | > 150 | > 150 | > 150 | > 150 | > 150 |
| 6 | 97.590 ± 20.504 | 124.909 ± 35.021 | 99.742 ± 17.001 | 68.376 ± 11.528 | > 150 |
| 5-Fu | 15.990 ± 0.121 | 32.223 ± 3.257 | 14.450 ± 2.193 | 24.450 ± 6.153 | > 150 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rong, Z.-J.; Hu, G.-S.; Lin, S.-Y.; Yan, T.; Li, N.; Zhao, Y.; Jia, J.-M.; Wang, A.-H. Constituents from the Seeds of Sophora Alopecuroides L. Molecules 2020, 25, 411. https://doi.org/10.3390/molecules25020411
AMA Style
Rong Z-J, Hu G-S, Lin S-Y, Yan T, Li N, Zhao Y, Jia J-M, Wang A-H. Constituents from the Seeds of Sophora Alopecuroides L. Molecules. 2020; 25(2):411. https://doi.org/10.3390/molecules25020411
Chicago/Turabian StyleRong, Zi-Jian, Gao-Sheng Hu, Shi-Yi Lin, Ting Yan, Na Li, Yue Zhao, Jing-Ming Jia, and An-Hua Wang. 2020. "Constituents from the Seeds of Sophora Alopecuroides L." Molecules 25, no. 2: 411. https://doi.org/10.3390/molecules25020411