Synthesis and Antiviral Activity of Novel 1,3,4-Thiadiazole Inhibitors of DDX3X

 , , , , ,
, , , , ,
Abstract
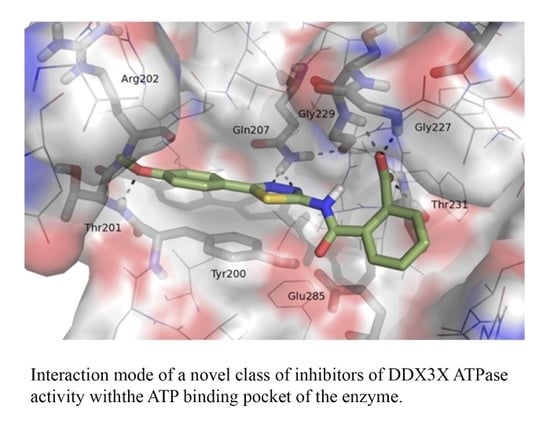
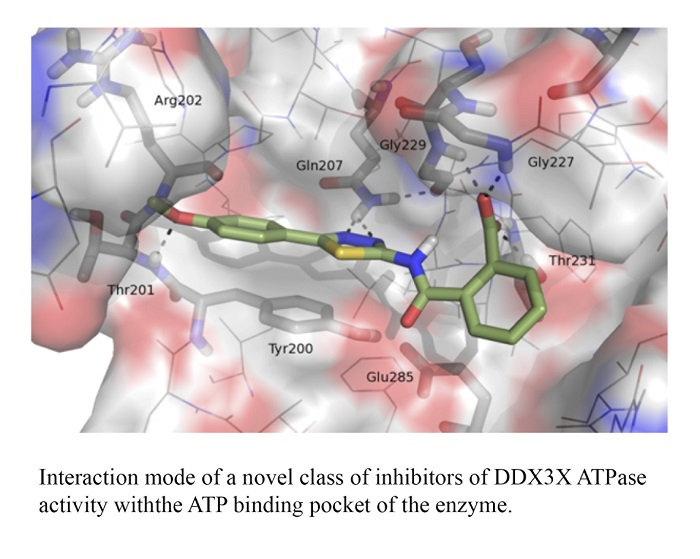
:
1. Introduction
2. Results
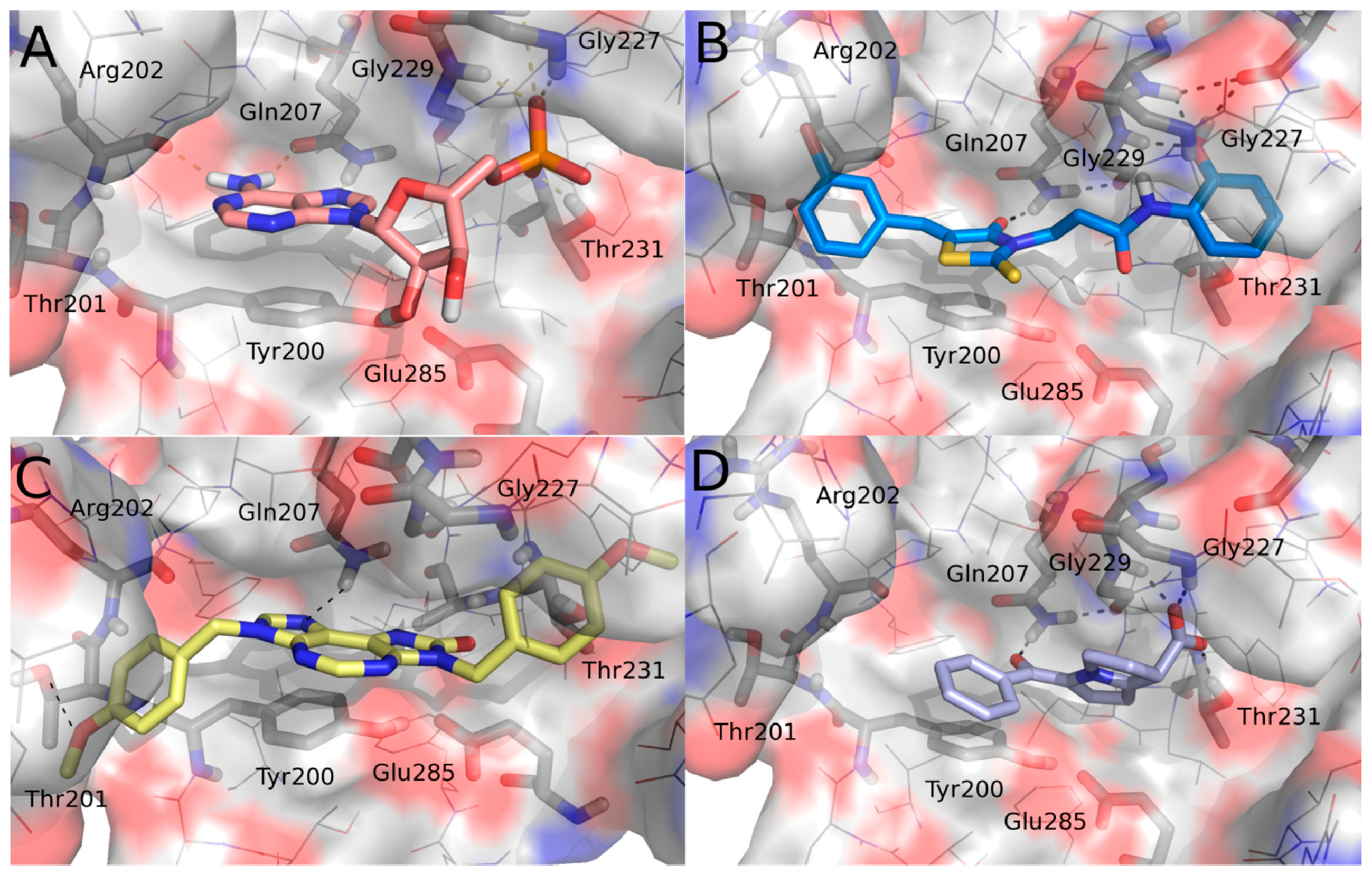
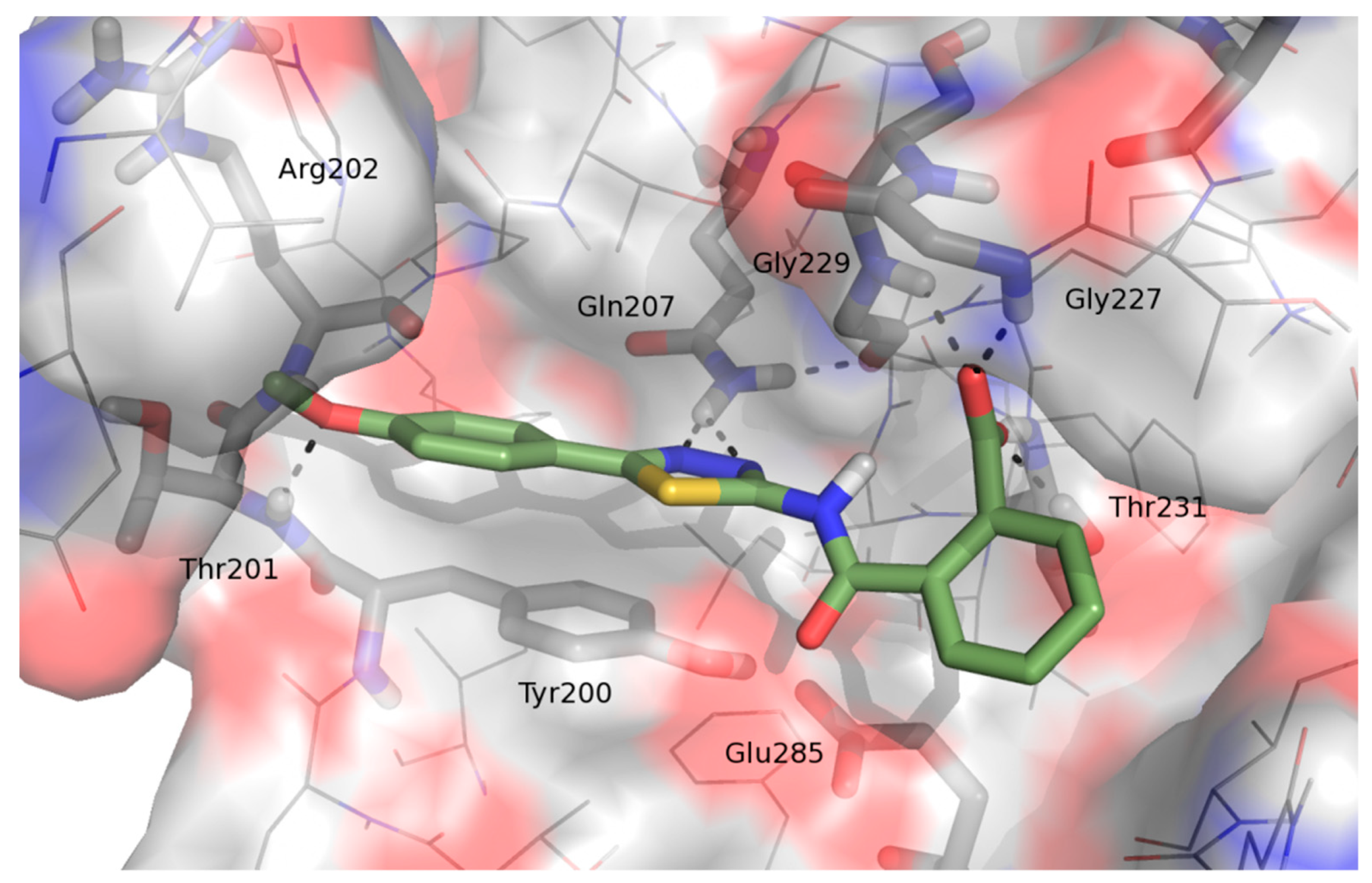
2.1. Molecular Modeling
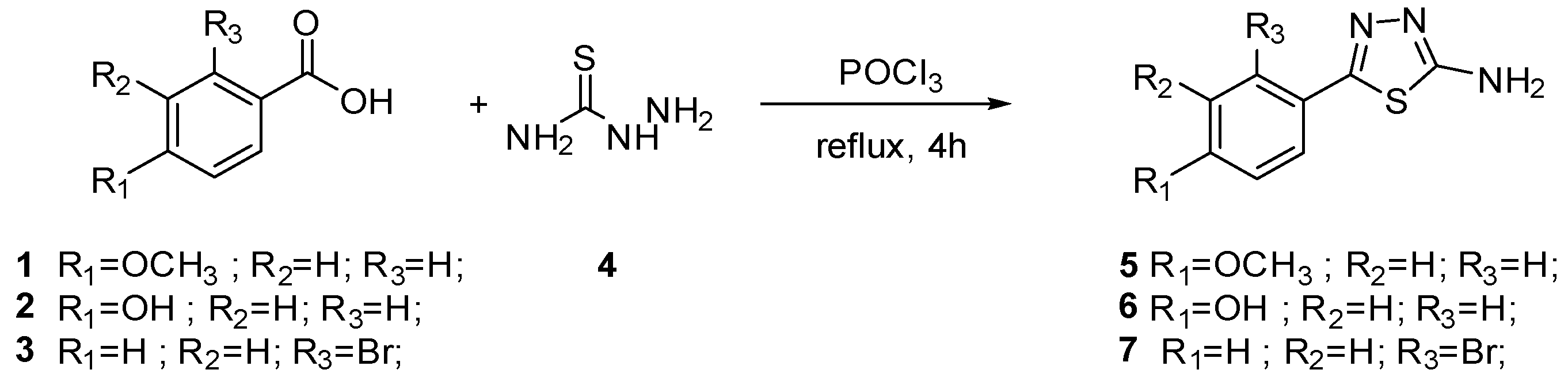
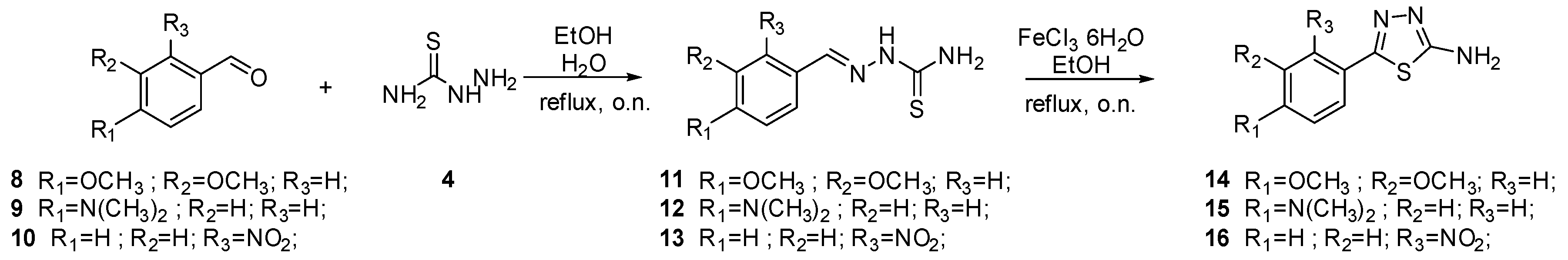
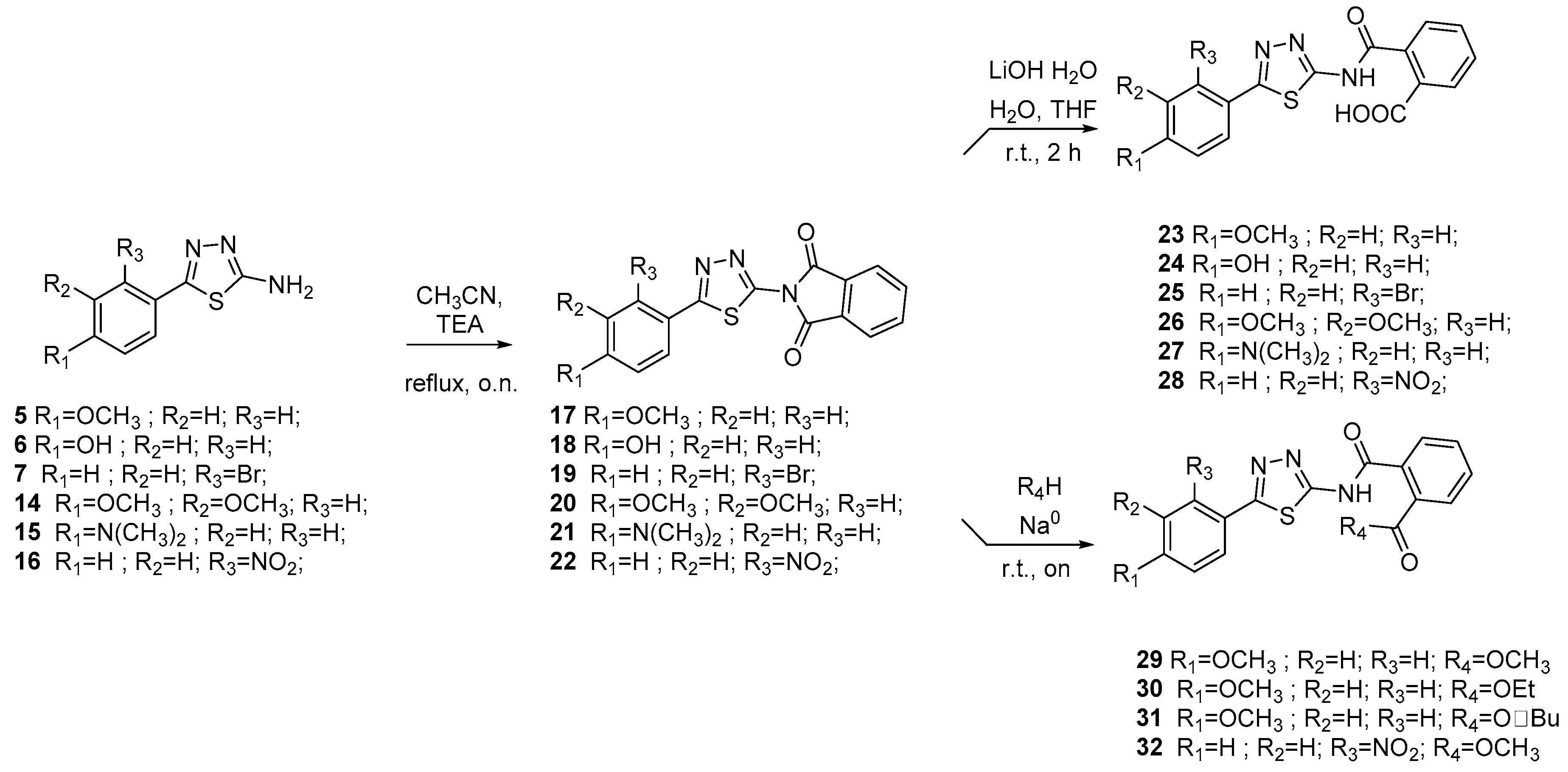
2.2. Chemistry
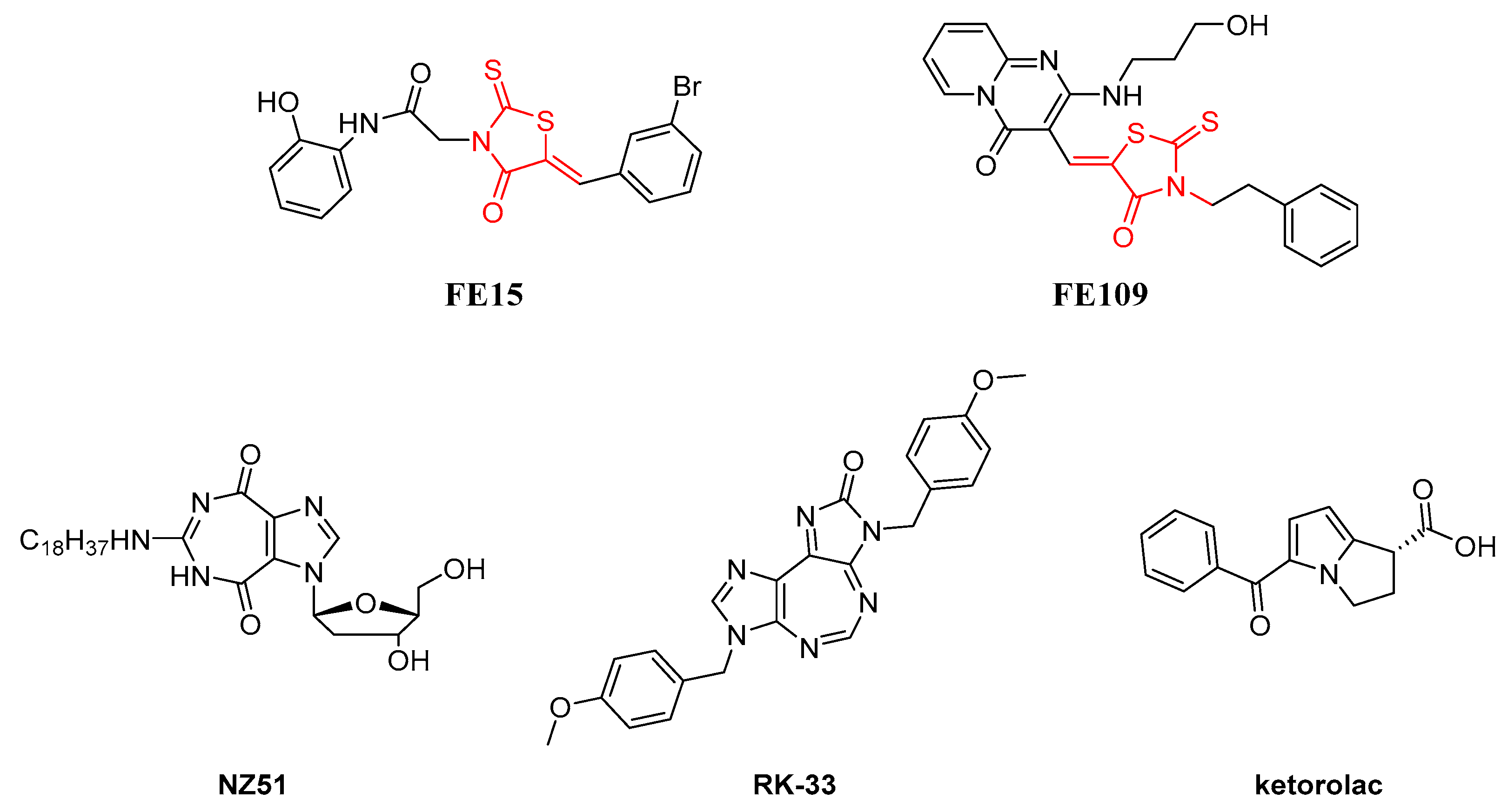
2.3. Biological Evaluation
2.4. ADME Assays
3. Experimental Section
3.1. General Procedures
3.1.1. General Procedure for the Synthesis of Amino Thiazoles 5–7
3.1.2. General Procedure for the Synthesis of Thiosemicarbazones 11–13
3.1.3. General Procedure for the Synthesis of Thiadiazoles 14–16
3.1.4. General Procedure for the Synthesis of Isoindoline-1,3-Diones 17–22
3.1.5. General Procedure for the Synthesis of Acids 23–28
3.1.6. General Procedure for the Synthesis of Esters 29–32
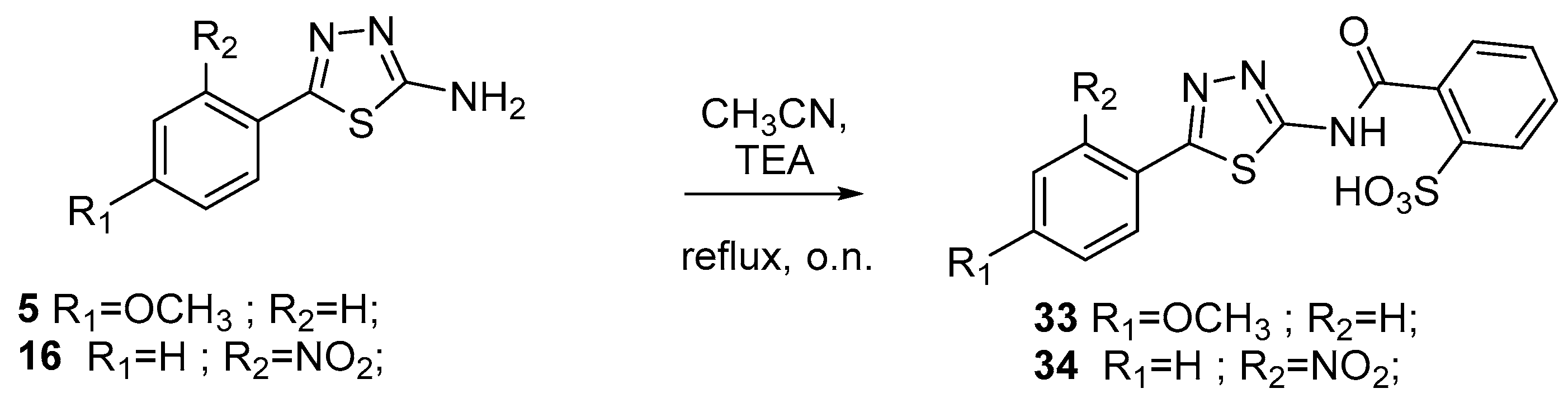
3.1.7. General Procedure for the Synthesis of Sulfonic Acids 33 and 34
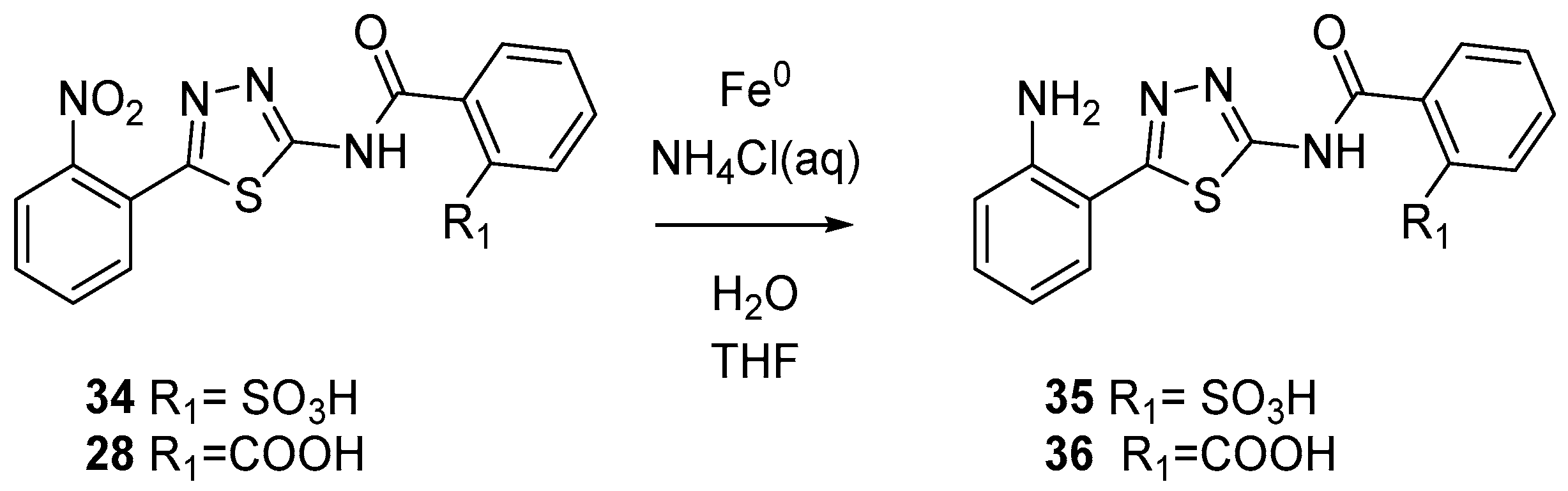
3.1.8. General Procedure for the Synthesis of Compounds 35 and 36
3.2. Enzymatic Assays
3.2.1. Protein Expression and Purification
3.2.2. ATPase Assay
3.3. Antiviral Assay
3.4. Cytotoxicity Assay
3.5. ADME Assay
3.5.1. Parallel Artificial Membrane Permeability Assay (PAMPA)
3.5.2. Water Solubility Assay
3.5.3. Microsomal Stability Assay
4. Discussion
Author Contributions
Notes
Funding
Acknowledgments
Conflicts of Interest
References
- Soto-Rifo, R.; Ohlmann, T. The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip. Rev. RNA 2013, 4, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Yedavalli, V.S.R.K.; Neuveut, C.; Chi, Y.-H.; Kleiman, L.; Jeang, K.-T. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 2004, 119, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Ishaq, M.; Hu, J.; Wu, X.; Fu, Q.; Yang, Y.; Liu, Q.; Guo, D. Knockdown of cellular RNA helicase DDX3 by short hairpin RNAs suppresses HIV-1 viral replication without inducing apoptosis. Mol. Biotechnol. 2008, 39, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Owsianka, A.M.; Patel, A.H. Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 1999, 257, 330–340. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kuroki, M.; Abe, K.I.; Dansako, H.; Ikeda, M.; Wakita, T.; Kato, N. DDX3 DEAD-Box RNA Helicase Is Required for Hepatitis C Virus RNA Replication. J. Virol. 2007, 81, 13922–13926. [Google Scholar] [CrossRef] [Green Version]
- Upadya, M.H.; Aweya, J.J.; Tan, Y.J. Understanding the interaction of hepatitis C virus with host DEAD-box RNA helicases. World J. Gastroenterol. 2014, 20, 2913–2926. [Google Scholar] [CrossRef]
- Chahar, H.S.; Chen, S.; Manjunath, N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436, 1–7. [Google Scholar] [CrossRef]
- Li, C.; Ge, L.; Li, P.; Wang, Y.; Dai, J.; Sun, M.; Huang, L.; Shen, Z.; Hu, X.; Ishag, H.; et al. Cellular DDX3 regulates Japanese encephalitis virus replication by interacting with viral un-translated regions. Virology 2014, 449, 70–81. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, S.G.; Kim, Y.; Song, K. Assignment of a human putative RNA helicase gene, DDX3, to human X chromosome bands p11.3→p11.23. Cytogenet. Cell Genet. 1998, 81, 178–179. [Google Scholar] [CrossRef]
- Högbom, M.; Collins, R.; van den Berg, S.; Jenvert, R.-M.; Karlberg, T.; Kotenyova, T.; Flores, A.; Hedestam, G.B.K.; Schiavone, L.H. Crystal Structure of Conserved Domains 1 and 2 of the Human DEAD-box Helicase DDX3X in Complex with the Mononucleotide AMP. J. Mol. Biol. 2007, 372, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Radi, M.; Falchi, F.; Garbelli, A.; Samuele, A.; Bernardo, V.; Paolucci, S.; Baldanti, F.; Schenone, S.; Manetti, F.; Maga, G.; et al. Discovery of the first small molecule inhibitor of human DDX3 specifically designed to target the RNA binding site: Towards the next generation HIV-1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2094–2098. [Google Scholar] [CrossRef] [PubMed]
- Fazi, R.; Tintori, C.; Brai, A.; Botta, L.; Selvaraj, M.; Garbelli, A.; Maga, G.; Botta, M. Homology Model-Based Virtual Screening for the Identification of Human Helicase DDX3 Inhibitors. J. Chem. Inf. Model. 2015, 55, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Brai, A.; Fazi, R.; Tintori, C.; Zamperini, C.; Bugli, F.; Sanguinetti, M.; Stigliano, E.; Esté, J.; Badia, R.; Franco, S.; et al. Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc. Natl. Acad. Sci. USA 2016, 113, 5388–5393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brai, A.; Martelli, F.; Riva, V.; Garbelli, A.; Fazi, R.; Zamperini, C.; Pollutri, A.; Falsitta, L.; Ronzini, S.; Maccari, L.; et al. DDX3X Helicase Inhibitors as a New Strategy To Fight the West Nile Virus Infection. J. Med. Chem. 2019, 62, 2333–2347. [Google Scholar] [CrossRef]
- Maga, G.; Falchi, F.; Garbelli, A.; Belfiore, A.; Witvrouw, M.; Manetti, F.; Botta, M. Pharmacophore modeling and molecular docking led to the discovery of inhibitors of human immunodeficiency Virus-1 replication targeting the human cellular aspartic acid−glutamic acid−alanine−aspartic acid box polypeptide 3. J. Med. Chem. 2008, 51, 6635–6638. [Google Scholar] [CrossRef]
- Yedavalli, V.S.R.K.; Zhang, N.; Cai, H.; Zhang, P.; Starost, M.F.; Hosmane, R.S.; Jeang, K.-T. Ring expanded nucleoside analogues inhibit RNA helicase and intracellular human immunodeficiency virus type 1 replication. J. Med. Chem. 2008, 51, 5043–5051. [Google Scholar] [CrossRef]
- Maga, G.; Falchi, F.; Radi, M.; Botta, L.; Casaluce, G.; Bernardini, M.; Irannejad, H.; Manetti, F.; Garbelli, A.; Samuele, A.; et al. Toward the discovery of novel Anti-HIV Drugs. Second-Generation inhibitors of the cellular ATPase DDX3 with improved Anti-HIV activity: Synthesis, structure-activity relationship analysis, cytotoxicity studies, and target validation. Chem. Med. Chem. 2011, 6, 1371–1389. [Google Scholar] [CrossRef]
- Xie, M.; Vesuna, F.; Botlagunta, M.; Bol, G.M.; Irving, A.; Bergman, Y.; Hosmane, R.S.; Kato, Y.; Winnard, P.T.; Raman, V. NZ51, a ring-expanded nucleoside analog, inhibits motility and viability of breast cancer cells by targeting the RNA helicase DDX3. Oncotarget 2015, 6, 29901–29913. [Google Scholar] [CrossRef]
- Bol, G.M.; Vesuna, F.; Xie, M.; Zeng, J.; Aziz, K.; Gandhi, N.; Levine, A.; Irving, A.; Korz, D.; Tantravedi, S.; et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol. Med. 2015, 7, 648–669. [Google Scholar] [CrossRef]
- Xie, M.; Vesuna, F.; Tantravedi, S.; Bol, G.M.; Heerma van Voss, M.R.; Nugent, K.; Malek, R.; Gabrielson, K.; van Diest, P.J.; Tran, P.T.; et al. RK-33 Radiosensitizes prostate cancer cells by blocking the RNA helicase DDX3. Cancer Res. 2016, 76, 6340–6350. [Google Scholar] [CrossRef]
- Kondaskar, A.; Kondaskar, S.; Kumar, R.; Fishbein, J.C.; Muvarak, N.; Lapidus, R.G.; Sadowska, M.; Edelman, M.J.; Bol, G.M.; Vesuna, F.; et al. Novel, Broad spectrum anticancer agents containing the tricyclic 5:7:5-fused diimidazodiazepine ring system. ACS Med. Chem. Lett. 2011, 2, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Samal, S.K.; Routray, S.; Veeramachaneni, G.K.; Dash, R.; Botlagunta, M. Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci. Rep. 2015, 5, 9982. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Tomašić, T.; Peterlin Mašič, L. Rhodanine as a scaffold in drug discovery: A critical review of its biological activities and mechanisms of target modulation. Expert Opin. Drug Discov. 2012, 7, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole-a promising structure in medicinal chemistry. Chem. Med. Chem. 2013, 8, 27–41. [Google Scholar] [CrossRef]
- Saladini, F.; Giannini, A.; Boccuto, A.; Vicenti, I.; Zazzi, M. Agreement between an in-house replication competent and a reference replication defective recombinant virus assay for measuring phenotypic resistance to HIV-1 protease, reverse transcriptase, and integrase inhibitors. J. Clin. Lab. Anal. 2018, 32, e22206. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 23, 24 and 29 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. ID | Structure | DDX3X ATPase | HIV-1 (NL4-3) | |
|---|---|---|---|---|
| Ki [b] (µM) | IC50 [b] (µM) | CC50 [c,d] (µM) | ||
| 17 |  | >50 | NT | NT |
| 23 |  | 1.3 ± 0.2 | >50 | >200 |
| 24 |  | 1.9 ± 0.4 | 2.8 ± 1.5 | 125 |
| 25 |  | NA | NT | NT |
| 26 |  | NA | NT | NT |
| 27 |  | 11.9 ± 1.9 | >50 | 130 |
| 28 |  | 18.3 ± 1.8 | 42.3 ± 5.2 | 74 |
| 29 |  | 15.6 | 10 | 45 |
| 30 |  | 20.1 ± 2.1 | >50 | >200 |
| 31 |  | 23.5 ± 2.5 | 16.0 ± 10 | 100 |
| 32 |  | NA | NT | NT |
| 33 |  | NA | NT | NT |
| 34 |  | 35.4 ± 3.5 | >50 | 110 |
| 35 |  | 20.1 ± 2.1 | >50 | 130 |
| 36 |  | NA | NT | NT |
| CPD ID | Metabolism a | AppP b | AqS c | |
|---|---|---|---|---|
 P 98.1% |  M1 1.9% | <0.1 (0%) | 3.3 (−5.03) | |
 P 98.8% |  M1 1.2% | <0.1 (0%) | 27.8 (−4.08) | |
 P 25.3% |  M1 61.7% |  M2 12.8% | 0.7 (36%) | 0.5 (−5.87) |
 P 75.7% |  M1 22.3 |  M2 2.0 | 2.1 (4.6%) | <0.1 (>−6) |
 P 53.8% |  M1 42.8% |  M2 3.3% | 6.1 (25.9%) | <0.1 (>−6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brai, A.; Ronzini, S.; Riva, V.; Botta, L.; Zamperini, C.; Borgini, M.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Boccuto, A.; et al. Synthesis and Antiviral Activity of Novel 1,3,4-Thiadiazole Inhibitors of DDX3X. Molecules 2019, 24, 3988. https://doi.org/10.3390/molecules24213988
Brai A, Ronzini S, Riva V, Botta L, Zamperini C, Borgini M, Trivisani CI, Garbelli A, Pennisi C, Boccuto A, et al. Synthesis and Antiviral Activity of Novel 1,3,4-Thiadiazole Inhibitors of DDX3X. Molecules. 2019; 24(21):3988. https://doi.org/10.3390/molecules24213988
Chicago/Turabian StyleBrai, Annalaura, Stefania Ronzini, Valentina Riva, Lorenzo Botta, Claudio Zamperini, Matteo Borgini, Claudia Immacolata Trivisani, Anna Garbelli, Carla Pennisi, Adele Boccuto, and et al. 2019. "Synthesis and Antiviral Activity of Novel 1,3,4-Thiadiazole Inhibitors of DDX3X" Molecules 24, no. 21: 3988. https://doi.org/10.3390/molecules24213988