3.1. Chemistry
IR spectra were recorded on a UR-20 spectrometer in the 400–3600 cm
−1 range in KBr.
1H-NMR spectra were recorded on a Bruker AVANCE 400 (400 MHz) spectrometer (Bruker BioSpin, Rheinstetten, Germany) with respect to the signals of residual protons of deuterated solvent (CDCl
3, DMSO-d
6).
13C-NMR spectra were recorded on a Bruker Avance 600 (151 MHz) spectrometer (Bruker BioSpin, Rheinstetten, Germany) relative to signals of residual protons of deuterated solvent (CDCl
3, DMSO-d
6). Elemental analysis was performed on a CHNS-O Elemental Analyser EuroEA3028-HT-OM (EuroVector S.p.A., Milan, Italy). The melting points were determined in glass capillaries on a Stuart SMP 10 instrument.
N-(4,4-diethoxybutyl)ureas
1a–
g were obtained as previously described [
38,
46].
The X-ray diffraction data for crystal of compound
8f were collected at 150 K on a Bruker AXS Smart Apex II CCD diffractometer in the ω and φ scan modes using graphite monochromated MoKα (λ 0.71073Å) radiation. The structure was solved by direct method and refined by the full matrix least-squares using the SHELXTL program [
71]. All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms were located from the Fourier electron density synthesis and were included in the refinement in the isotropic riding model approximation. All figures were made using OLEX2 [
72] and Mercury [
73]. Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Center (1941912,
www.ccdc.ac.uk).
6-Chloro-4-((3-hydroxyphenyl)amino)-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide (4). To the solution of benzofuroxan 3 (0.40 g, 1.6 mmol) in DMSO (3 mL) a solution of 3-aminophenol (0.35 g, 3.2 mmol) in DMSO (3 mL) was added at room temperature. The reaction mixture was stirred at room temperature for 2 h, reagents consumption was monitored by TLC (eluent: toluene/ethylacetate, 2/1). Then the reaction mixture was poured in water (100 mL), precipitate was filtered off, washed with water, and dried. Crude product was purified by column chromatography (eluent: toluene/ethylacetate, 2/1) and recrystallized from chloroform/hexane (3/1) to give target compound 4 as dark solid. Yield 93%, m.p. 128–130 °C; IR (ν, cm−1): 1563, 1628, 3094, 3320, 3447; 1H-NMR (400 MHz, CHCl3, δ ppm) 4.91 (s, 1H, NH), 6.73–6.75 (m, 1H, Ar-H), 6.80–6.84 (m, 2H, Ar-H), 6.92 (s, 1H, Ar-H), 7.27 (s, 1H, Ar-H), 8.49 (s, 1H, OH); 13C-NMR (151 MHz, CHCl3, δ ppm) 102.7, 111.8, 113.0, 114.8, 117.1, 128.4, 130.4, 130.6, 132.6, 138.8, 146.1, 156.3; Elemental analysis: calc. for C12H7ClN4O5 (322.5): C 44.67; H 2.19; Cl 10.99; N 17.36; found C 44.49; H 2.32; Cl 10.83; N 17.44. ESI m/z: [M + H]+: calc. for C12H8ClN4O5 323; found 323.
1-(4,4-Diethoxybutyl)-3-(2-(dimethylamino)ethyl)urea (1h). To a solution of N1,N1-dimethylethane-1,2-diamine (0.97 g, 11.0 mmol) in dichloromethane (11 mL) 1,1’-carbonyldiimidazole (2.0 g, 12.3 mmol) was added. The reaction mixture was stirred for 48 hours at room temperature. Then 4,4-diethoxybutan-1-amine (1.77 g, 11.0 mmol) was added and reaction mixture was stirred for another 48 h at room temperature. The reaction mixture was extracted with water (3 × 10 mL), the organic layer was separated, and solvent was removed in vacuum to give target compound 1h as yellow oil. Yield 64%; 1H-NMR (400 MHz, CHCl3, δ ppm) 1.10 (t, 6H, J = 7.1 Hz, CH3), 1.40–1.49 (m, 2H, CH2), 1.53–1.60 (m, 2H, CH2), 2.13 (s, 6H, CH3), 2.31 (t, 2H, J = 5.9 Hz, CH2), 3.04–3.11 (m, 2H, CH2), 3.12–3.20 (m, 2H, CH2), 3.36–3.45 (m, 2H, CH2), 3.51–3.60 (m, 2H, CH2), 4.39 (t, 1H, J = 5.6 Hz, CH), 5.37 (s, 1H,NH), 5.55 (s, 1H,NH); 13C-NMR (151 MHz, CHCl3, δ ppm) 15.2, 25.5, 31.1, 38.1, 40.0, 45.2, 59.3, 61.2, 102.8, 159.1.
General method for the synthesis of pyrrolidine-1-carboxamides 5–8. To a mixture of appropriate C-nucleophile (1.61 mmol) and chloroform (5 mL), urea 1 (1.61 mmol) and trifluoroacetic acid (0.18 g, 1.61 mmol; 0.36 g, 3.22 mmol in the case of urea 1h) were added. The reaction mixture was stirred for 24 h at room temperature, the solvent was removed in vacuum, and the residue was washed thoroughly with diethyl ether and dried in vacuum to give title compound.
2-(6-Hydroxybenzo[d][1,3]dioxol-5-yl)pyrrolidine-1-carboxamide (5a). Beige solid, yield 93%, m.p. 206–207 °C; IR (ν, cm−1): 1534, 1637, 2986, 3174, 3294; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.69–1.77 (m, 1H, CH2), 1.78–1.88 (m, 2H, CH2), 2.06–2.16 (m, 1H, CH2), 3.29–3.36 (m, 1H, CH2), 3.46–3.52 (m, 1H, CH2), 4.93–4.99 (m, 1H, CH), 5.48–5.80 (br s, 2H, NH2), 5.86 (dd, 2H, J = 4.3 Hz, 1.0 Hz, CH2), ), 6.41 (s, 1H, Ar-H), 6.47 (s, 1H, Ar-H); 9.07–9.86 (br s, 1H, OH). 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.8, 33.2, 46.9, 55.3, 98.4, 100.9, 106.2, 122.9, 140.1, 146.3, 149.1, 157.9; Elemental analysis: calc. for C12H14N2O4 (250): C, 57.59; H, 5.64; N, 11.19; found C, 57.70; H, 5.71; N, 11.01; ESI m/z: [M + H]+: calc. for C12H15N2O4 251; found 251.
2-(6-Hydroxybenzo[d][1,3]dioxol-5-yl)-N-phenylpyrrolidine-1-carboxamide (5b). Beige solid, yield 98%, m.p. 190–191 °C; IR (ν, cm−1): 1596, 1627, 2997, 3050; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.72–1.91 (m, 3H, CH2), 2.12–2.22 (m, 1H, CH2), 3.48–3.56 (m, 1H, CH2), 3.72–3.80 (m, 1H, CH2), 5.14–5.20 (m, 1H, CH), 5.85 (s, 2H, CH2), 6.44 (s, 1H, Ar-H), 6.52 (s, 1H, Ar-H), 6.90 (t, 1H, J = 7.4 Hz, Ar-H), 7.20 (t, 2H, J = 7.8 Hz, Ar-H), 7.43 (d, 2H, J = 8.1 Hz, Ar-H), 7.97 (s, 1H,NH), 9.33 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.7, 33.3, 47.1, 55.7, 98.2, 101.0, 106.1, 119.8, 122.1, 122.7, 128.7, 140.2, 140.9, 146.4, 148.8, 154.1; Elemental analysis: calc. for C18H18N2O4 (326): C, 66.25; H, 5.56; N, 8.58; found C, 66.31; H, 5.70; N, 8.35; ESI m/z: [M + H]+: calc. for C18H19N2O4 327; found 327.
2-(6-Hydroxybenzo[d][1,3]dioxol-5-yl)-N-(4-methoxyphenyl)pyrrolidine-1-carboxamide (5c). Beige solid, yield 91%, m.p. 112–114 °C; IR (ν, cm−1): 1597, 1627, 2971, 2989, 3037; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.72–1.92 (m, 3H, CH2), 2.10–2.27 (m, 1H, CH2), 3.45–3.56 (m, 2H, CH2), 3.68 (s, 3H, CH3), 5.12–5.22 (m, 1H, CH), 5.85 (s, 2H, CH2), 6.46 (s, 1H, Ar-H), 6.52 (s, 1H, Ar-H), 6.79 (d, 2H, J = 9.1 Hz, Ar-H), 7.33 (d, 2H, J = 9.0 Hz, Ar-H), 7.85 (s, 1H,NH), 9.39 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.7, 33.2, 47.0, 55.6, 55.6, 98.3, 100.9, 106.2, 114.0, 121.8, 128. 8, 133.9, 140.2, 146.4, 148.9, 154.5, 154.9; Elemental analysis: calc. for C19H20N2O5 (356): C, 64.04; H, 5.66; N, 7.86; found C, 64.21; H, 5.87; N, 7.94; ESI m/z: [M + H]+: calc. for C19H21N2O5 357; found 357.
N-(4-Bromophenyl)-2-(6-hydroxybenzo[d][1,3]dioxol-5-yl)pyrrolidine-1-carboxamide (5d). White solid, yield 95%, m.p. 164 °C; IR (ν, cm−1): 1595, 1627, 2848, 2978, 3047; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.71–1.94 (m, 3H, CH2), 2.11–2.23 (m, 1H, CH2), 3.46–3.57 (m, 1H, CH2), 3.70–3.81 (m, 1H, CH2), 5.14–5.24 (m, 1H, CH), 5.85 (s, 2H, CH2), 6.44 (s, 1H, Ar-H), 6.49 (s, 1H, Ar-H), 7.37 (d, 2H, J = 8.8 Hz, Ar-H), 7.44 (d, 2H, J = 8.7 Hz, Ar-H), 8.17 (s, 1H,NH), 9.30 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.7, 33.2, 47.1, 55.9, 98.2, 100.9, 106.1, 113.5, 121.6, 122.6, 131.5, 140.1, 140.4, 146.3, 148.8, 153.9; Elemental analysis: calc. for C18H17BrN2O4 (405): C, 53.35; H, 4.23; Br, 19.72; N, 6.91; found C, 53.41; H, 4.33; Br, 19.79; N, 6.79; ESI m/z: [M + H]+: calc. for C12H8ClN4O5 323; found 323; ESI m/z: [M + H]+: calc. for C18H18BrN2O4 406; found 406.
N-(4-Fluorophenyl)-2-(6-hydroxybenzo[d][1,3]dioxol-5-yl)pyrrolidine-1-carboxamide (5e). White solid, yield 91%, m.p. 184–185 °C; IR (ν, cm−1): 1595, 1638, 2883, 2948, 2989, 3164; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.72–1.83 (m, 2H, CH2), 1.88–1.98 (m, 1H, CH2), 2.11–2.28 (m, 1H, CH2), 3.48–3.59 (m, 1H, CH2), 3.68–3.78 (m, 1H, CH2), 5.17–5.27 (m, 1H, CH), 6.57 (d, 2H, J = 7.9 Hz, CH2), 6.64 (s, 1H, Ar-H), 6.85 (d, 1H, J = 8.2 Hz, CH2), 6.99–7.07 (m, 2H, Ar-H), 7.33 (s, 1H, Ar-H), 7.40–7.48 (m, 2H, Ar-H), 8.09 (s, 1H, Ar-H), 9.72 (s, 1H,NH), 9.84 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.6, 32.8, 47.1, 55.9, 102.2, 110.2, 113.7, 115.1 (d, J = 22.0 Hz), 121.6 (d, J = 7.6 Hz), 126.1, 130.5, 133.2, 137.8 (d, J = 124.5 Hz), 148.3, 154.2, 157.5 (d, J = 137.9 Hz); Elemental analysis: calc. for C18H17FN2O4 (344): C, 62.79; H, 4.98; N, 8.14; found C, 62.93; H, 5.09; N, 8.30; ESI m/z: [M + H]+: calc. for C18H18FN2O4 345; found 345.
N-Hexyl-2-(6-hydroxybenzo[d][1,3]dioxol-5-yl)pyrrolidine-1-carboxamide (5f). White solid, yield 72%, m.p. 91–93 °C; IR (ν, cm−1): 1535, 1624, 2720, 2855, 2929, 3115; 1H-NMR (400 MHz, CDCl3, δ ppm) 0.87 (t, 3H, J = 6.9 Hz, CH3), 1.23–1.31 (m, 6H, CH2), 1.42–1.51 (m, 2H, CH2), 2.04–2.13 (m, 2H, CH2), 2.18–2.32 (m, 2H, CH2), 3.10–3.16 (m, 1H, CH2), 3.19–3.28 (m, 1H, CH2), 3.40–3.54 (m, 2H, CH2), 5.12–5.21 (m, 1H, CH), 5.85 (d, 2H, J = 12.4 Hz, CH2), 6.47 (s, 1H, Ar-H), 6.61 (s, 1H, Ar-H); 13C-NMR (151 MHz, CDCl3, δ ppm) 14.0, 22.6, 24.9, 26.5, 30.1, 31.5, 32.7, 40.9, 46.4, 55.0, 99.9, 100.9, 105.3, 120.8, 141.0, 147.3, 150.4, 158.0; Elemental analysis: calc. for C18H26N2O4 (334): C, 64.65; H, 7.84; N, 8.38; found C, 64.75; H, 8.06; N, 8.25; ESI m/z: [M + H]+: calc. for C18H27N2O4 335; found 335.
N-Cyclohexyl-2-(6-hydroxybenzo[d][1,3]dioxol-5-yl)pyrrolidine-1-carboxamide (5g). White solid, yield 84%, m.p. 159–160 °C; IR (ν, cm−1): 1528, 1624, 2720, 2855, 2929, 3113, 3403; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.03–1.12 (m, 2H, CH2), 1.13–1.25 (m, 3H, CH2), 1.47–1.66 (m, 4H, CH2), 1.69–1.78 (m, 2H, CH2), 1.79–1.90 (m, 2H, CH2), 2.07–2.19 (m, 1H, CH2), 3.31–3.43 (m, 2H, CH2), 3.47–3.54 (m, 1H, CH2), 4.95–5.04 (m, 1H, CH), 5.85 (d, 2H, J = 8.2 Hz, CH2), 6.42 (s, 1H, Ar-H), 6.47 (s, 1H, Ar-H); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.9, 25.2, 25.8, 33.5, 33.6, 46.7, 49.1, 55.0, 98.3, 100.9, 106.2, 122.7, 140.3, 146.5, 149.1, 156.3; Elemental analysis: calc. for C18H24N2O4 (332): C, 65.04; H, 7.28; N, 8.43; found C, 65.16; H, 7.42; N, 8.35; ESI m/z: [M + H]+: calc. for C18H25N2O4 333; found 333.
2-(2-(6-Hydroxybenzo[d][1,3]dioxol-5-yl)pyrrolidine-1-carboxamido)-N,N-dimethylethan-1-aminium 2,2,2-trifluoroacetate (5h). White solid, yield 75%, m.p. 171–172 °C; IR (ν, cm−1): 1527, 1626, 2838, 2929, 3113, 3394; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.69–1.80 (m, 2H, CH2), 1.81–1.89 (m, 1H, CH2), 2.04–2.09 (m, 1H, CH2), 2.79 (s, 6H, CH3), 3.06–3.16 (m, 2H, CH2), 3.31–3.37 (m, 3H, CH2), 3.53–3.60 (m, 1H, CH2), 4.99–5.05 (m, 1H, CH), 5.85 (d, 2H, J = 11.0 Hz, CH2), 6.42 (s, 1H, Ar-H), 6.44 (s, 1H, Ar-H), 9.37 (s, 1H,NH), 9.63 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.4, 33.2, 35.9, 43.1, 46.7, 55.9, 57.9, 98.3, 100.9, 106.1, 117.7 (q, J = 300.4 Hz), 122.8, 139.9, 146.2, 148.9, 157.1, 158.6 (q, J = 31.0 Hz); Elemental analysis: calc. for C18H24F3N3O6 (435): C, 49.66; H, 5.56; N, 9.65; found C, 49.85; H, 5.67; N, 9.80; ESI m/z: [M − CF3CO2]+: calc. for C16H24N3O4 322; found 322.
6-Chloro-4-((3-hydroxy-4-(1-(phenylcarbamoyl)pyrrolidin-2-yl)phenyl)amino)-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide (6b). Dark solid, yield 87%, m.p. 165–170 °C with decomposition; IR (ν, cm−1): 753, 1383, 1559, 1627, 3073, 3336, 3396; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.80 (m, 2H, CH2), 1.91 (m, 1H, CH2), 2.20 (m, 1H, CH2), 3.53–3.54 (m, 1H, CH2), 3.75 (m, 1H, CH2), 5.22–5.23 (m, 1H, CH), 6.57 (d, 1H, J = 7.7 Hz, Ar-H), 6.64 (s, 1H, Ar-H), 6.87 (d, 1H, J = 8.2 Hz, Ar-H), 6.91 (t, 1H, J = 7.40 Hz, Ar-H), 7.20 (t, 2H, J = 7.9 Hz, Ar-H), 7.33 (s, 1H, Ar-H), 7.43 (d, 2H, J = 7.1 Hz, Ar-H), 7.99 (s, 1H, NH), 9.74 (s, 1H, NH), 9.84 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.7, 33.0, 47.1, 55.8, 102.3, 110.3, 113.9, 114.3, 119.9, 122.1, 126.2, 127.1, 128.1, 128.7, 130.6, 133.3, 138.5, 140.9, 148.3, 154.2, 154.4; Elemental analysis: calc. for C23H19ClN6O6 (510.9): C 54.07; H 3.75; Cl 6.94; N 16.45; found C 54.18; H 3.83; Cl 6.85; N 16.32; ESI m/z: [M + H]+: calc. for C23H20ClN6O6 511.9; found 512.
6-Chloro-4-((3-hydroxy-4-(1-((4-methoxyphenyl)carbamoyl)pyrrolidin-2-yl)phenyl)amino)-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide (6c). Dark solid, yield 89%, m.p. 196–202 °C with decomposition; IR (ν, cm−1): 750, 1377, 1557, 1628, 3075, 3306, 3391; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.73–1.84 (m, 2H, CH2), 1.86–1.94 (m, 1H, CH2), 2.12–2.23 (m, 1H, CH2), 3.47–3.53 (m, 1H, CH2), 3.69 (s, 3H, CH3), 3.69–3.74 (m, 1H, CH2), 5.16–5.23 (m, 1H, CH), 6.57 (d, 1H, J = 7.6 Hz, Ar-H), 6.63 (s, 1H, Ar-H), 6.78 (d, 2H, J = 9.02 Hz, Ar-H), 6.87 (d, 1H, J = 8.3 Hz, Ar-H), 7.31 (s, 1H, Ar-H), 7.33 (d, 2H, J = 4.8 Hz, Ar-H), 7.86 (br s, 1H, NH), 9.73 (br s, 1H, NH), 9.83 (nr s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.7, 32.1, 32.9, 47.1, 55.6, 102.2, 107.0, 110.3, 114.0, 121.8, 121.9, 123.0, 124.5, 127.1, 130.6, 133.9, 138.5, 141.3, 146.7, 148.2, 154.5, 154.9; Elemental analysis: calc. for C24H21ClN6O7 (540.9): C 53.29; H 3.91; Cl 6.55; N 15.54; found C 53.38; H 3.81; Cl 6.42; N 15.67; ESI m/z: [M + H]+: calc. for C24H22ClN6O7 541.9; found 542.
4-((4-(1-((4-Bromophenyl)carbamoyl)pyrrolidin-2-yl)-3-hydroxyphenyl)amino)-6-chloro-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide (6d). Dark solid, yield 95%, m.p. 166–170 °C with decomposition; IR (ν, cm−1): 753, 1362, 1560, 1629, 3100, 3297, 3382; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.74–1.84 (m, 2H, CH2), 1.84–1.97 (m, 1H, CH2), 2.15–2.24 (m, 1H, CH2), 3.47–3.57 (m, 1H, CH2), 3.68–3.77 (m, 1H, CH2), 5.18–5.25 (m, 1H, CH), 6.56 (d, 1H, J = 8.2 Hz, Ar-H), 6.63 (s, 1H, Ar-H), 6.85 (d, 1H, J = 8.24 Hz, Ar-H), 7.33 (s, 1H, Ar-H), 7.36 (d, 2H, J = 8.8 Hz, Ar-H), 7.44 (t, 2H, J = 8.8 Hz, Ar-H), 8.19 (br s, 1H, NH), 9.71 (br s, 1H, NH), 9.83 (br s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.6, 32.9, 47.1, 56.0, 110.2, 113.5, 113.8, 114.3, 120.8, 121.7, 126.1, 127.1, 130.6, 131.4, 132.0, 138.5, 139.5, 140.4, 148.3, 153.9, 154.4; Elemental analysis: calc. for C23H18BrClN6O6 (589.8): C 46.84; H 3.08; Cl 6.01; N 14.25; found C 46.92; H 3.12; Cl 6.05; N 14.34; ESI m/z: [M + H]+: calc. for C23H19BrClN6O6 590.8; found 591.
6-Chloro-4-((4-(1-((4-fluorophenyl)carbamoyl)pyrrolidin-2-yl)-3-hydroxyphenyl)amino)-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide (6e). Dark solid, yield 76%, m.p. 175–177 °C with decomposition; IR (ν, cm−1): 1348, 1564, 1623, 3247, 3404; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.65–1.83 (m, 2H, CH2), 1.86–1.98 (m, 1H, CH2), 2.11–2.27 (m, 1H, CH2), 3.49–3.58 (m, 1H, CH2), 3.69–3.79 (m, 1H, CH2), 5.16–5.25 (m, 1H, CH), 6.57 (dd, 1H, J = 8.2 Hz, J = 2.1 Hz, Ar-H), 6.64 (d, 1H, J = 2.1 Hz, Ar-H), 6.85 (d, 1H, J = 8.1 Hz, Ar-H), 6.98–7.07 (m, 2H, Ar-H), 7.33 (s, 1H, Ar-H), 7.41–7.49 (m, 2H, Ar-H), 8.09 (s, 1H, NH), 9.72 (s, 1H,NH), 9.84 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.6, 32.8, 47.1, 55.9, 102.2, 110.2, 113.7, 114.2, 115.10 (d, J = 22.0 Hz), 121.6 (d, J = 7.6 Hz), 126.1, 127.1, 128.0, 130.5, 133.2, 137.2 (d, J = 2.1 Hz), 138.4, 148.3, 154.2, 157.7 (d, J = 237.9 Hz); Elemental analysis: calc. for C23H18ClFN6O6 (528.9): C, 52.23; H, 3.43; Cl, 6.70; N, 15.89; found C, 52.00; H, 3.61; Cl, 6.87; N, 16.07; ESI m/z: [M + H]+: calc. for C23H19ClFN6O6 529.9; found 530.
6-Chloro-4-((4-(1-(hexylcarbamoyl)pyrrolidin-2-yl)-3-hydroxyphenyl)amino)-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide (6f). Dark solid, yield 44%, m.p. 121–122 °C with decomposition; IR (ν, cm−1): 722, 1347, 1563, 1630, 3106, 3194, 3426; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 0.84 (t, 3H, J = 6.7 Hz, CH3), 1.17–1.26 (m, 6H, CH2), 1.31–1.38 (m, 2H, CH2), 1.71–1.79 (m, 2H, CH2), 1.80–1.90 (m, 1H, CH2), 2.07–2.18 (m, 1H, CH2), 2.90–3.02 (m, 2H, CH2), 3.42–3.54 (m, 2H, CH2), 4.99–5.06 (m, 1H, CH), 5.85 (br s, 1H, NH), 6.55 (d, 1H, J = 7.2 Hz, Ar-H), 6.61 (s, 1H, Ar-H), 6.79 (d, 1H, J = 8.3 Hz, Ar-H), 7.34 (s, 1H, Ar-H), 9.01 (br s, 1H, NH), 9.85 (br s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 14.4, 22.5, 23.7, 26.5, 30.4, 31.5, 32.9, 46.7, 55.3, 102.3, 110.4, 113.7, 126.3, 127.1, 128.3, 130.6, 133.3, 134.9, 138.5, 148.3, 154.6, 157.0; Elemental analysis: calc. for C23H27ClN6O6 (518.9): C 53.23; H 5.24; Cl 6.83; N 16.19; found C 53.35; H 5.33; Cl 6.72; N 16.12; ESI m/z: [M + H]+: calc. for C23H28ClN6O6 519.9; found 520.
6-Chloro-4-((4-(1-(cyclohexylcarbamoyl)pyrrolidin-2-yl)-3-hydroxyphenyl)amino)-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide (6g). Dark solid, yield 67%, m.p. 170–175 °C with decomposition; IR (ν, cm−1): 632, 1348, 1564, 1623, 2934, 3247, 3404; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 0.99–1.23 (m, 6H, CH2), 1.50–1.79 (m, 7H, CH2), 2.30–2.35 (m, 1H, CH2), 2.64–2.69 (m, 1H, CH2), 3.27–3.33 (m, 1H, CH2), 3.33–3.39 (m, 1H, CH), 4.95–5.02 (m, 1H, CH), 6.57 (d, 1H, J = 8.05 Hz, Ar-H), 6.61 (s, 1H, Ar-H), 6.82 (d, 1H, J = 8.13 Hz, Ar-H), 7.31 (s, 1H, Ar-H), 7.93 (br s, 1H, NH), 9.83 (br s, 1H, NH), 9.94 (br s, 1H, OH). 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.8, 25.3, 25.8, 33.6, 46.8, 49.2, 55.0, 65.5, 102.3, 110.4, 114.0, 126.5, 127.1, 128.7, 129.4, 130.6, 133.4, 138.7, 148.3, 154.7, 156.3; Elemental analysis: calc. for C23H25ClN6O6 (516.9): C 53.44; H 4.87; Cl 6.86; N 16.26; found C 53.33; H 4.75; Cl 6.79; N 16.21; ESI m/z: [M + H]+: calc. for C23H26ClN6O6 517.9; found 518.
6-Chloro-4-((4-(1-((2-(dimethylammonio)ethyl)carbamoyl)pyrrolidin-2-yl)-3-hydroxyphenyl)amino)-5-nitrobenzo[c][1,2,5]oxadiazole 1-oxide 2,2,2-trifluoroacetate (6h). Beige solid, yield 57%, m.p. 211–212 °C with decomposition; IR (ν, cm−1): 1349, 1566, 1623, 3175, 3275, 3400; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.70–1.79 (m, 1H, CH2), 1.84–1.91 (m, 1H, CH2), 2.08–2.19 (m, 1H, CH2), 2.79 (s, 6H, CH3), 3.07–3.13 (m, 2H, CH2), 3.32–3.38 (m, 3H, CH2), 3.51–3.58 (m, 1H, CH2), 5.04–5.12 (m, 1H, CH), 6.54 (d, 1H, J = 7.9 Hz, Ar-H), 6.58 (s, 1H, Ar-H), 6.63 (s, 1H, Ar-H), 7.35 (s, 1H, NH), 9.76 (s, 1H,NH), 9.83 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 23.3, 32.9, 36.0, 43.1, 46.7, 55.9, 58.0, 102.3, 110.1, 113.5, 114.3, 114.7, 117.7 (q, J = 300.3 Hz), 126.1, 127.1, 130.5, 133.4, 138.5, 140.3, 148.3, 154.5, 157.1, 158.2, 158.5 (q, J = 31.0 Hz); Elemental analysis: calc. for C23H25ClF3N7O8 (619.9): C, 44.56; H, 4.06; Cl, 5.72; N, 15.82; found C, 44.79; H, 3.81; Cl, 5.87; N, 15.99; ESI m/z: [M − CF3CO2]+: calc. for C21H25ClN7O6 506.9; found 507.
2-(4-Hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)-N-phenylpyrrolidine-1-carboxamide (7b). White solid, yield 69%, m.p. 190–191 °C; IR (ν, cm−1): 1588, 1612, 1692, 2630, 2687, 2872, 2961; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.76–1.87 (m, 1H, CH2), 1.95–2.06 (m, 2H, CH2), 2.08–2.21 (m, 1H, CH2), 2.13 (s, 3H, CH3), 3.50–3.65 (m, 2H, CH2), 4.99-5.09 (m, 1H, CH), 5.98 (s, 1H, Ar-H), 6.88 (t, 1H, J = 7.3 Hz, Ar-H), 7.18 (t, 2H, J = 7.8 Hz, Ar-H), 7.42 (d, 2H, J = 8.1 Hz, Ar-H), 7.81 (s, 1H, NH), 11.54 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 19.7, 25.4, 30.8, 47.4, 52.1, 100.7, 118.1, 119.4, 121.8, 128.7, 141.1, 153.6, 161.3, 163.8, 166.1; Elemental analysis: calc. for C17H18N2O4 (314): C, 64.96; H, 5.77; N, 8.91; found C, 65.22; H, 5.89; N, 8.80; ESI m/z: [M + H]+: calc. for C17H19N2O4 315; found 315.
2-(4-Hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)-N-(4-methoxyphenyl)pyrrolidine-1-carboxamide (7c). White solid, yield 87%, m.p. 168–169 °C; IR (ν, cm−1): 1579, 1612, 1690, 2631, 2678, 2872, 3006; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.75–1.86 (m, 1H, CH2), 1.97–2.18 (m, 3H, CH2), 2.13 (s, 3H, CH3), 3.46–3.59 (m, 2H, CH2), 3.68 (s, 3H, CH3), 4.96–5.03 (m, 1H, CH), 5.97 (s, 1H, Ar-H), 6.78 (d, 2H, J = 8.8 Hz, Ar-H), 7.31 (d, 2H, J = 9.1 Hz, Ar-H), 7.69 (s, 1H, NH), 11.54 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 19.7, 25.4, 30.7, 47.3, 52.1, 55.6, 100.7, 102.9, 114.0, 121.2, 134.1, 154.0, 154.7, 161.3, 163.8, 166.2; Elemental analysis: calc. for C18H20N2O5 (344): C, 62.78; H, 5.85; N, 8.13; found C, 62.89; H, 5.98; N, 8.06; ESI m/z: [M + H]+: calc. for C18H21N2O5 345; found 315.
N-(4-Bromophenyl)-2-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)pyrrolidine-1-carboxamide (7d). White solid, yield 75%, m.p. 194–195 °C; IR (ν, cm−1): 1578, 1612, 1679, 2654, 2815, 2877, 2989; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.76–1.86 (m, 1H, CH2), 1.92–2.04 (m, 2H, CH2), 2.06–2.17 (m, 1H, CH2), 2.12 (s, 3H, CH3), 3.50–3.61 (m, 2H, CH2), 4.97–5.05 (m, 1H, CH), 5.96 (s, 1H, Ar-H), 7.35 (d, 2H, J = 8.7 Hz, Ar-H), 7.43 (d, 2H, J = 8.5 Hz, Ar-H), 8.03 (s, 1H, NH), 11.42 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 19.7, 25.4, 30.8, 47.4, 52.3, 100.7, 102.7, 113.1, 121.2, 131.5, 140.6, 153.3, 161.2, 163.7, 166.0; Elemental analysis: calc. for C17H17BrN2O4 (392): C, 51.92; H, 4.36; Br, 20.32; N, 7.12; found C, 52.09; H, 4.50; Br, 20.48; N, 7.31; ESI m/z: [M + H]+: calc. for C17H18BrN2O4 393; found 393.
N-(4-Fluorophenyl)-2-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)pyrrolidine-1-carboxamide (7e). White solid, yield 58%, m.p. 177–178 °C; IR (ν, cm−1): 1602, 1631, 1689, 2631, 2689, 2881, 3066; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.74–1.87 (m, 1H, CH2), 1.91–2.14 (m, 3H, CH2), 2.12 (s, 3H, CH3), 3.50–3.63 (m, 2H, CH2), 4.95–5.07 (m, 1H, CH), 5.97 (s, 1H, Ar-H), 6.98–7.04 (m, 2H, Ar-H), 7.39–7.49 (m, 2H, Ar-H), 7.92 (s, 1H, NH), 11.62 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 19.7, 25.4, 30.8, 47.3, 52.2, 100.6, 102.7, 115.1 (d, J = 22.0 Hz), 121.0 (d, J = 7.4 Hz), 137.4 (d, J = 2.5 Hz), 153.6, 157.5 (d, J = 237.6 Hz), 161.2, 163.7, 166.0; Elemental analysis: calc. for C17H17FN2O4 (332): C, 61.44; H, 5.16; N, 8.43; found 61.55; H, 4.89; N, 8.27; ESI m/z: [M + H]+: calc. for C17H18FN2O4 333; found 333.
N-Hexyl-2-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)pyrrolidine-1-carboxamide (7f). White solid, yield 47%, m.p. 137–138 °C; IR (ν, cm−1): 1592, 1690, 2631, 2686, 2935, 3079; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 0.84 (t, 3H, J = 7.0 Hz, CH3), 1.16–1.27 (m, 6H, CH2), 1.30–1.38 (m, 2H, CH2), 1.70–1.80 (m, 1H, CH2), 1.95–2.10 (m, 3H, CH2), 2.13 (s, 3H, CH3), 2.87–2.96 (m, 1H, CH2), 2.98–3.07 (m, 1H, CH2), 3.31–3.35 (m, 1H, CH2), 3.36–3.41 (m, 1H, CH2), 4.80–4.88 (m, 1H, CH), 5.70 (s, 1H, NH), 5.96 (s, 1H, Ar-H); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 14.4, 19.7, 22.5, 25.3, 26.5, 30.3, 30.4, 31.5, 35.6, 47.0, 51.9, 101.0, 103.0, 156.9, 161.4, 163.5, 167.0; Elemental analysis: calc. for C17H26N2O4 (322): C, 63.33; H, 8.13; N, 8.69; found C, 63.50; H, 8.31; N, 8.87; ESI m/z: [M + H]+: calc. for C17H27N2O4 323; found 323.
N-Cyclohexyl-2-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)pyrrolidine-1-carboxamide (7g). Beige solid, yield 60%, m.p. 184–185 °C; IR (ν, cm−1): 1593, 1692, 2631, 2686, 2934, 3079; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 0.99–1.14 (m, 3H, CH2), 1.16–1.28 (m, 2H, CH2), 1.48–1.54 (m, 1H, CH2), 1.55–1.66 (m, 3H, CH2), 1.69–1.77 (m, 2H, CH2), 1.99–2.09 (m, 3H, CH2), 2.12–2.22 (m, 1H, CH2), 2.14 (s, 3H, CH3), 3.36–3.39 (m, 1H, CH2), 3.40–3.47 (m, 1H, CH2), 4.78–4.87 (m, 1H, CH), 5.36 (s, 1H, NH), 5.97 (s, 1H, Ar-H), 12.01 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 19.7, 25.1, 25.3, 25.8, 30.3, 33.6, 47.1, 49.1, 51.6, 100.8, 102.8, 156.1, 161.7, 163.6, 167.0; Elemental analysis: calc. for C17H24N2O4 (320): C, 63.73; H, 7.55; N, 8.74; found C, 63.87; H, 7.76; N, 8.59; ESI m/z: [M + H]+: calc. for C17H25N2O4 321; found 321.
2-(2-(4-Hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)pyrrolidine-1-carboxamido)-N,N-dimethylethan-1-aminium 2,2,2-trifluoroacetate (7h). White solid, yield 77%, m.p. 146–147 °C; IR (ν, cm−1): 1592, 1692, 2683, 2985, 3064; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.73–1.81 (m, 1H, CH2), 1.83–1.90 (m, 1H, CH2), 1.96–2.08 (m, 2H, CH2), 2.12 (s, 3H, CH3), 2.78 (s, 6H, CH3), 3.05–3.12 (m, 2H, CH2), 3.21–3.30 (m, 1H, CH2), 3.32–3.39 (m, 3H, CH2), 4.85–4.94 (m, 1H, CH), 5.98 (s, 1H, Ar-H), 6.22 (s, 1H, NH), 9.56 (s, 1H, OH), 11.70 (s, 1H, NH+); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 19.7, 25.2, 31.3, 36.0, 43.1, 46.9, 52.4, 58.1, 100.8, 102.9, 177.7 (q, J = 31.2 Hz), 156.8, 158.6 (q, J = 299.4 Hz), 160.9, 163.6, 166.2; Elemental analysis: calc. for C17H24F3N3O6 (423): C, 48.23; H, 5.71; N, 9.92; found C, 48.30; H, 5.85; N, 10.14; ESI m/z: [M − CF3CO2]+: calc. for C15H24N3O4 310; found 310.
2-(4-Hydroxy-2-oxo-2H-chromen-3-yl)-N-phenylpyrrolidine-1-carboxamide (8b). Beige solid, yield 51%, m.p. 179–180 °C; IR (ν, cm−1): 1595, 1616, 1695, 2853, 2930, 3075. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.87–1.98 (m, 1H, CH2), 2.13–2.27 (m, 3H, CH2), 3.64–3.71 (m, 2H, CH2), 5.23–5.29 (m, 1H, CH), 6.91 (t, 1H, J = 7.4 Hz, Ar-H), 7.19 (t, 3H, J = 7.9 Hz, Ar-H), 7.32–7.37 (m, 2H, Ar-H, NH), 7.42 (d, 2H, J = 8.0 Hz, Ar-H), 7.59 (t, 1H, J = 8.0 Hz, Ar-H), 7.94 (d, 1H, J = 7.9 Hz, Ar-H), 8.26 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 25.8, 29.6, 47.8, 53.2, 106.5, 116.5, 118.1, 120.2, 122.3, 124.0, 124.3, 128.7, 132.5, 140.6, 152.7, 154.9, 161.3, 162.2; Elemental analysis: calc. for C20H18N2O4 (350): C, 68.56; H, 5.18; N, 8.00; found C, 68.70; H, 5.40; N, 7.89; ESI m/z: [M + H]+: calc. for C20H19N2O4 351; found 351.
2-(4-Hydroxy-2-oxo-2H-chromen-3-yl)-N-(4-methoxyphenyl)pyrrolidine-1-carboxamide (8c). Beige solid, yield 45%, m.p. 152–153 °C; IR (ν, cm−1): 1596, 1617, 1697, 2847, 2984, 3036; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.81–2.00 (m, 1H, CH2), 2.12–2.23 (m, 2H, CH2), 2.24–2.34 (m, 1H, CH2), 3.59–3.66 (m, 2H, CH2), 3.69 (s, 3H, CH3), 5.18–5.28 (m, 1H, CH), 6.79 (d, 2H, J = 9.1 Hz, Ar-H), 7.25–7.38 (m, 5H, Ar-H, NH), 7.59 (t, 1H, J = 7.0 Hz, Ar-H), 7.92 (d, 1H, J = 6.7 Hz, Ar-H), 8.19 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 25.8, 29.4, 47.7, 53.2, 55.6, 106.5, 114.0, 116.4, 120.4, 122.3, 123.9, 124.3, 132.5, 133.3, 152.7, 155.1, 155.5, 161.2, 162.6; Elemental analysis: calc. for C21H20N2O5 (380): C, 66.53; H, 5.50; N, 7.49; found C, 66.53; H, 5.50; N, 7.49; ESI m/z: [M + H]+: calc. for C21H21N2O5 381; found 381.
N-(4-Bromophenyl)-2-(4-hydroxy-2-oxo-2H-chromen-3-yl)pyrrolidine-1-carboxamide (8d). Beige solid, yield 68%, m.p. 179 °C; IR (ν, cm−1): 1595, 1617, 2797, 2837, 2987, 3078; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.83–1.86 (m, 1H, CH2), 2.08–2.27 (m, 3H, CH2), 3.62–3.72 (m, 2H, CH2), 5.23–5.31 (m, 1H, CH), 7.30–7.40 (m, 5H, Ar-H, NH), 7.43 (d, 2H, J = 8.90 Hz), 7.59 (t, 1H, J = 8.2 Hz, Ar-H), 7.94 (d, 1H, J = 8.3 Hz, Ar-H), 8.39 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 25.8, 29.8, 47.8, 53.2, 106.5, 113.6, 116.5, 120.0, 121.8, 123.9, 124.3, 131.5, 132.4, 140.2, 152.7, 154.3, 161.3, 161.9; Elemental analysis: calc. for C20H17BrN2O4 (429): C, 55.96; H, 3.99; Br, 18.61; N, 6.53; found C, 56.14; H, 4.18; Br, 18.73; N, 6.70; ESI m/z: [M + H]+: calc. for C20H18BrN2O4 430; found 430.
N-(4-Fluorophenyl)-2-(4-hydroxy-2-oxo-2H-chromen-3-yl)pyrrolidine-1-carboxamide (8e). Beige solid, yield 51%, m.p. 165 °C; IR (ν, cm−1): 1589, 1624, 1697, 2847, 2983, 3036, 3106; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.92–1.98 (m, 1H, CH2), 1.12–1.28 (m, 3H, CH2), 3.61–3.70 (m, 2H, CH2), 5.22–5.29 (m, 1H, CH), 6.99–7.05 (m, 2H, Ar-H), 7.29–7.40 (m, 3H, Ar-H, NH), 7.40–7.47 (m, 2H, Ar-H), 7.59 (t, 1H, J = 8.5 Hz, Ar-H), 7.93 (d, 1H, J = 6.9 Hz, Ar-H), 8.33 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 25.7, 29.6, 47.7, 53.1, 106.5, 115.2 (d, J = 22.0 Hz), 116.4, 117.0, 121.9 (d, J = 7.7 Hz), 123.9, 124.2, 132.4, 136.9 (d, J = 2.5 Hz), 152.7, 154.8, 157.8 (d, J = 138.1 Hz), 161.3, 162.2; Elemental analysis: calc. for C20H17FN2O4 (368): C, 65.21; H, 4.65; N, 7.60; found C, 65.42; H, 4.78; N, 7.83; ESI m/z: [M + H]+: calc. for C20H18FN2O4 369; found 369.
N-Hexyl-2-(4-hydroxy-2-oxo-2H-chromen-3-yl)pyrrolidine-1-carboxamide (8f). Beige solid, yield 34%, m.p. 124-125 °C; IR (ν, cm−1): 1554, 1614, 1687, 2858, 2929, 2953, 3075, 3374; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 0.79 (t, 3H, J = 6.8 Hz, CH3), 1.19–1.22 (m, 5H, CH2), 1.30–1.35 (m, 1H, CH2), 1.37–1.41 (m, 1H, CH2), 1.85–1.95 (m, 1H, CH2), 1.07–1.16 (m, 1H, CH2), 2.20–2.17 (m, 1H, CH2), 2.40–2.48 (m, 1H, CH2), 2.94–3.02 (m, 2H, CH2), 3.03–3.10 (m, 1H, CH2), 3.31–3.39 (m, 1H, CH2), 3.41–3.49 (m, 1H, CH2), 5.06–5.14 (m, 1H, CH), 6.57 (s, 1H, NH); 7.29–7.33 (m, 2H, Ar-H), 7.55–7.60 (m, 1H, Ar-H), 7.87 (d, 1H, J = 7.8 Hz, Ar-H); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 14.3, 22.5, 25.7, 26.5, 28.8, 30.1, 31.5, 40.7, 47.4, 53.4, 106.3, 116.3, 117.2, 124.1, 124.2, 132.6, 153.0, 158.8, 161.1, 164.5; Elemental analysis: calc. for C20H26N2O4 (358): C, 67.02; H, 7.31; N, 7.82; found C, 67.29; H, 7.55; N, 7.99; ESI m/z: [M + H]+: calc. for C20H27N2O4 359; found 359.
Crystal data: C20H26N2O4, M = 358.43, colorless crystal 0.12 × 0.15 × 0.15 mm3, triclinic, space group P-1, Z = 6, a = 12.9336(12), b = 14.2803(13), c = 16.0108(14) Å, α = 70.825(2), β = 84.411(2), γ = 87.932(2)°, V = 2779.8(4) Å3, ρcalc = 1.285 g/cm3, μ = 0.9 mm−1, 33,903 reflections collected (±h, ±k, ±l), 14,791 independent (Rint 0.0849) and 7433 observed reflections [I ≥ 2σ(I)], 703 refined parameters, R = 0.0673, wR2 = 0.1723, max. residual electron density was 0.667 (−0.528) eÅ−3.
N-Cyclohexyl-2-(4-hydroxy-2-oxo-2H-chromen-3-yl)pyrrolidine-1-carboxamide (8g). Beige solid, yield 68%, m.p. 166–167 °C; IR (ν, cm−1): 1549, 1616, 1694, 2475, 2853, 2935, 3075, 3373; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.01–1.10 (m, 1H, CH2), 1.14–1.26 (m, 4H, CH2), 1.48–1.56 (m, 1H, CH2), 1.59–1.67 (m, 2H, CH2), 1.68–1.78 (m, 2H, CH2), 1.84–1.94 (m, 1H, CH2), 2.06–2.16 (m, 1H, CH2), 2.20–2.29 (m, 1H, CH2), 2.39–2.48 (m, 1H, CH2), 3.35–3.50 (m, 3H, CH2, CH), 5.04–5.13 (m, 1H, CH), 6.26 (s, 1H, OH), 7.30–7.35 (m, 2H, Ar-H), 7.59 (td, 1H, J = 7.8 Hz, J = 1.6 Hz, Ar-H) 7.88 (dd, 1H, J = 8.3 Hz, J = 1.6 Hz, Ar-H); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 25.4, 25.7, 28.7, 33.3, 33.5, 47.5, 49.9, 53.4, 106.2, 116.3, 117.2, 124.1, 124.2, 132.6, 153.0, 158.1, 161.1, 164.5; Elemental analysis: calc. for C20H24N2O4 (356): C, 67.40; H, 6.79; N, 7.86; found C, 67.54; H, 6.89; N, 7.73; ESI m/z: [M + H]+: calc. for C20H25N2O4 357; found 357.
2-(2-(4-Hydroxy-2-oxo-2H-chromen-3-yl)pyrrolidine-1-carboxamido)-N,N-dimethylethan-1-aminium 2,2,2-trifluoroacetate (8h). Beige solid, yield 73%, m.p. 147–148 °C; IR (ν, cm−1): 1544, 1615, 1684, 2718, 2876, 2957, 3038, 3368; 1H-NMR (400 MHz, DMSO-d6, δ ppm) 1.83–1.94 (m, 1H, CH2), 2.08–2.22 (m, 2H, CH2), 2.24–2.35 (m, 1H, CH2), 2.78 (s, 6H, CH3), 3.06–3.15 (m, 2H, CH2), 3.26–3.33 (m, 1H, CH2), 3.35–3.41 (m, 1H, CH2), 3.43–3.49 (m, 2H, CH2), 5.08–5.16 (m, 1H, CH), 6.75 (s, 1H, NH), 7.29–7.38 (m, 2H, Ar-H), 7.59 (t, 1H, J = 8.2 Hz, Ar-H) 7.92 (d, 1H, J = 7.9 Hz, Ar-H) 9.62 (s, 1H, OH); 13C-NMR (151 MHz, DMSO-d6, δ ppm) 25.6, 29.7, 36.0, 43.0, 47.2, 53.3, 57.4, 106.2, 116.2, 116.9 (q, J = 199.9 Hz), 117.4, 124.1, 124.2, 132.4, 152.9, 157.9, 158.7 (q, J = 32.1 Hz), 161.3, 163.5; Elemental analysis: calc. for C20H24F3N3O6 (458): C, 52.29; H, 5.27; N, 9.15; found C, 52.48; H, 5.16; N, 8.97; ESI m/z: [M + H]+: calc. for C18H24N3O4 346; found 346.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
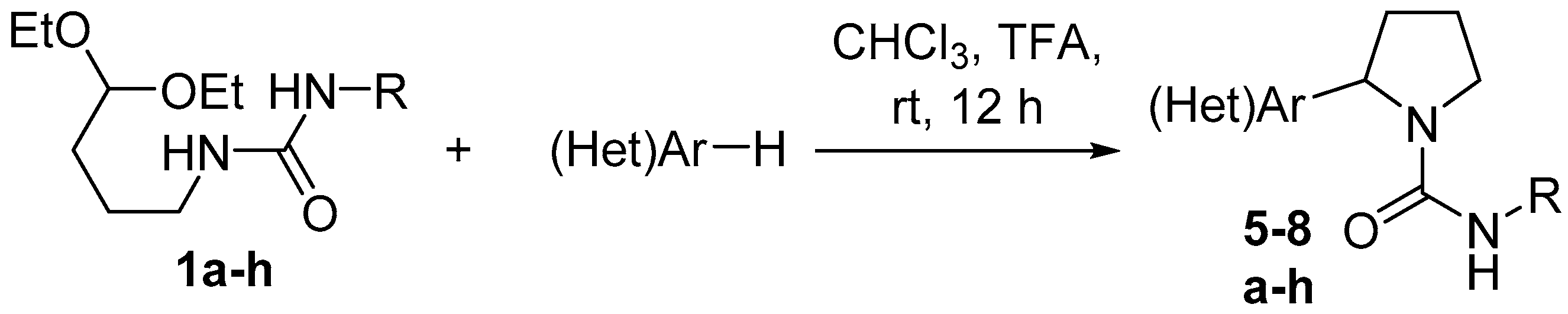{kind=link}



