
Bioactivity of Methoxylated and Methylated 1-Hydroxynaphthalene-2-Carboxanilides: Comparative Molecular Surface Analysis †

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
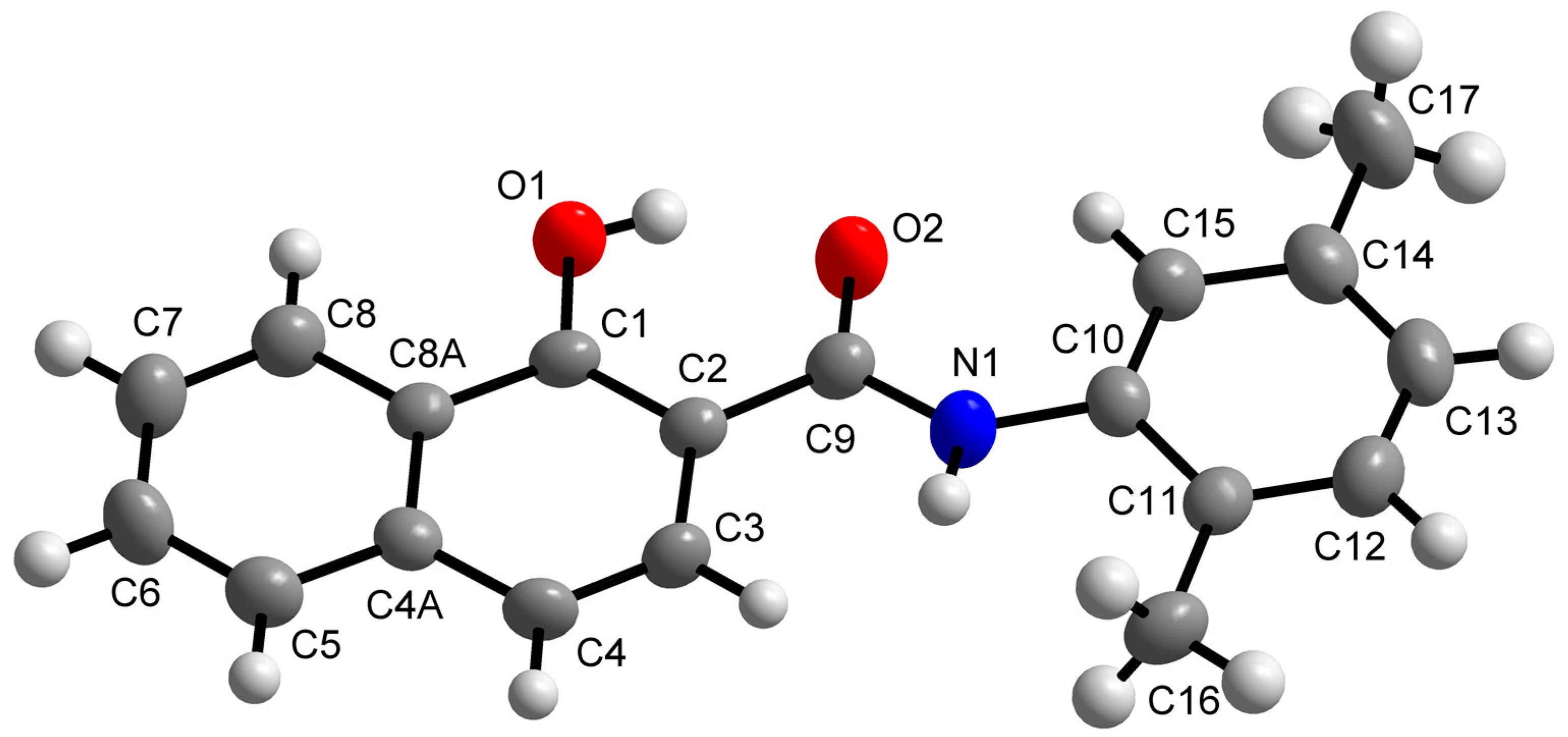
2.2. X-ray Crystallography
2.3. In Silico clogP Estimation and Experimental Lipophilicity Specification
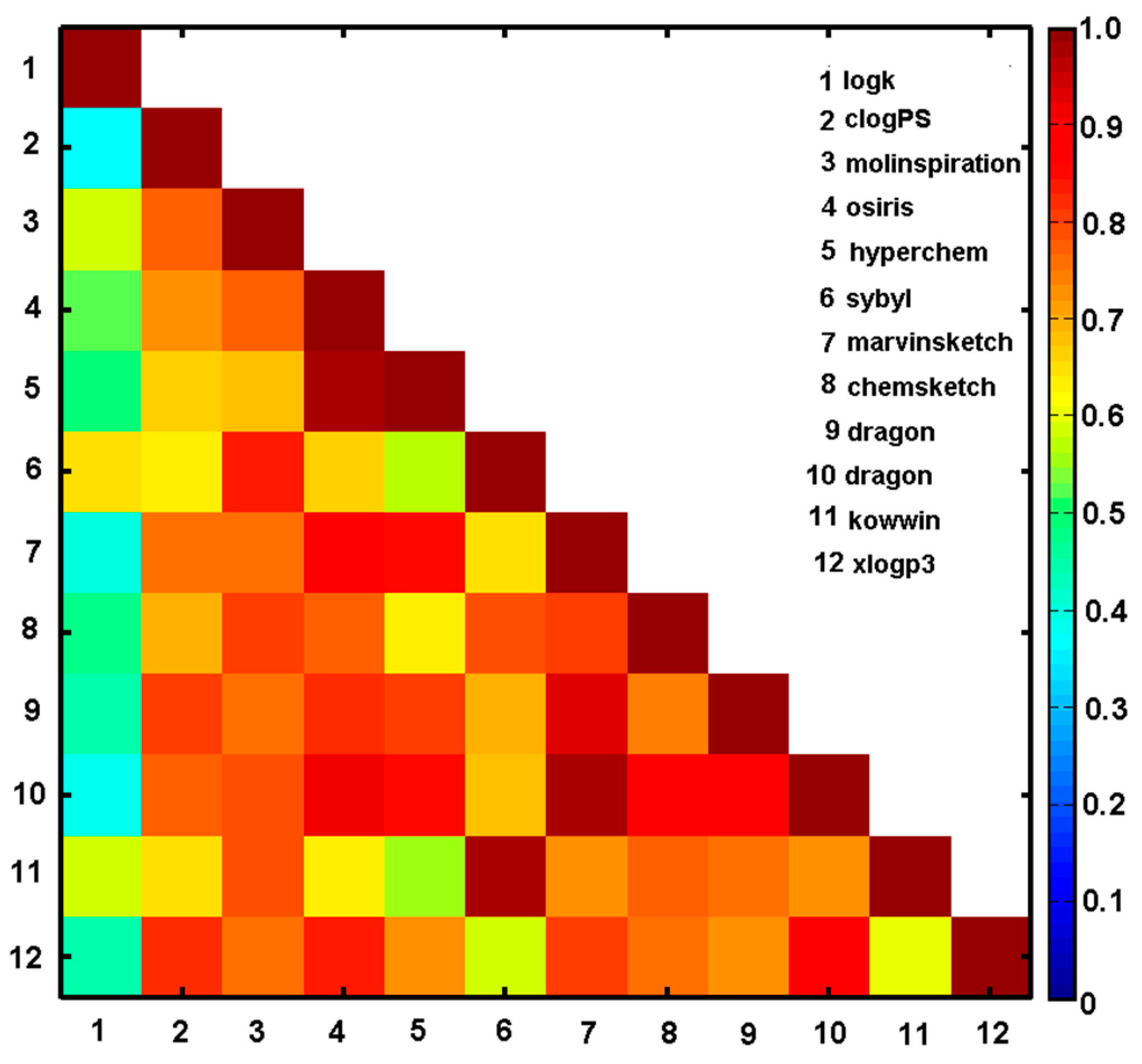
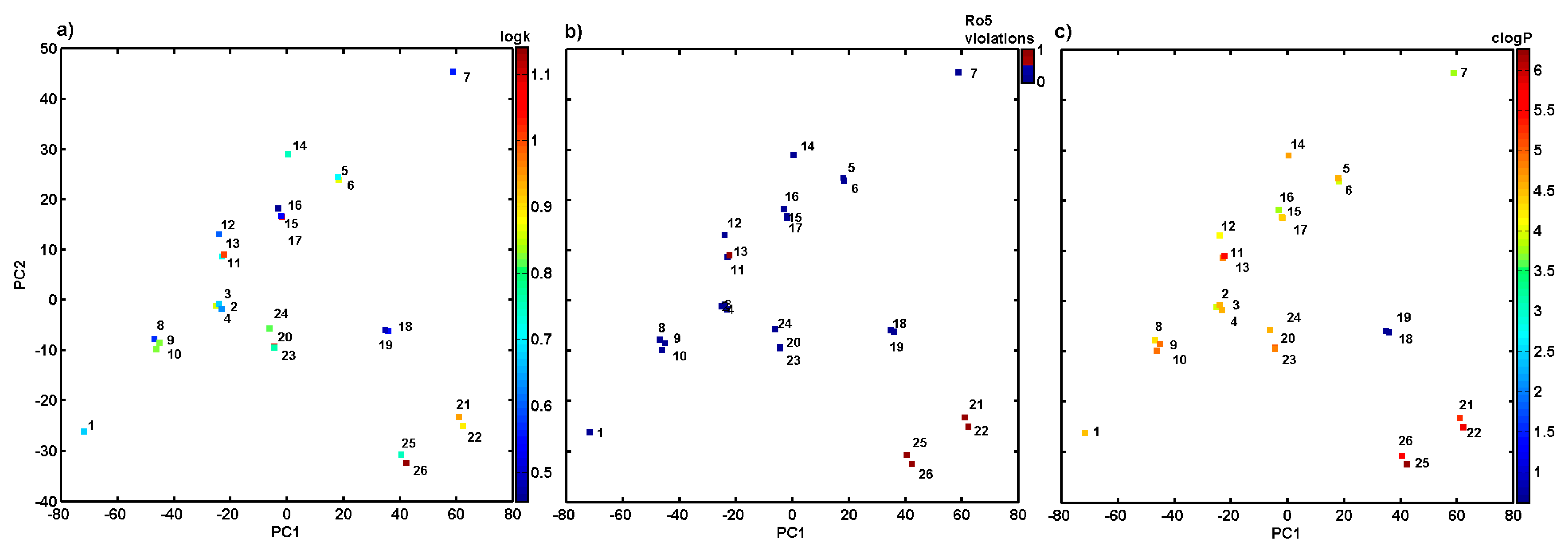
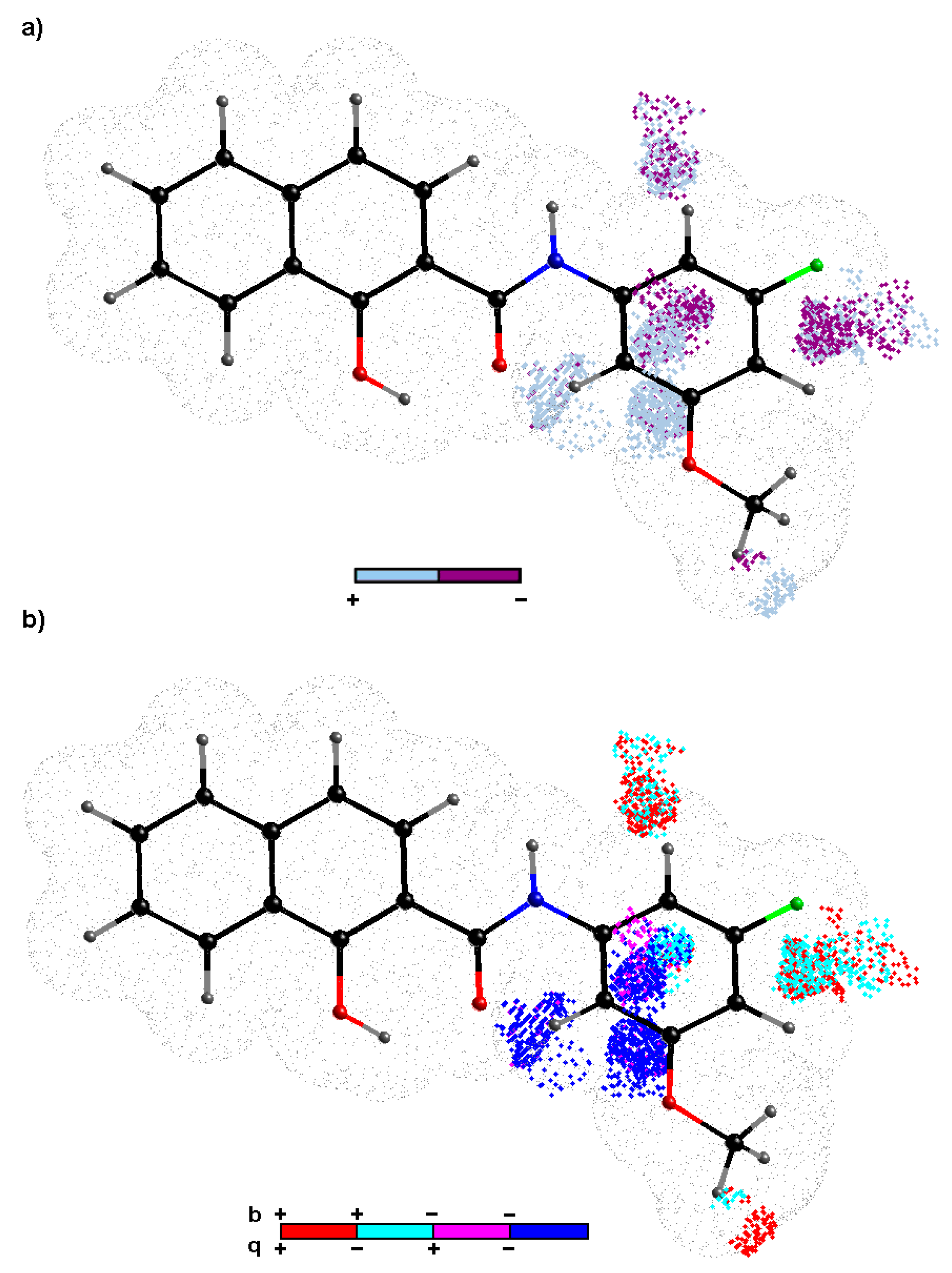
2.4. Variable-Oriented Similarity Evaluation
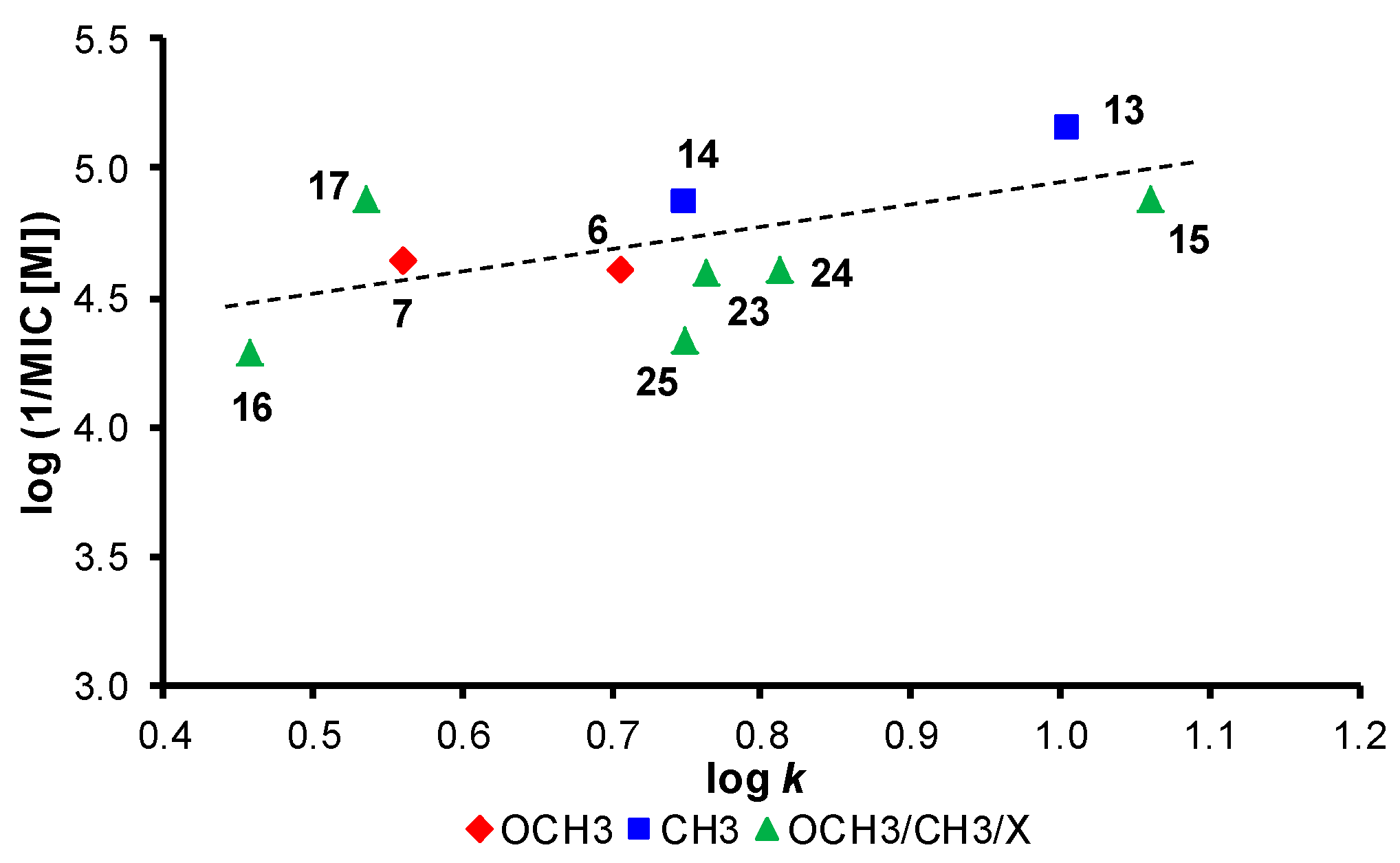
2.5. Biological Screening
2.5.1. In Vitro Antistaphylococcal Susceptibility Testing
2.5.2. In Vitro Antimycobacterial Testing
2.5.3. In Vitro Cytotoxicity Assay
2.5.4. Inhibition of Photosynthetic Electron Transport (PET) in Spinach Chloroplasts
2.6. Probability-Oriented Pharmacophore Modelling
3. Experimental Section
3.1. Chemistry—General Information
3.1.1. General Information
3.1.2. Synthesis
3.1.3. Lipophilicity Determination by HPLC (Capacity Factor k/Calculated log k)
3.1.4. X-ray Crystallography
3.2. Biological Testing
3.2.1. In Vitro Antibacterial Evaluation
3.2.2. In Vitro Antimycobacterial Evaluation
3.2.3. MTT Assay
3.2.4. In Vitro Cytotoxicity Assay
3.2.5. Study of Inhibition of Photosynthetic Electron Transport (PET) in Spinach Chloroplasts
3.3. Theoretical Calculations
3.3.1. Molecular Modelling
3.3.2. In Silico Lipophilicity Estimation, PCA and PLS analysis
3.3.3. Iterative PLS-based Variable Elimination
- Stage 1. Standard PLS analysis with LOO-CV to evaluate the performance of the PLS model
- Stage 2. Elimination of a matrix column with the lowest abs(mean(b)/std(b)) value
- Stage 3. Standard PLS analysis of the new matrix without the column eliminated in stage 2
- Stage 4. Recurrent repetition of steps 1–3 to maximize the LOO parameter
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Bobal, P.; Kollar, P.; Cizek, A.; Kralova, K.; et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013, 21, 6531–6541. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Gonec, T.; Bobal, P.; Kauerova, T.; Oravec, M.; Kollar, P.; et al. Antibacterial and herbicidal activity of ring-substituted 3-hydroxynaphthalene-2-carboxanilides. Molecules 2013, 18, 7977–7997. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Govender, R.; Keltosova, S.; Chambel, B.; Pereira, D.; Kollar, P.; Imramovsky, A.; et al. Antibacterial and herbicidal activity of ring-substituted 2-hydroxynaphthalene-1-carboxanilides. Molecules 2013, 18, 9397–9419. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Zadrazilova, I.; Nevin, E.; Kauerova, T.; Pesko, M.; Kos, J.; Oravec, M.; Kollar, P.; Coffey, A.; O’Mahony, J.; et al. Synthesis and biological evaluation of N-alkoxyphenyl-3-hydroxynaphthalene-2-carboxanilides. Molecules 2015, 20, 9767–9787. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Nevin, E.; Soral, M.; Kushkevych, I.; Gonec, T.; Bobal, P.; Kollar, P.; Coffey, A.; O’Mahony, J.; Liptaj, T.; et al. Synthesis and antimycobacterial properties of ring-substituted 6-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2015, 23, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Pospisilova, S.; Kauerova, T.; Kos, J.; Dohanosova, J.; Oravec, M.; Kollar, P.; Coffey, A.; Liptaj, T.; Cizek, A.; et al. N-Alkoxyphenylhydroxynaphthalenecarboxamides and their antimycobacterial activity. Molecules 2016, 21, 1068. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Pospisilova, S.; Holanova, L.; Stranik, J.; Cernikova, A.; Pudelkova, V.; Kos, J.; Oravec, M.; Kollar, P.; Cizek, A.; et al. Synthesis and antimicrobial evaluation of 1-[(2-substituted phenyl)carbamoyl]naphthalen-2-yl carbamates. Molecules 2016, 21, 1189. [Google Scholar] [CrossRef]
- Kos, J.; Kapustikova, I.; Clements, C.; Gray, A.I.; Jampilek, J. 3-Hydroxynaphthalene- 2-carboxanilides and their antitrypanosomal activity. Monatsh. Chem. 2018, 149, 887–892. [Google Scholar] [CrossRef]
- Kauerova, T.; Kos, J.; Gonec, T.; Jampilek, J.; Kollar, P. Antiproliferative and pro-apoptotic effect of novel nitro-substituted hydroxynaphthanilides on human cancer cell lines. Int. J. Mol. Sci. 2016, 17, 1219. [Google Scholar] [CrossRef]
- Spaczynska, E.; Mrozek-Wilczkiewicz, A.; Malarz, K.; Kos, J.; Gonec, T.; Oravec, M.; Gawecki, R.; Bak, A.; Dohanosova, J.; Kapustikova, I.; et al. Design and synthesis of anticancer 1-hydroxynaphthalene-2-carboxanilides with a p53 independent mechanism of action. Sci. Rep. 2019, 9, 6387. [Google Scholar] [CrossRef] [Green Version]
- Miro-Canturri, A.; Ayerbe-Algaba, R.; Smani, Y. Drug repurposing for the treatment of bacterial and fungal infections. Front. Microbiol. 2019, 10, 41. [Google Scholar] [CrossRef]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C.; et al. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef]
- Imperi, F.; Massai, F.; Ramachandran Pillai, C.; Longo, F.; Zennaro, E.; Rampioni, G.; Visca, P.; Leoni, L. New life for an old drug: The anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob. Agents Chemother. 2013, 57, 996–1005. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pesko, M.; Kralova, K.; Vejsova, M.; Stolarikova, J.; Vinsova, J.; Jampilek, J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy- N-[2-(arylamino)-1-alkyl-2-oxoethyl]benzamides. Molecules 2011, 16, 2414–2430. [Google Scholar] [CrossRef]
- Pauk, K.; Zadrazilova, I.; Imramovsky, A.; Vinsova, J.; Pokorna, M.; Masarikova, M.; Cizek, A.; Jampilek, J. New derivatives of salicylamides: Preparation and antimicrobial activity against various bacterial species. Bioorg. Med. Chem. 2013, 21, 6574–6581. [Google Scholar] [CrossRef]
- Zadrazilova, I.; Pospisilova, S.; Pauk, K.; Imramovsky, A.; Vinsova, J.; Cizek, A.; Jampilek, J. In vitro bactericidal activity of 4- and 5-chloro-2-hydroxy-N-[1-oxo-1-(phenylamino)alkan-2-yl]- benzamides against MRSA. BioMed Res. Int. 2015, 2015, 349534. [Google Scholar] [CrossRef]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Ferriz, J.M.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef]
- Walters, W.P.; Green, J.; Weiss, J.R.; Murcko, M.A. What do medicinal chemists actually make? A 50-year retrospective. J. Med. Chem. 2011, 54, 6405–6416. [Google Scholar] [CrossRef]
- Polanski, J.; Gasteiger, J. Computer representation of chemical compounds. In Handbook of Computational Chemistry; Leszczynski, J., Kaczmarek-Kedziera, A., Puzyn, T., Papadopoulos, M., Reis, H., Shukla, M., Eds.; Springer: Cham, Germany, 2017. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH Verlag: Weinheim, Germany, 2000. [Google Scholar]
- Martel, S.; Gillerat, F.; Carosati, E.; Maiarelli, D.; Tetko, I.V.; Mannhold, R.; Carrupt, P.-A. Large, chemically diverse dataset of logP measurements for benchmarking studies. Eur. J. Pharm. Sci. 2013, 48, 21–29. [Google Scholar] [CrossRef]
- Polanski, J.; Tkocz, A. Between descriptors and properties: Understanding the ligand efficiency trends for G protein-coupled receptor and kinase structure–activity data sets. J. Chem. Inf. Model. 2017, 57, 1321–1329. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Environ. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pesko, M.; Ferriz, J.M.; Kralova, K.; Vinsova, J.; Jampilek, J. Photosynthesis—Inhibiting efficiency of 4-chloro-2-(chlorophenylcarbamoyl)phenyl alkylcarbamates. Bioorg. Med. Chem. Lett. 2011, 21, 4564–4567. [Google Scholar] [CrossRef]
- Pesko, M.; Kos, J.; Kralova, K.; Jampilek, J. Inhibition of photosynthetic electron transport by 6-hydroxynaphthalene-2-carboxanilides. Indian J. Chem. B 2015, 54, 1511–1517. [Google Scholar]
- Jampilek, J.; Kralova, K.; Pesko, M.; Kos, J. Ring-substituted 8-hydroxyquinoline- 2-carboxanilides as photosystem II inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3862–3865. [Google Scholar] [CrossRef]
- Gonec, T.; Kralova, K.; Pesko, M.; Jampilek, J. Antimycobacterial N-alkoxyphenylhydroxy- naphthalenecarboxamides affecting photosystem II. Bioorg. Med. Chem. Lett. 2017, 27, 1881–1885. [Google Scholar] [CrossRef]
- Bowyer, J.R.; Camilleri, P.; Vermaas, W.F.J. Herbicides, Topics in Photosynthesis; Baker, N.R., Percival, M.P., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 10, pp. 27–85. [Google Scholar]
- Draber, W.; Tietjen, K.; Kluth, J.F.; Trebst, A. Herbicides in photosynthesis research. Angew. Chem. Int. Ed. 1991, 30, 1621–1633. [Google Scholar] [CrossRef]
- Otevrel, J.; Mandelova, Z.; Pesko, M.; Guo, J.; Kralova, K.; Sersen, F.; Vejsova, M.; Kalinowski, D.S.; Kovacevic, Z.; Coffey, A.; et al. Investigating spectrum of biological activity of ring-substituted salicylanilides and carbamoylphenylcarbamates. Molecules 2010, 15, 8122–8142. [Google Scholar] [CrossRef]
- Gonec, T.; Kos, J.; Pesko, M.; Dohanosova, J.; Oravec, M.; Liptaj, T.; Kralova, K.; Jampilek, J. Halogenated 1-hydroxynaphthalene-2-carboxanilides affecting photosynthetic electron transport in photosystem II. Molecules 2017, 22, 1709. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hann, M.M.; Oprea, T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef]
- Heinrichs, M.; May, R.; Heider, F.; Reimers, T.; Sy, S.B.; Peloquin, C.; Derendorf, H. Mycobacterium tuberculosis Strains H37Ra and H37Rv have equivalent minimum inhibitory concentrations to most antituberculosis drugs. Int. J. Mycobacteriol. 2018, 7, 156–161. [Google Scholar] [CrossRef]
- Honda, J.R.; Virdi, R.; Chan, E.D. Global environmental nontuberculous mycobacteria and their contemporaneous man-made and natural niches. Front. Microbiol. 2018, 9, 2029. [Google Scholar] [CrossRef]
- Measuring Cell Viability/Cytotoxicity. Dojindo EU GmbH, Munich, Germany. Available online: https://www.dojindo.eu.com/Protocol/Dojindo-Cell-Proliferation-Protocol.pdf (accessed on 18 June 2019).
- Grela, E.; Kozłowska, J.; Grabowiecka, A. Current methodology of MTT assay in bacteria—A review. Acta Histochem. 2018, 120, 303–311. [Google Scholar] [CrossRef]
- Bueno, J. Antitubercular in vitro drug discovery: Tools for begin the search. In Understanding Tuberculosis—New Approaches to Fighting Against Drug Resistance; IntechOpen: Rijeka, Croatia, 2012; pp. 147–168. [Google Scholar]
- Suffness, M.; Douros, J. Current status of the NCI plant and animal product program. J. Nat. Prod. 1982, 45, 1–14. [Google Scholar] [CrossRef]
- Shultz, M.D. Setting expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. [Google Scholar] [CrossRef]
- De Marco, R.; Bedini, A.; Spampinato, S.; Gentilucci, L. Synthesis of tripeptides containing d -trp substituted at the indole ring, assessment of opioid receptor binding and in vivo central antinociception. J. Med. Chem. 2014, 57, 6861–6866. [Google Scholar] [CrossRef]
- Fajkusova, D.; Pesko, M.; Keltosova, S.; Guo, J.; Oktabec, Z.; Vejsova, M.; Kollár, P.; Coffey, A.; Csollei, J.; Kralova, K.; et al. Anti-infective and herbicidal activity of N-substituted 2-aminobenzothiazoles. Bioorg. Med. Chem. 2012, 20, 7059–7068. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Mod. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Kubinyi, H. Hansch Analysis and Related Approaches; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1993. [Google Scholar]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2003, 46, 2310–2318. [Google Scholar] [CrossRef]
- Kapustikova, I.; Gonec, T.; Kos, J.; Spaczynska, E.; Oravec, M.; Dohanosova, J.; Liptaj, T.; Musiol, R.; Jampilek, J. Preparation and hydro-lipophilic properties of methoxylated and methylated 1-hydroxynaphthalene-2-carboxanilides. In Proceedings of the 22nd International Electronic Conference on Synthetic Organic Chemistry (ECSOC-22), 15 November–15 December 2018; MDPI: Basel, Switzerland, 2019; Volume 9, p. 43. [Google Scholar] [CrossRef]
- Bruker. Apex3; Bruker AXS Inc.: Madison, WI, USA, 2015. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond Version 4.5.3.; Crystal Impact GbR: Bonn, Germany, 2018. [Google Scholar]
- Abate, G.; Mshana, R.N.; Miörner, H. Evaluation of a colorimetric assay based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) for rapid detection of rifampicin resistance in Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 1998, 2, 1011–1016. [Google Scholar]
- Polanski, J.; Gieleciak, R.; Magdziarz, T.; Bak, A. GRID formalism for the comparative molecular surface analysis: Application to the CoMFA benchmark steroids, azo dyes, and HEPT derivatives. J. Chem. Inf. Comput. Sci. 2004, 44, 1423–1435. [Google Scholar] [CrossRef]
- Gieleciak, R.; Magdziarz, T.; Bak, A.; Polanski, J. Modeling robust QSAR. 1. Coding molecules in 3D-QSAR—From a point to surface sectors and molecular volumes. J. Chem. Inf. Model. 2005, 45, 1447–1455. [Google Scholar] [CrossRef]
- Smolinski, A.; Drobek, L.; Dombek, V.; Bąk, A. Modeling of experimental data on trace elements and organic compounds content in industrial waste dumps. Chemosphere 2016, 162, 189–198. [Google Scholar] [CrossRef]
- Stanton, D.T. QSAR and QSPR model interpretation using partial least squares (PLS) analysis. Curr. Comput. Drug Des. 2012, 8, 107–127. [Google Scholar] [CrossRef]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef]
- Centner, V.; Massart, D.-L.; De Noord, O.E.; De Jong, S.; Vandeginste, B.M.; Sterna, C. Elimination of uninformative variables for multivariate calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef]
Sample Availability: Samples of compounds are available from authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
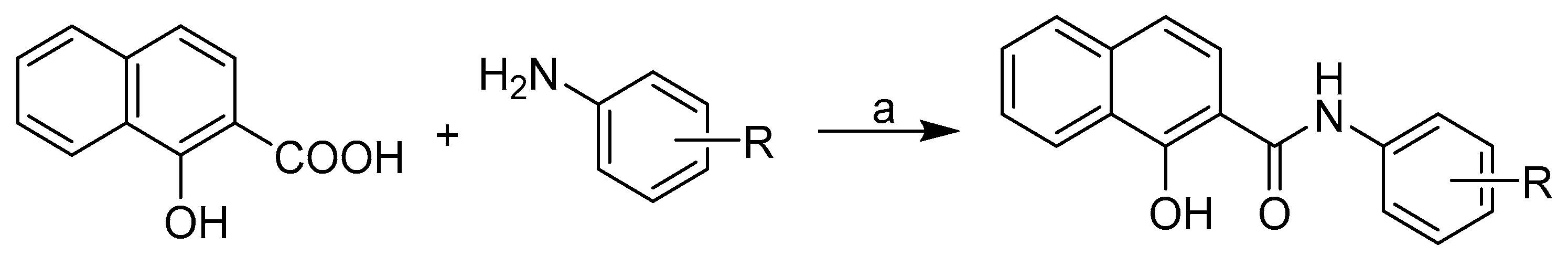{kind=link}
| Comp. | R | log k | MIC [μM] | Tox LD50 [μM] | PET IC50 [μM] | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SA | MRSA 63718 | MRSA SA 630 | MRSA SA 3202 | MT | MK | |||||
| 1 | H a | 0.6769 | >972 | >972 | 243 | 122 | 486 | 15.2 | >30 | 31.3 |
| 2 | 2-OCH3 a | 0.8584 | >873 | >873 | >873 | >873 | 436 | >873 | – | 199 |
| 3 | 3-OCH3 a | 0.6713 | 109 | >873 | >873 | >873 | 436 | >873 | – | 23.5 |
| 4 | 4-OCH3 a | 0.6284 | 873 | >873 | >873 | >873 | 436 | >873 | – | 79.5 |
| 5 | 2,5-OCH3 b | 0.8712 | 791 | 395 | 395 | 197 | 396 | 24.7 | – | 201 |
| 6 | 3,5-OCH3 b | 0.7048 | 12.3 | 24.7 | 24.7 | 24.7 | 24.7 | 24.7 | >30 | 13.4 |
| 7 | 3,4,5-OCH3 b | 0.5603 | 724 | 724 | 724 | 724 | 22.6 | 90.5 | – | 468 |
| 8 | 2-CH3 a | 0.5650 | 115 | 231 | 231 | 231 | 462 | 115 | – | 62.8 |
| 9 | 3-CH3 a | 0.8235 | 115 | 923 | 923 | 462 | 462 | 28.8 | – | 20.0 |
| 10 | 4-CH3 a | 0.8294 | 923 | 923 | 923 | 923 | 462 | 57.7 | – | 28.7 |
| 11 | 2,5-CH3 b | 0.7155 | 13.7 | 27.5 | 27.5 | 27.5 | 110 | 220 | >30 | 52.4 |
| 12 | 2,6-CH3 b | 0.5990 | 55.0 | 110 | 110 | 110 | 110 | 220 | – | 60.3 |
| 13 | 3,5-CH3 b | 1.0030 | 3.43 | 6.85 | 6.85 | 6.85 | 6.86 | 27.4 | >30 | 8.19 |
| 14 | 2,4,6-CH3 b | 0.7477 | >838 | >838 | >838 | >838 | 13.1 | 104 | – | 295 |
| 15 | 2-OCH3-5-CH3 b | 1.0603 | >832 | >832 | >832 | >832 | 13.0 | 52.0 | – | 588 |
| 16 | 2-OCH3-6-CH3 b | 0.4574 | >832 | >832 | >832 | >832 | 52.0 | 104 | – | 747 |
| 17 | 2-CH3-5-OCH3 b | 0.5362 | 52.0 | 832 | 104 | 208 | 13.0 | 104 | – | 142 |
| 18 | 2-OCH3-4-NO2 b | 0.5434 | 756 | 756 | 756 | 756 | 378 | 756 | – | 495 |
| 19 | 2-OCH3-5-NO2 b | 0.4827 | 756 | 756 | 756 | 756 | 378 | 756 | – | 155 |
| 20 | 2-OCH3-5-Br | 1.0432 | 687 | 687 | 687 | 687 | 344 | 687 | – | 569 |
| 21 | 2-OCH3-5-CF3 b | 0.9428 | 708 | 708 | 708 | 708 | 354 | 708 | – | 26.4 |
| 22 | 3-CF3-4-OCH3 b | 0.8915 | 354 | 354 | 354 | 5.53 | 354 | 88.5 | – | 18.5 |
| 23 | 3-F-5-OCH3 | 0.7629 | 1.60 | 3.21 | 3.21 | 3.21 | 25.6 | 12.0 | >30 | 10.1 |
| 24 | 2-Cl-5-OCH3 b | 0.8130 | 781 | 781 | 781 | 781 | 24.4 | 781 | – | 60.9 |
| 25 | 2-CH3-5-CF3 b | 0.7479 | 741 | 741 | 741 | 741 | 46.3 | 92.6 | – | 36.7 |
| 26 | 3-CF3-4-CH3 b | 1.1411 | 741 | 741 | 741 | 741 | 371 | 741 | – | 16.3 |
| AMP | – | – | 5.72 | 45.8 | 45.8 | 45.8 | – | – | – | – |
| INH | – | – | – | – | – | – | 36.6 | 233 | – | – |
| DCMU | – | – | – | – | – | – | – | – | – | 2.1 |
| Formula | C19H17NO2 |
|---|---|
| Formula weight | 291.33 |
| Temperature | 120(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P21/n |
| Unit cell dimensions | a = 6.887(4) Å, α = 90° |
| b = 16.134(11) Å, β = 99.81(2)° | |
| c = 13.620(8) Å, γ = 90° | |
| Volume | 1491.3(15) Å3 |
| Z | 4 |
| Density (calculated) | 1.298 g/cm3 |
| Absorption coefficient | 0.084 mm−1 |
| F(000) | 616 |
| Crystal size | 0.240 × 0.200 × 0.180 mm |
| Theta range for data collection | 1.974 to 25.074° |
| Index ranges | −8 ≤ h ≤ 8, −19 ≤ k ≤ 19, −16 ≤ l ≤16 |
| Reflections collected | 14255 |
| Independent reflections | 2635 [R(int) = 0.1005] |
| Completeness to θ | 25.074° (99.7%) |
| Absorption correction | Semi-empirical from equivalents |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2635/0/201 |
| Goodness-of-fit on F2 | 1.065 |
| Final R indices [I > 2σ(I)] | R1 = 0.0586, wR2 = 0.1246 |
| R indices (all data) | R1 = 0.1246, wR2 = 0.1506 |
| Largest diff. peak and hole | 0.196 and −0.228 e.Å−3 |
| No. | logP a | miLogP b | ClogP c | ClogP d | ClogP e | ClogP f | ClogP g | MlogP h | AlogP i | ClogP j | ClogP k |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3.98 | 4.27 | 3.65 | 3.51 | 4.43 | 3.75 | 4.50 | 3.91 | 3.26 | 4.47 | 4.20 |
| 2 | 4.27 | 4.28 | 3.58 | 3.25 | 3.93 | 3.59 | 4.40 | 3.61 | 3.24 | 3.99 | 4.17 |
| 3 | 4.25 | 4.30 | 3.58 | 3.25 | 4.52 | 3.59 | 4.66 | 3.61 | 3.24 | 4.55 | 4.17 |
| 4 | 4.23 | 4.33 | 3.58 | 3.25 | 4.52 | 3.59 | 4.45 | 3.61 | 3.24 | 4.55 | 4.17 |
| 5 | 3.95 | 4.31 | 3.51 | 3.00 | 3.96 | 3.44 | 4.61 | 3.06 | 3.22 | 4.07 | 4.15 |
| 6 | 4.02 | 4.31 | 3.51 | 3.00 | 4.55 | 3.44 | 4.70 | 3.06 | 3.22 | 4.63 | 4.15 |
| 7 | 3.47 | 3.90 | 3.44 | 2.75 | 3.81 | 3.28 | 4.46 | 2.51 | 3.21 | 3.98 | 4.12 |
| 8 | 4.44 | 4.67 | 3.99 | 3.97 | 4.29 | 4.26 | 4.96 | 4.15 | 3.74 | 4.46 | 4.57 |
| 9 | 4.47 | 4.69 | 3.99 | 3.97 | 4.94 | 4.26 | 4.96 | 4.15 | 3.74 | 5.02 | 4.57 |
| 10 | 4.47 | 4.72 | 3.99 | 3.97 | 4.94 | 4.26 | 4.96 | 4.15 | 3.74 | 5.02 | 4.57 |
| 11 | 4.72 | 5.09 | 4.34 | 4.44 | 4.79 | 4.78 | 5.42 | 4.38 | 4.23 | 5.00 | 4.93 |
| 12 | 4.69 | 4.13 | 4.34 | 4.44 | 4.14 | 4.78 | 5.42 | 4.38 | 4.23 | 4.44 | 4.93 |
| 13 | 4.74 | 5.09 | 4.34 | 4.44 | 5.44 | 4.78 | 5.42 | 4.38 | 4.23 | 5.57 | 4.93 |
| 14 | 4.54 | 4.53 | 4.68 | 4.91 | 4.64 | 5.29 | 5.88 | 4.61 | 4.71 | 4.99 | 5.30 |
| 15 | 4.57 | 4.70 | 3.93 | 3.72 | 4.43 | 4.11 | 4.86 | 3.84 | 3.73 | 4.54 | 4.54 |
| 16 | 4.50 | 4.68 | 3.93 | 3.72 | 3.78 | 4.11 | 4.86 | 3.84 | 3.73 | 3.98 | 4.54 |
| 17 | 4.59 | 4.70 | 3.93 | 3.72 | 4.37 | 4.11 | 5.12 | 3.84 | 3.73 | 4.54 | 4.54 |
| 18 | 4.10 | 4.21 | 2.66 | 1.27 | 4.21 | 3.53 | 4.79 | 3.39 | 3.13 | 4.53 | 4.00 |
| 19 | 4.07 | 4.21 | 2.66 | 1.27 | 4.21 | 3.53 | 4.95 | 3.39 | 3.13 | 4.53 | 4.00 |
| 20 | 4.07 | 5.06 | 4.31 | 4.04 | 4.99 | 4.36 | 5.86 | 3.95 | 3.99 | 4.88 | 4.87 |
| 21 | 4.82 | 5.15 | 4.43 | 4.14 | 5.23 | 4.47 | 6.44 | 4.18 | 4.18 | 4.95 | 5.06 |
| 22 | 4.82 | 5.15 | 4.43 | 4.14 | 5.82 | 4.47 | 6.26 | 4.18 | 4.18 | 5.52 | 5.06 |
| 23 | 4.65 | 4.42 | 3.69 | 3.39 | 4.86 | 3.74 | 5.20 | 3.73 | 3.44 | 4.75 | 4.27 |
| 24 | 5.14 | 4.93 | 4.19 | 3.77 | 4.58 | 4.20 | 5.26 | 3.84 | 3.90 | 4.63 | 6.08 |
| 25 | 5.12 | 5.54 | 4.85 | 4.86 | 5.63 | 5.14 | 6.31 | 4.99 | 4.68 | 5.42 | 5.45 |
| 26 | 5.07 | 5.54 | 4.85 | 4.86 | 6.28 | 5.14 | 6.31 | 4.99 | 4.68 | 5.98 | 5.45 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michnová, H.; Pospíšilová, Š.; Goněc, T.; Kapustíková, I.; Kollár, P.; Kozik, V.; Musioł, R.; Jendrzejewska, I.; Vančo, J.; Trávníček, Z.; et al. Bioactivity of Methoxylated and Methylated 1-Hydroxynaphthalene-2-Carboxanilides: Comparative Molecular Surface Analysis. Molecules 2019, 24, 2991. https://doi.org/10.3390/molecules24162991
Michnová H, Pospíšilová Š, Goněc T, Kapustíková I, Kollár P, Kozik V, Musioł R, Jendrzejewska I, Vančo J, Trávníček Z, et al. Bioactivity of Methoxylated and Methylated 1-Hydroxynaphthalene-2-Carboxanilides: Comparative Molecular Surface Analysis. Molecules. 2019; 24(16):2991. https://doi.org/10.3390/molecules24162991
Chicago/Turabian StyleMichnová, Hana, Šárka Pospíšilová, Tomáš Goněc, Iva Kapustíková, Peter Kollár, Violetta Kozik, Robert Musioł, Izabela Jendrzejewska, Ján Vančo, Zdeněk Trávníček, and et al. 2019. "Bioactivity of Methoxylated and Methylated 1-Hydroxynaphthalene-2-Carboxanilides: Comparative Molecular Surface Analysis" Molecules 24, no. 16: 2991. https://doi.org/10.3390/molecules24162991