Structural and Biological Characterizations of Novel High-Affinity Fluorescent Probes with Overlapped and Distinctive Binding Regions on CXCR4


Abstract
:1. Introduction
2. Results
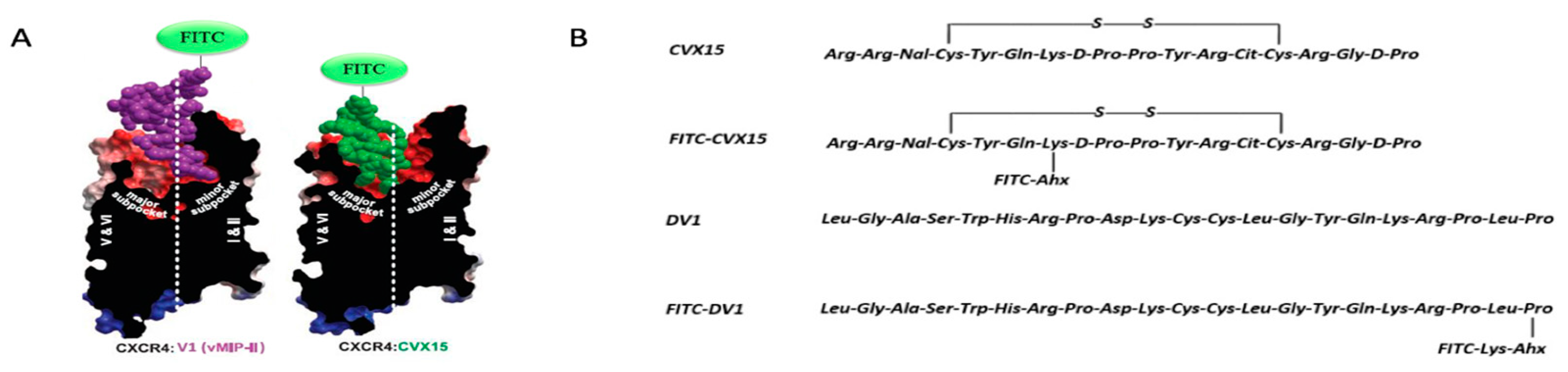
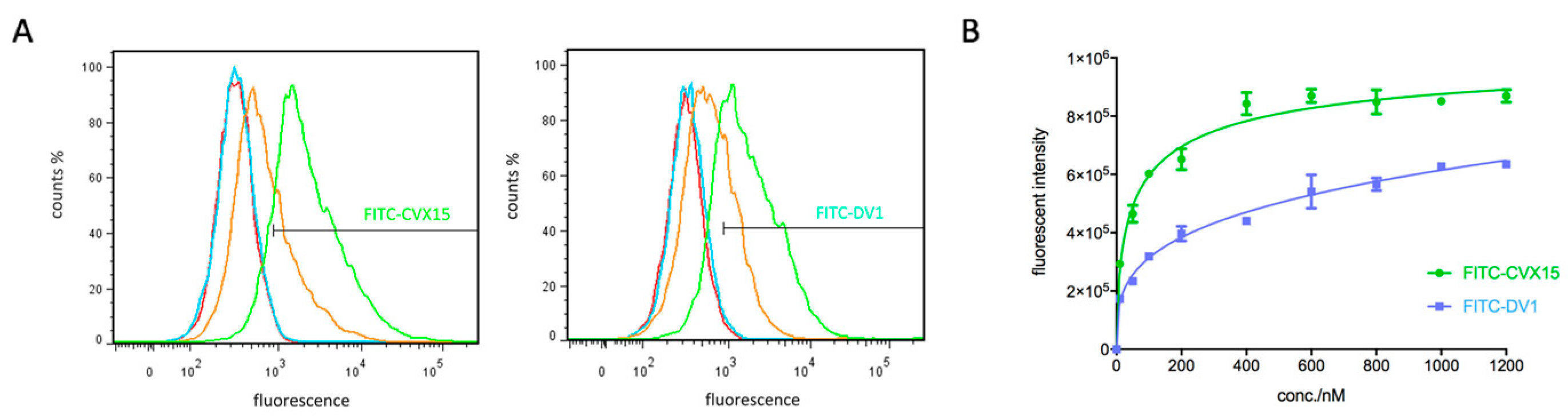
2.1. Synthesis and Characterization of New Peptidic CXCR4 Probes
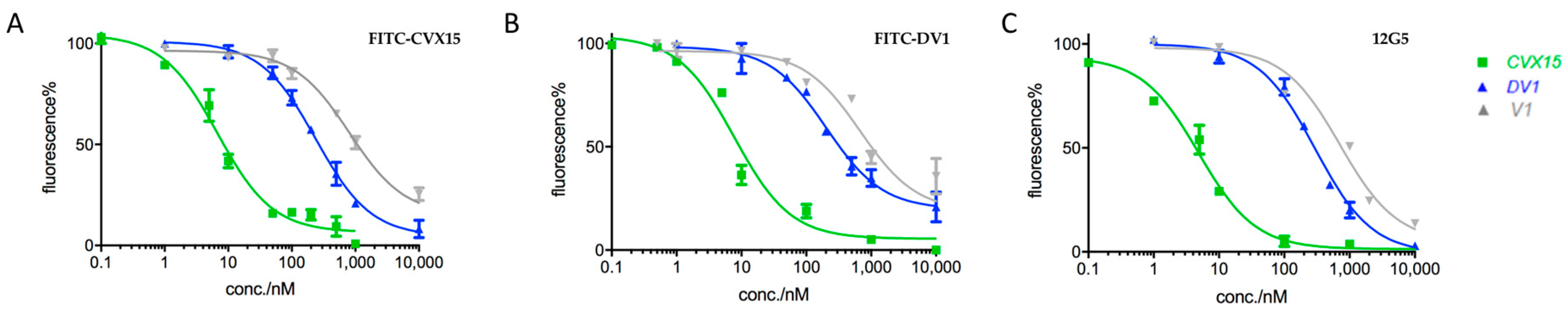
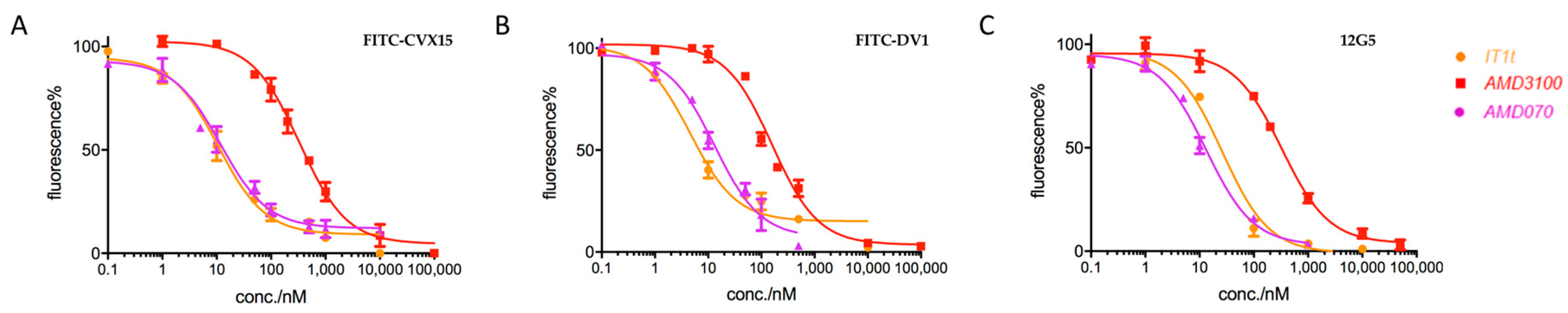
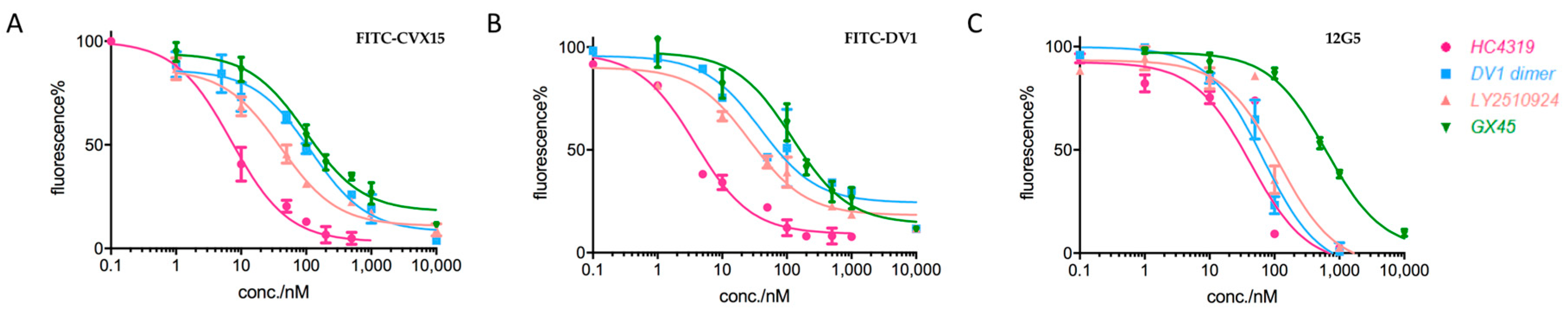
2.2. Applications in CXCR4-Ligand Competitive Binding Assays
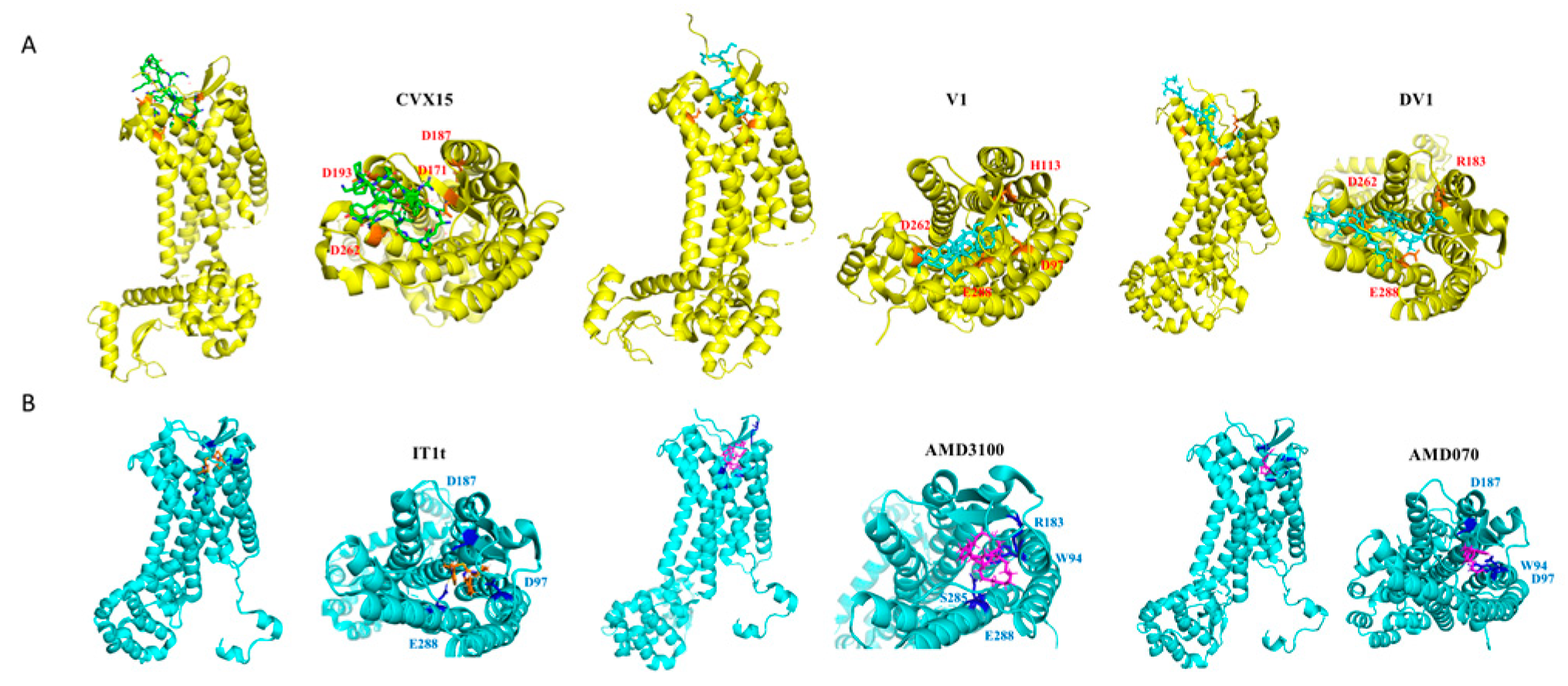
2.3. Prediction of Ligand Binding Modes Based on FITC-HAP Paired Probes
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemical Synthesis and Evaluation of FITC-CVX15 and FITC-DV1
5.2. Determination of Saturation Curves on FITC-CVX15 and FITC-DV1
5.3. 12G5 Antibody-Based Competitive Binding Assay
5.4. FITC-CVX15/FITC-DV1-Based Competitive Binding Assays
5.5. Flow Cytometry Analysis on Binding of Peptide Probes
5.6. Imaging Analysis by Confocal Laser Scanning Microscope
5.7. Hardware and Software
5.8. Molecular Docking Studies of CXCR4-Ligand Interactions In Silico
5.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CXCR4 | CXC-type chemokine receptor 4 |
| HIV-1 | Human immunodeficiency virus type 1 |
| GPCR | G protein-coupled receptor |
| JAK | Janus kinase |
| STAT | Signal transducer, and activator of transcription |
| PPIs | Protein-protein interactions |
| HAPs | High-affinity peptides |
| PE | Phycoerythrin |
| MALDI-TOF | Matrix-assisted laser desorption ionization time-of-flight |
| HPLC | High Performance Liquid Chromatography |
| FCM | Flow cytometry |
| Ahx | Aminocaproic acid |
| BSA | Bovine serum albumin |
| PBS | Phosphate-buffered saline |
| SFXC | Surflex-Dock algorithm |
References
- Bleul, C.C.; Fuhlbrigge, R.C.; Casasnovas, J.M.; Aiuti, A.; Springer, T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 1996, 184, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.T.; Duggineni, S.; Xu, Y.; Huang, Z.; An, J. Drug discovery research targeting the CXC chemokine receptor 4 (CXCR4). J. Med. Chem. 2012, 55, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biard-Piechaczyk, M.; Robert-Hebmann, V.; Roland, J.; Coudronnière, N.; Devaux, C. Role of CXCR4 in HIV-1-induced apoptosis of cells with a CD4+, CXCR4+ phenotype. Immunol. Lett. 1999, 70, 1–3. [Google Scholar] [CrossRef]
- Hesselgesser, J.; Taub, D.; Baskar, P.; Greenberg, M.; Hoxie, J.; Kolson, D.L.; Horuk, R. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr. Biol. 1998, 8, 595–598. [Google Scholar] [CrossRef]
- Pelchen-Matthews, A.; Signoret, N.; Klasse, P.J.; Fraile-Ramos, A.; Marsh, M. Chemokine receptor trafficking and viral replication. Immunol. Rev. 1999, 168, 33–49. [Google Scholar] [CrossRef]
- Crazzolara, R.; Bernhard, D. CXCR4 chemokine receptors, histone deacetylase inhibitors and acute lymphoblastic leukemia. Leuk Lymphoma. 2005, 46, 1545–1551. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Oishi, S.; Fujii, N. Peptide and peptidomimetic ligands for CXC chemokine receptor 4 (CXCR4). Org. Biomol. Chem. 2012, 10, 5720–5731. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Masuda, M.; Murakami, T.; Koyanagi, Y.; Matsumoto, A.; Fujii, N.; Yamamoto, N. Anti-human immunodeficiency virus activity of a novel synthetic peptide, T22 ([Tyr-5,12, Lys-7]polyphemusin II): A possible inhibitor of virus-cell fusion. Antimicrob. Agents Chemother 1992, 36, 1249–1255. [Google Scholar] [CrossRef]
- Tamamura, H.; Kuroda, M.; Masuda, M.; Otaka, A.; Funakoshi, S.; Nakashima, H.; Yamamoto, N.; Waki, M.; Matsumoto, A.; Lancelin, J.M.; et al. A comparative study of the solution structures of tachyplesin I and a novel anti-HIV synthetic peptide, T22 ([Tyr5,12, Lys7]-polyphemusin II), determined by nuclear magnetic resonance. Biochim. Biophys. Acta 1993, 1163, 209–216. [Google Scholar] [CrossRef]
- Tamamura, H.; Ishihara, T.; Otaka, A.; Murakami, T.; Ibuka, T.; Waki, M.; Matsumoto, A.; Yamamoto, N.; Fujii, N. Analysis of the interaction of an anti-HIV peptide, T22 ([Tyr5, 12, Lys7]-polyphemusin II), with gp120 and CD4 by surface plasmon resonance. Biochim. Biophys. Acta 1996, 1298, 37–44. [Google Scholar] [CrossRef]
- Arakaki, R.; Tamamura, H.; Premanathan, M.; Kanbara, K.; Ramanan, S.; Mochizuki, K.; Baba, M.; Fujii, N.; Nakashima, H. T134, a small-molecule CXCR4 inhibitor, has no cross-drug resistance with AMD3100, a CXCR4 antagonist with a different structure. J. Virol. 1999, 73, 1719–1723. [Google Scholar]
- Tamamura, H.; Xu, Y.; Hattori, T.; Zhang, X.; Arakaki, R.; Kanbara, K.; Omagari, A.; Otaka, A.; Ibuka, T.; Yamamoto, N.; et al. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: A strong anti-HIV peptide T140. Biochem. Biophys. Res. Commun. 1998, 253, 877–882. [Google Scholar] [CrossRef]
- Fujii, N.; Oishi, S.; Hiramatsu, K.; Araki, T.; Ueda, S.; Tamamura, H.; Otaka, A.; Kusano, S.; Terakubo, S.; Nakashima, H.; et al. Molecular-size reduction of a potent CXCR4-chemokine antagonist using orthogonal combination of conformation- and sequence-based libraries. Angew Chem. Int. Ed. Engl. 2003, 42, 3251–3253. [Google Scholar] [CrossRef]
- DeMarco, S.J.; Henze, H.; Lederer, A.; Moehle, K.; Mukherjee, R.; Romagnoli, B.; Robinson, J.A.; Brianza, F.; Gombert, F.O.; Lociuro, S.; et al. Discovery of novel, highly potent and selective beta-hairpin mimetic CXCR4 inhibitors with excellent anti-HIV activity and pharmacokinetic profiles. Bioorg. Med. Chem. 2006, 14, 8396–8404. [Google Scholar] [CrossRef]
- O’Brien, W.A.; Sumner-Smith, M.; Mao, S.H.; Sadeghi, S.; Zhao, J.Q.; Chen, I.S. Anti-human immunodeficiency virus type 1 activity of an oligocationic compound mediated via gp120 V3 interactions. J. Virol. 1996, 70, 2825–2831. [Google Scholar] [Green Version]
- Crump, M.P.; Gong, J.H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16, 6996–7007. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Zhou, N.; Luo, J.; Hall, J.W.; Huang, Z. The role of positively charged residues in CXCR4 recognition probed with synthetic peptides. Biochem. Biophys. Res. Commun. 1999, 263, 691–695. [Google Scholar] [CrossRef]
- Zhou, N.; Luo, Z.; Luo, J.; Hall, J.W.; Huang, Z. A novel peptide antagonist of CXCR4 derived from the N-terminus of viral chemokine vMIP-II. Biochemistry 2000, 39, 3782–3787. [Google Scholar] [CrossRef]
- Zhou, N.; Luo, Z.; Luo, J.; Fan, X.; Cayabyab, M.; Hiraoka, M.; Liu, D.; Han, X.; Pesavento, J.; Dong, C.Z.; et al. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J. Biol. Chem. 2002, 277, 17476–17485. [Google Scholar] [CrossRef]
- Huang, Z. Structure, function and modulation of chemokine receptors: Members of the g-protein-coupled receptor superfamily. Mini Rev. Med. Chem. 2002, 2, 373–383. [Google Scholar] [CrossRef]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef]
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 347, 1117–1122. [Google Scholar] [CrossRef]
- Banisadr, G.; Dicou, E.; Berbar, T.; Rostène, W.; Lombet, A.; Haour, F. Characterization and visualization of [125I] stromal cell-derived factor-1alpha binding to CXCR4 receptors in rat brain and human neuroblastoma cells. J. Neuroimmunol. 2000, 110, 151–160. [Google Scholar] [CrossRef]
- De Silva, R.A.; Peyre, K.; Pullambhatla, M.; Fox, J.J.; Pomper, M.G.; Nimmagadda, S. Imaging CXCR4 expression in human cancer xenografts: Evaluation of monocyclam 64Cu-AMD3465. J. Nucl. Med. 2011, 52, 986–993. [Google Scholar] [CrossRef]
- Jacobson, O.; Weiss, I.D.; Szajek, L.; Farber, J.M.; Kiesewetter, D.O. 64Cu-AMD3100--a novel imaging agent for targeting chemokine receptor CXCR4. Bioorg. Med. Chem. 2009, 17, 1486–1493. [Google Scholar] [CrossRef]
- Jacobson, O.; Weiss, I.D.; Szajek, L.P.; Niu, G.; Ma, Y.; Kiesewetter, D.O.; Peled, A.; Eden, H.S.; Farber, J.M.; Chen, X. Improvement of CXCR4 tracer specificity for PET imaging. J. Control. Release 2012, 157, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Poty, S.; Gourni, E.; Désogère, P.; Boschetti, F.; Goze, C.; Maecke, H.R.; Denat, F. AMD3100: A Versatile Platform for CXCR4 Targeting (68)Ga-Based Radiopharmaceuticals. Bioconjug Chem. 2016, 27, 752–761. [Google Scholar] [CrossRef]
- Misra, P.; Lebeche, D.; Ly, H.; Schwarzkopf, M.; Diaz, G.; Hajjar, R.J.; Schecter, A.D.; Frangioni, J.V. Quantitation of CXCR4 expression in myocardial infarction using 99mTc-labeled SDF-1alpha. J. Nucl. Med. 2008, 49, 963–969. [Google Scholar] [CrossRef]
- Jacobson, O.; Weiss, I.D.; Szajek, L.P.; Niu, G.; Ma, Y.; Kiesewetter, D.O.; Farber, J.M.; Chen, X. PET imaging of CXCR4 using copper-64 labeled peptide antagonist. Theranostics 2011, 1, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, O.; Weiss, I.D.; Kiesewetter, D.O.; Farber, J.M.; Chen, X. PET of tumor CXCR4 expression with 4-18F-T140. J. Nucl. Med. 2010, 51, 1796–1804. [Google Scholar] [CrossRef]
- Amor-Coarasa, A.; Kelly, J.; Ponnala, S.; Vedvyas, Y.; Nikolopoulou, A.; Williams, C., Jr.; Jin, M.M.; David, W.J.; Babich, J.W. [(18)F]RPS-544: A PET tracer for imaging the chemokine receptor CXCR4. Nucl. Med. Biol. 2018, 60, 37–44. [Google Scholar] [CrossRef]
- Hanaoka, H.; Mukai, T.; Tamamura, H.; Mori, T.; Ishino, S.; Ogawa, K.; Iida, Y.; Doi, R.; Fujii, N.; Saji, H. Development of a 111In-labeled peptide derivative targeting a chemokine receptor, CXCR4, for imaging tumors. Nucl. Med. Biol. 2006, 33, 489–494. [Google Scholar] [CrossRef]
- Nimmagadda, S.; Pullambhatla, M.; Pomper, M.G. Immunoimaging of CXCR4 expression in brain tumor xenografts using SPECT/CT. J. Nucl. Med. 2009, 50, 1124–1130. [Google Scholar] [CrossRef]
- Hartimath, S.V.; Domanska, U.M.; Walenkamp, A.M.E.; Dierckx, R.A.J.O.; de Vries, E.F.J. [(99)mTc]O(2)-AMD3100 as a SPECT tracer for CXCR4 receptor imaging. Nucl. Med. Biol. 2013, 40, 507–517. [Google Scholar] [CrossRef]
- Hartimath, S.V.; van Waarde, A.; Dierckx, R.A.J.O.; de Vries, E.F.J. Evaluation of N-[(11)C]methyl-AMD3465 as a PET tracer for imaging of CXCR4 receptor expression in a C6 glioma tumor model. Mol. Pharm. 2014, 11, 3810–3817. [Google Scholar] [CrossRef]
- Gourni, E.; Demmer, O.; Schottelius, M.; D'Alessandria, C.; Schulz, S.; Dijkgraaf, I.; Schumacher, U.; Schwaiger, M.; Kessler, H.; Wester, H.J. PET of CXCR4 expression by a (68)Ga-labeled highly specific targeted contrast agent. J. Nucl. Med. 2011, 52, 1803–1810. [Google Scholar] [CrossRef]
- Herrmann, K.; Lapa, C.; Wester, H.J.; Schottelius, M.; Schiepers, C.; Eberlein, U.; Bluemel, C.; Keller, U.; Knop, S.; Kropf, S.; et al. Biodistribution and radiation dosimetry for the chemokine receptor CXCR4-targeting probe 68Ga-pentixafor. J. Nucl. Med. 2015, 56, 410–416. [Google Scholar] [CrossRef]
- Dar, A.; Goichberg, P.; Shinder, V.; Kalinkovich, A.; Kollet, O.; Netzer, N.; Margalit, R.; Zsak, M.; Nagler, A.; Hardan, I.; et al. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat. Immunol. 2005, 6, 1038–1046. [Google Scholar] [CrossRef]
- Hatse, S.; Princen, K.; Liekens, S.; Vermeire, K.; De Clercq, E.; Schols, D. Fluorescent CXCL12AF647 as a novel probe for nonradioactive CXCL12/CXCR4 cellular interaction studies. Cytometry 2004, 61, 178–188. [Google Scholar] [CrossRef]
- van den Berg, N.S.; Buckle, T.; Kuil, J.; Wesseling, J.; van Leeuwen, F.W. Immunohistochemical detection of the CXCR4 expression in tumor tissue using the fluorescent peptide antagonist Ac-TZ14011-FITC. Transl. Oncol. 2011, 4, 234–240. [Google Scholar] [CrossRef]
- Nomura, W.; Tanabe, Y.; Tsutsumi, H.; Tanaka, T.; Ohba, K.; Yamamoto, N.; Tamamura, H. Fluorophore labeling enables imaging and evaluation of specific CXCR4-ligand interaction at the cell membrane for fluorescence-based screening. Bioconjug. Chem. 2008, 19, 1917–1920. [Google Scholar] [CrossRef]
- Oishi, S.; Masuda, R.; Evans, B.; Ueda, S.; Goto, Y.; Ohno, H.; Hirasawa, A.; Tsujimoto, G.; Wang, Z.; Peiper, S.C.; et al. Synthesis and application of fluorescein- and biotin-labeled molecular probes for the chemokine receptor CXCR4. Chembiochem 2008, 9, 1154–1158. [Google Scholar] [CrossRef]
- Zhan, W.; Liang, Z.; Zhu, A.; Kurtkaya, S.; Shim, H.; Snyder, J.P.; Liotta, D.C. Discovery of small molecule CXCR4 antagonists. J. Med. Chem. 2007, 50, 5655–5664. [Google Scholar] [CrossRef]
- Baribaud, F.; Edwards, T.G.; Sharron, M.; Brelot, A.; Heveker, N.; Price, K.; Mortari, F.; Alizon, M.; Tsang, M.; Doms, R.W. Antigenically distinct conformations of CXCR4. J. Virol. 2001, 75, 8957–8967. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Q.; Gao, M.; Yang, X.; Huang, Z.; An, J. A novel CXCR4-selective high-affinity fluorescent probe and its application in competitive binding assays. Biochemistry 2014, 53, 4881–4883. [Google Scholar] [CrossRef]
- Carnec, X.; Quan, L.; Olson, W.C.; Hazan, U.; Dragic, T. Anti-CXCR4 monoclonal antibodies recognizing overlapping epitopes differ significantly in their ability to inhibit entry of human immunodeficiency virus type 1. J. Virol. 2005, 79, 1930–1933. [Google Scholar] [CrossRef]
- Peng, S.B.; Zhang, X.; Paul, D.; Kays, L.M.; Gough, W.; Stewart, J.; Uhlik, M.T.; Chen, Q.; Hui, Y.H.; Zamek-Gliszczynski, M.J.; et al. Identification of LY2510924, a novel cyclic peptide CXCR4 antagonist that exhibits antitumor activities in solid tumor and breast cancer metastatic models. Mol. Cancer Ther. 2015, 14, 480–490. [Google Scholar] [CrossRef]
- Cox, B.D.; Prosser, A.R.; Katzman, B.M.; Alcaraz, A.A.; Liotta, D.C.; Wilson, L.J.; Snyder, J.P. Anti-HIV small-molecule binding in the peptide subpocket of the CXCR4:CVX15 crystal structure. Chembiochem 2014, 15, 1614–1620. [Google Scholar] [CrossRef]
- Liu, D.; Madani, N.; Li, Y.; Cao, R.; Choi, W.T.; Kawatkar, S.P.; Lim, M.Y.; Kumar, S.; Dong, C.Z.; Wang, J.; et al. Crystal structure and structural mechanism of a novel anti-human immunodeficiency virus and D-amino acid-containing chemokine. J. Virol. 2007, 81, 11489–11498. [Google Scholar] [CrossRef]
- Spitzer, R.; Jain, A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput. Aided. Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef]
Sample Availability: Samples of FITC-CVX15 and FITC-DV1 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Name\IC50 (nM) | FITC-CVX15 | FITC-DV1 | 12G5 Ab |
|---|---|---|---|---|
| Type 1 | CVX15 | 6.2 | 6.8 | 6.0 |
| V1 | 687.4 | 676.5 | 712.5 | |
| DV1 | 238.2 | 203.6 | 296.9 | |
| Type 2 | IT1t | 19.3 | 6.0 | 26.2 |
| AMD3100 | 314.5 | 104.9 | 324.3 | |
| AMD070 | 14.7 | 5.0 | 12.2 | |
| Type 3 | HC4319 | 7.1 | 5.0 | 46.6 |
| DV1 dimer | 29.4 | 25.6 | 67.0 | |
| LY2510924 | 35.4 | 33.9 | 117.4 | |
| GX45 | 137.4 | 139.1 | 628.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, S.; Meng, Q.; Schooley, R.T.; An, J.; Xu, Y.; Huang, Z. Structural and Biological Characterizations of Novel High-Affinity Fluorescent Probes with Overlapped and Distinctive Binding Regions on CXCR4. Molecules 2019, 24, 2928. https://doi.org/10.3390/molecules24162928
Zhu S, Meng Q, Schooley RT, An J, Xu Y, Huang Z. Structural and Biological Characterizations of Novel High-Affinity Fluorescent Probes with Overlapped and Distinctive Binding Regions on CXCR4. Molecules. 2019; 24(16):2928. https://doi.org/10.3390/molecules24162928
Chicago/Turabian StyleZhu, Siyu, Qian Meng, Robert T. Schooley, Jing An, Yan Xu, and Ziwei Huang. 2019. "Structural and Biological Characterizations of Novel High-Affinity Fluorescent Probes with Overlapped and Distinctive Binding Regions on CXCR4" Molecules 24, no. 16: 2928. https://doi.org/10.3390/molecules24162928