Discovery of (5-Phenylfuran-2-yl)methanamine Derivatives as New Human Sirtuin 2 Inhibitors

Abstract
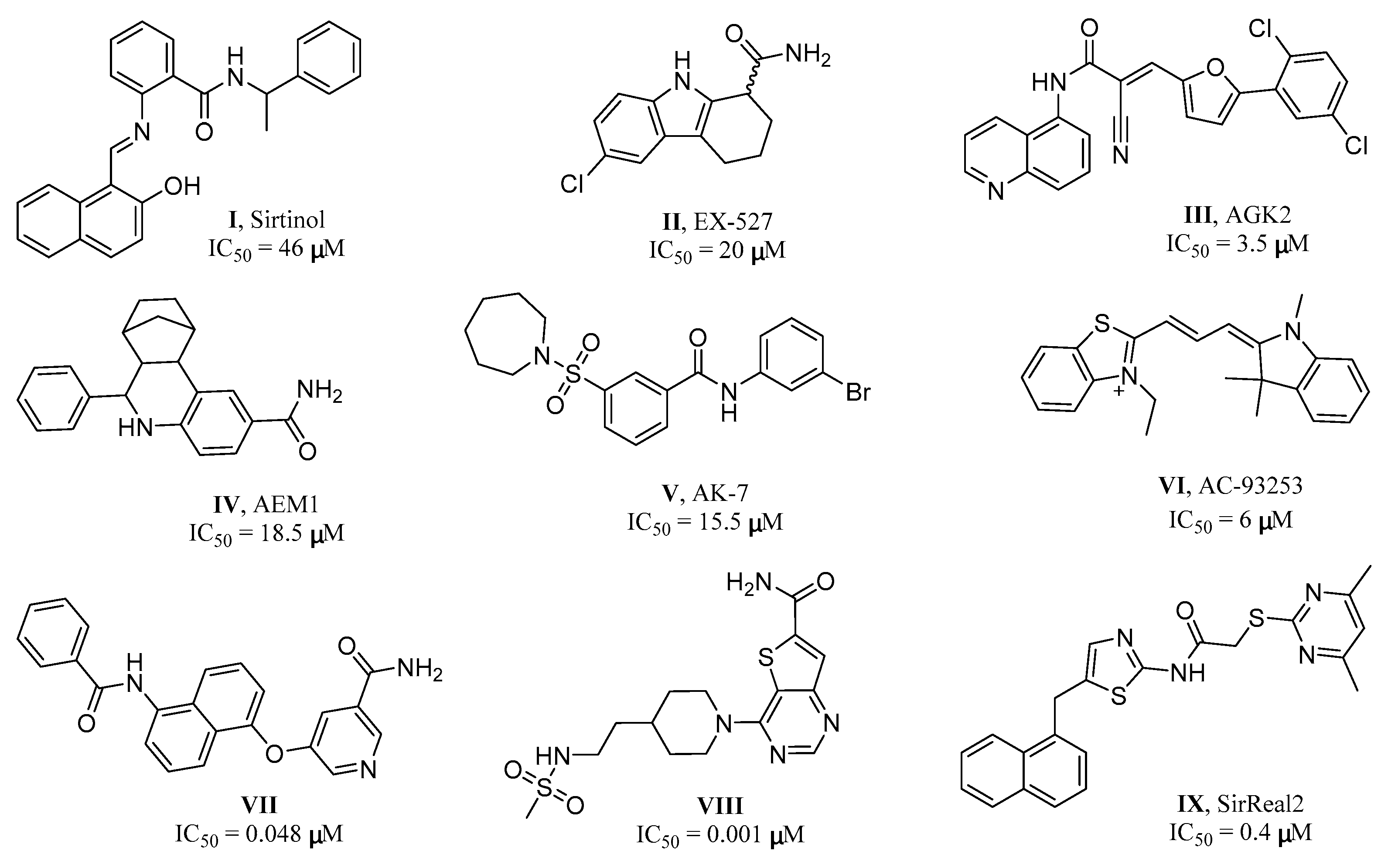
:1. Introduction
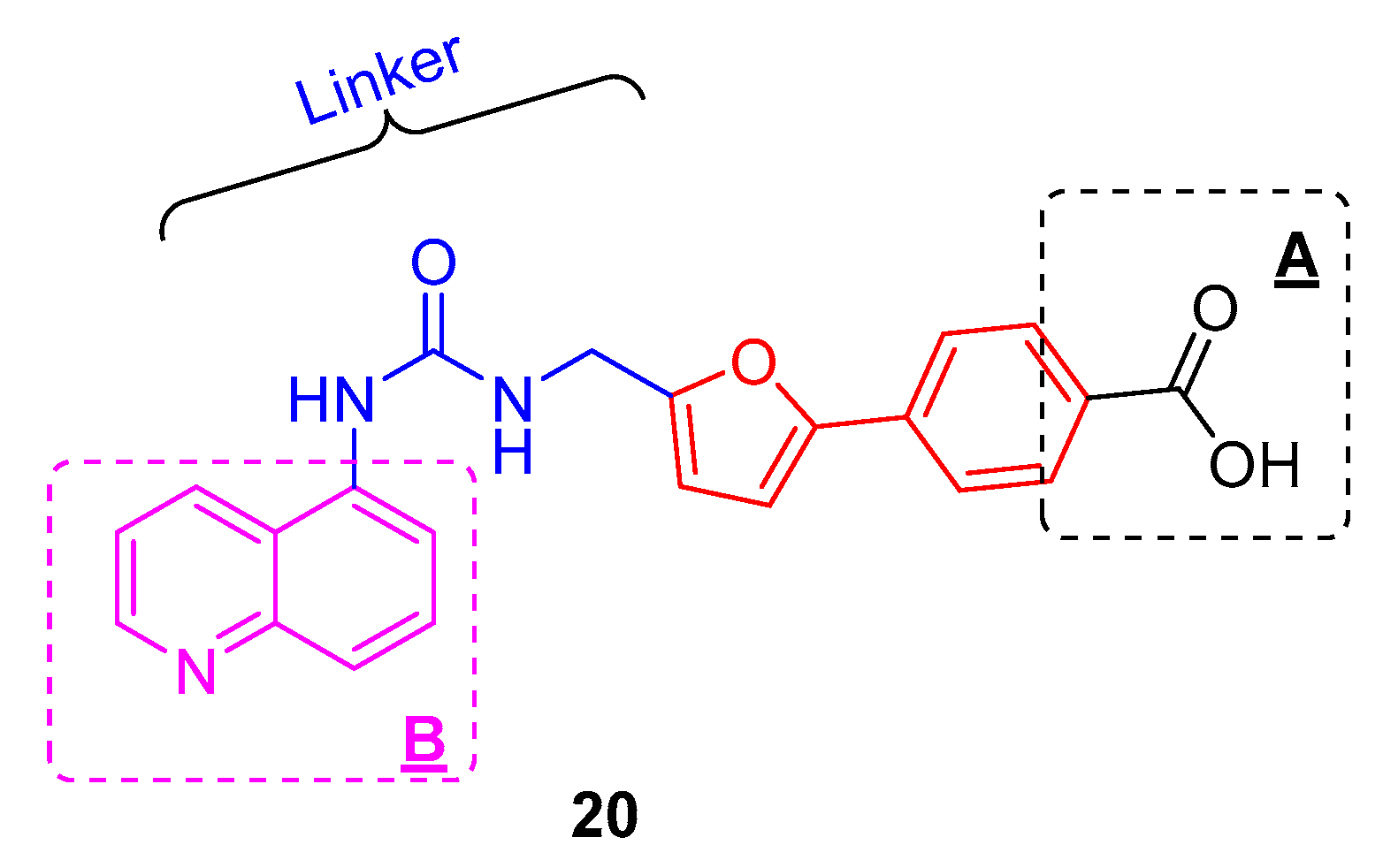
2. Results and Discussion
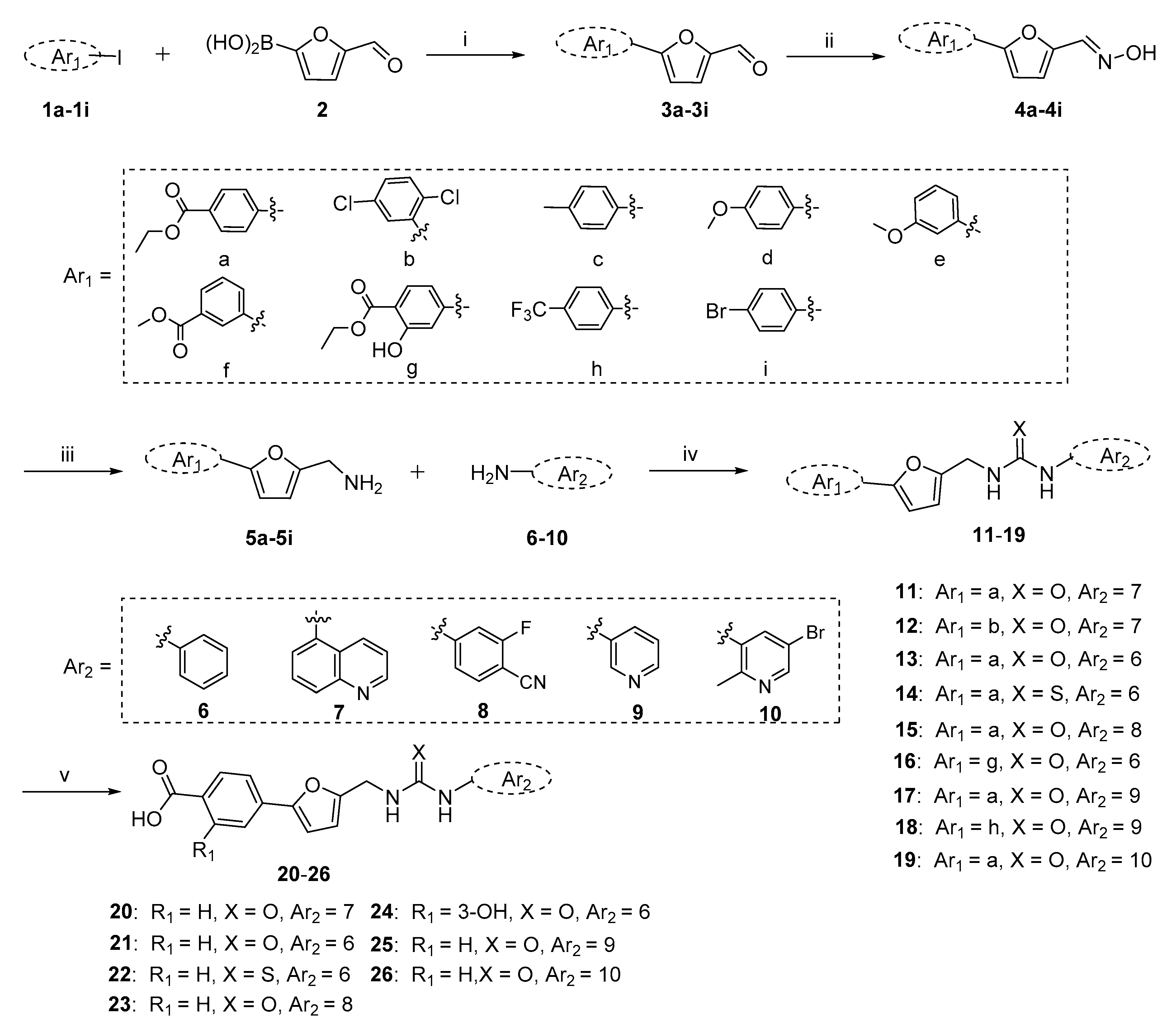
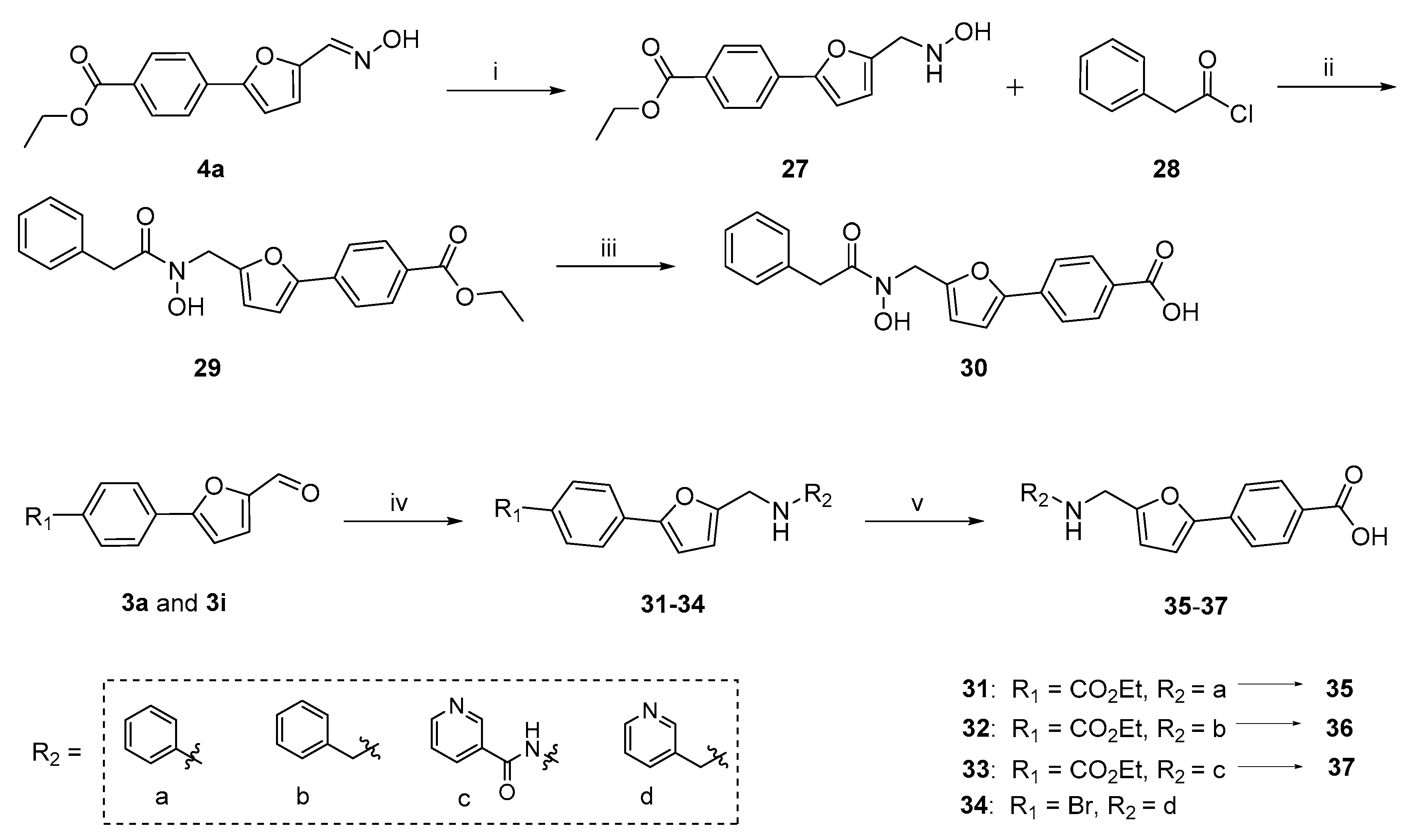
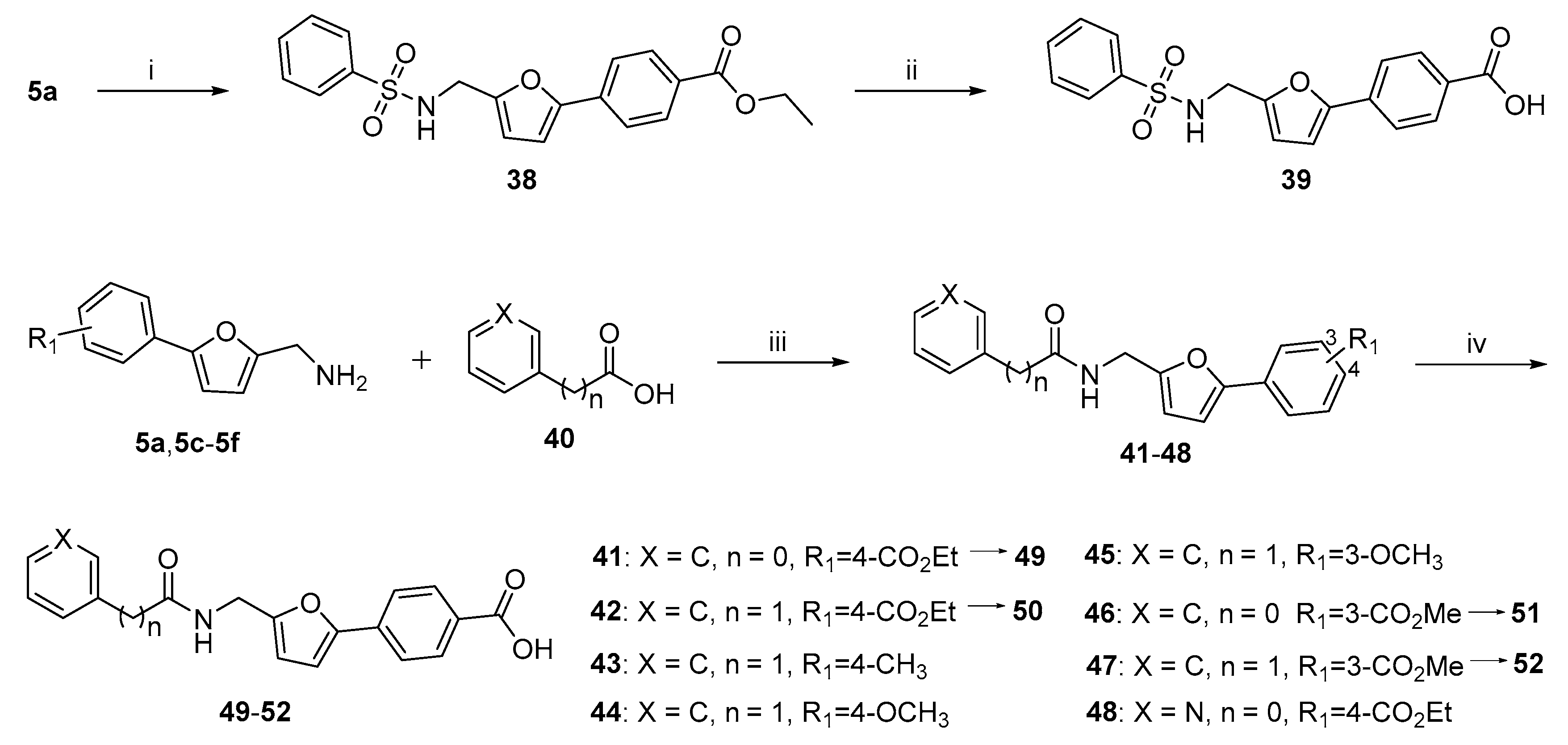
2.1. Chemistry
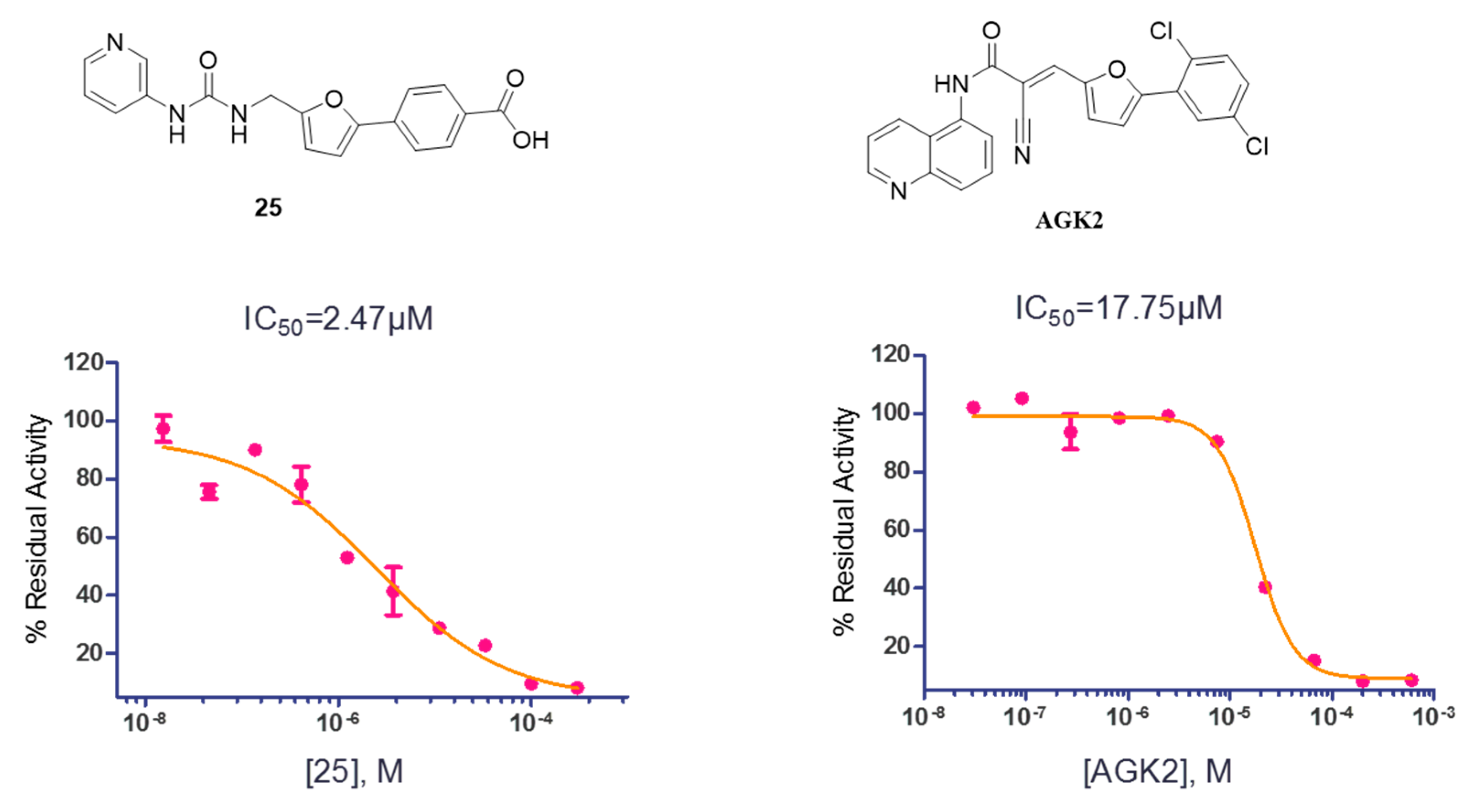
2.2. SAR Studies with SIRT2
3. Materials and Methods
3.1. Synthesis
3.1.1. General Procedure for the Preparation of Key Intermediates 5a–5i
3.1.2. Hantzsch-Involved Reductive Amination Used for Compounds 31–34
3.2. Inhibition Assays
3.3. Molecular Docking Assays
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bhalla, K.N. Epigenetic and Chromatin Modifiers as Targeted Therapy of Hematologic Malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 Family of Protein Deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ji, S.; Yu, Z.-J.; Wang, H.-L.; Cheng, X.; Li, W.-J.; Jing, L.; Yu, Y.; Chen, Q.; Yang, L.-L.; et al. Structure-based discovery of new selective small-molecule sirtuin 5 inhibitors. Chem. Biol. Drug Des. 2018, 91, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D. Old Enzymes, New Tricks: Sirtuins Are NAD+-Dependent De-acylases. Cell Metab. 2011, 14, 718–719. [Google Scholar] [CrossRef] [Green Version]
- Jing, H.; Lin, H. Sirtuins in Epigenetic Regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ma, X.; He, Y.; Yuan, C.; Chen, Q.; Li, G.; Chen, X. Sirtuin 5: A review of structure, known inhibitors and clues for developing new inhibitors. Sci. China Life Sci. 2016, 60, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Goto, Y.; Nishimasu, H.; Morimoto, J.; Ishitani, R.; Dohmae, N.; Takeda, N.; Nagai, R.; Komuro, I.; Suga, H.; et al. Structural basis for potent inhibition of SIRT2 deacetylase by a macrocyclic peptide inducing dynamic structural change. Structure 2014, 22, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, J.; Liao, M.; Hu, M.; Li, W.; Ouyang, H.; Wang, X.; Ye, T.; Zhang, Y.; Ouyang, L. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 2019, 161, 48–77. [Google Scholar] [CrossRef]
- Das, C.; Lucia, M.S.; Hansen, K.C.; Tyler, J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009, 459, 113–117. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Peck, B.; Chen, C.-Y.; Ho, K.-K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.-D.; Lam, E.W.-F. SIRT Inhibitors Induce Cell Death and p53 Acetylation through Targeting Both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 Regulates Adipocyte Differentiation through FoxO1 Acetylation/Deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Matsumori, H.; Nakayama, Y.; Osaki, M.; Kojima, H.; Kurimasa, A.; Ito, H.; Mori, S.; Katoh, M.; Oshimura, M.; et al. SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 2011, 16, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Luscher, B.; Hottiger, M.O. Correction: SIRT2 regulates NF-kappaB-dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2019, 132, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Song, C.; Wang, X.; Zhang, G.; Wang, Y.; Jiang, X.; Sun, Q.; Huang, L.; Xiang, R.; Hu, Y.; et al. Discovery of New SIRT2 Inhibitors by Utilizing a Consensus Docking/Scoring Strategy and Structure–Activity Relationship Analysis. J. Chem. Inf. Model. 2017, 57, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Dryden, S.C.; Nahhas, F.A.; Nowak, J.E.; Goustin, A.-S.; Tainsky, M.A. Role for Human SIRT2 NAD-Dependent Deacetylase Activity in Control of Mitotic Exit in the Cell Cycle. Mol. Cell. Biol. 2003, 23, 3173–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Hiratsuka, M.; Osaki, M.; Oshimura, M. The Molecular Biology of Mammalian SIRT Proteins: SIRT2 Functions on Cell Cycle Regulation. Cell Cycle 2007, 6, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Machado de Oliveira, R.; Sarkander, J.; Kazantsev, A.; Outeiro, T. SIRT2 as a Therapeutic Target for Age-Related Disorders. Front. Pharmacol. 2012, 3, 1–9. [Google Scholar] [CrossRef]
- Beirowski, B.; Gustin, J.; Armour, S.M.; Yamamoto, H.; Viader, A.; North, B.J.; Michán, S.; Baloh, R.H.; Golden, J.P.; Schmidt, R.E.; et al. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, E952–E961. [Google Scholar] [CrossRef]
- Eskandarian, H.A.; Impens, F.; Nahori, M.-A.; Soubigou, G.; Coppée, J.-Y.; Cossart, P.; Hamon, M.A. A Role for SIRT2-Dependent Histone H3K18 Deacetylation in Bacterial Infection. Science 2013, 341, 1238858. [Google Scholar] [CrossRef] [PubMed]
- Pais, T.F.; Szegő, É.M.; Marques, O.; Miller-Fleming, L.; Antas, P.; Guerreiro, P.; de Oliveira, R.M.; Kasapoglu, B.; Outeiro, T.F. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 2013, 32, 2603–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Alam, H.B.; Liu, B.; Bronson, R.T.; Nikolian, V.C.; Wu, E.; Chong, W.; Li, Y. Selective Inhibition of SIRT2 Improves Outcomes in a Lethal Septic Model. Curr. Mol. Med. 2015, 15, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Vassilopoulos, A.; Wang, R.-H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 Maintains Genome Integrity and Suppresses Tumorigenesis through Regulating APC/C Activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Zhu, Y.M.; Ozden, O.; Kim, H.S.; Jiang, H.Y.; Deng, C.X.; Gius, D.; Vassilopoulos, A. SIRT2 is a tumor suppressor that connects aging, acetylome, cell cycle signaling, and carcinogenesis. Transl. Cancer Res. 2012, 1, 15–21. [Google Scholar] [PubMed]
- Grozinger, C.M.; Chao, E.D.; Blackwell, H.E.; Moazed, D.; Schreiber, S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001, 276, 38837–38843. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, H.; Yu, X.; Peng, T.; Wang, G.; Wen, X.; Sun, Y.; Liu, S.; Zhang, S.; Hu, L.; et al. Synthesis and Evaluation of Novel Benzofuran Derivatives as Selective SIRT2 Inhibitors. Molecules 2017, 22, 1348. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Pirrie, L.; McCarthy, A.R.; Major, L.L.; Morkūnaitė, V.; Zubrienė, A.; Matulis, D.; Lain, S.; Lebl, T.; Westwood, N.J. Discovery and Validation of SIRT2 Inhibitors Based on Tenovin-6: Use of a 1H-NMR Method to Assess Deacetylase Activity. Molecules 2012, 17, 12206–12224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, G.; Breitenbucher, F.; Schuler, M.; Ehrenhofer-Murray, A.E. A Novel Sirtuin 2 (SIRT2) Inhibitor with p53-dependent Pro-apoptotic Activity in Non-small Cell Lung Cancer. J. Biol. Chem. 2014, 289, 5208–5216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.M.; Balabadra, U.; Xiang, Z.; Woodman, B.; Meade, S.; Amore, A.; Maxwell, M.M.; Reeves, S.; Bates, G.P.; Luthi-Carter, R.; et al. A Brain-Permeable Small Molecule Reduces Neuronal Cholesterol by Inhibiting Activity of Sirtuin 2 Deacetylase. ACS Chem. Biol. 2011, 6, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Xu, N.; Malyukova, A.; Scarlett, C.J.; Sun, Y.T.; Zhang, X.D.; Ling, D.; Su, S.P.; Nelson, C.; Chang, D.K.; et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 2012, 20, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Kamal, Z.; Ai, T.; Xu, Y.; More, S.S.; Wilson, D.J.; Chen, L. Discovery of Potent and Selective Sirtuin 2 (SIRT2) Inhibitors Using a Fragment-Based Approach. J. Med. Chem. 2014, 57, 8340–8357. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-Based Development of an Affinity Probe for Sirtuin2. Angew. Chem. Int. Ed. 2016, 55, 2252–2256. [Google Scholar] [CrossRef]
- Yang, L.-L.; Wang, H.-L.; Zhong, L.; Yuan, C.; Liu, S.-Y.; Yu, Z.-J.; Liu, S.; Yan, Y.-H.; Wu, C.; Wang, Y.; et al. X-ray crystal structure guided discovery of new selective, substrate-mimicking sirtuin 2 inhibitors that exhibit activities against non-small cell lung cancer cells. Eur. J. Med. Chem. 2018, 155, 806–823. [Google Scholar] [CrossRef]
- Yang, L.-L.; Xu, W.; Yan, J.; Su, H.-L.; Yuan, C.; Li, C.; Zhang, X.; Yu, Z.-J.; Yan, Y.-H.; Yu, Y.; et al. Crystallographic and SAR analyses reveal the high requirements needed to selectively and potently inhibit SIRT2 deacetylase and decanoylase. MedChemComm 2019, 10, 164–168. [Google Scholar] [CrossRef]
- Galleano, I.; Schiedel, M.; Jung, M.; Madsen, A.; Olsen, C. A Continuous, Fluorogenic Sirtuin 2 Deacylase Assay: Substrate Screening and Inhibitor Evaluation. J. Med. Chem. 2016, 59, 1021–1031. [Google Scholar] [CrossRef]
- Yang, L.; Ma, X.; Yuan, C.; He, Y.; Li, L.; Fang, S.; Xia, W.; He, T.; Qian, S.; Xu, Z.; et al. Discovery of 2-((4,6-dimethylpyrimidin-2-yl)thio)-N-phenylacetamide derivatives as new potent and selective human sirtuin 2 inhibitors. Eur. J. Med. Chem. 2017, 134, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Robinson, E.; Hom, K.; Yu, W.; Nguyen, N.; Li, Y.; Zong, Q.; Wilks, A.; Xue, F. Structure-based design and biological evaluation of inhibitors of the pseudomonas aeruginosa heme oxygenase (pa-HemO). Bioorg. Med. Chem. Lett. 2018, 28, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Denton, T.T.; Cerny, M.A.; Zhang, X.; Johnson, E.F.; Cashman, J.R. Synthetic Inhibitors of Cytochrome P-450 2A6: Inhibitory Activity, Difference Spectra, Mechanism of Inhibition, and Protein Cocrystallization. Eur. J. Med. Chem. 2006, 49, 6987–7001. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Pavletich, N.P. Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 2001, 8, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Structure | Inhibition% 1/SIRT2-p2270 | cLogP | cLogS | |
|---|---|---|---|---|---|
| @ 100 μM | @ 10 μM | ||||
| 20 |  | 63 ± 5 | 35 ± 3 | 3.05 | −4.04 |
| 12 |  | 60 ± 3 | 33 ± 3 | 5.14 | −4.43 |
| 21 |  | 46 ± 4 | 33 ± 3 | 2.85 | −3.98 |
| 22 |  | 44 ± 5 | 23 ± 3 | 3.60 | −4.22 |
| 30 |  | 13 ± 2 | −2 ± 3 | 2.99 | −4.05 |
| 35 |  | 24 ± 3 | 1 ± 2 | 4.27 | −4.13 |
| 39 |  | 38 ± 3 | 11 ± 2 | 1.85 | −4.13 |
| 49 |  | 32 ± 3 | 8 ± 1 | 3.12 | −4.14 |
| 50 |  | 30 ± 2 | 5 ± 2 | 3.34 | −4.25 |
| 32 |  | 9 ± 2 | −2 ± 2 | 4.17 | −4.54 |
| 36 |  | 15 ± 2 | 3 ± 2 | 2.48 | −4.29 |
| 43 |  | 3 ± 2 | 2 ± 3 | 4.18 | −4.46 |
| 44 |  | 20 ± 3 | 8 ± 2 | 3.79 | −4.36 |
| 45 |  | 19 ± 2 | 5 ± 2 | 3.76 | −4.36 |
| 46 |  | 15 ± 2 | 3 ± 3 | 3.46 | −4.3 |
| 51 |  | 18 ± 3 | 13 ± 3 | 3.09 | −4.12 |
| 47 |  | 16 ± 3 | 5 ± 3 | 3.67 | −4.4 |
| 52 |  | 18 ± 2 | 2 ± 3 | 3.31 | −4.24 |
| AGK2 | 80 ± 6 | 30 ± 5 | 5.65 | −4.25 | |
| ID | Structure | Inhibition% 1/SIRT2-p2270 | cLogP | cLogS | |
|---|---|---|---|---|---|
| @ 100 μM | @ 10 μM | ||||
| 23 |  | 35 ± 4 | 10 ± 3 | 3.15 | −3.58 |
| 24 |  | 43 ± 2 | 13 ± 2 | 3.21 | −3.82 |
| 25 |  | 99 ± 2 | 90 ± 3 | 1.63 | −3.63 |
| 26 |  | 9 ± 2 | −5 ± 3 | 3.05 | −3.83 |
| 17 |  | 50 ± 4 | 37 ± 3 | 2.85 | −3.83 |
| 18 |  | 40 ± 5 | 23 ± 2 | 3.54 | −4.12 |
| 33 |  | 20 ± 3 | 3 ± 2 | 3.28 | −4.08 |
| 37 |  | 30 ± 5 | 5 ± 2 | 2.56 | −3.83 |
| 48 |  | 18 ± 2 | 0 ± 2 | 2.82 | −4.11 |
| 34 |  | 3 ± 2 | −2 ± 3 | 3.19 | −4.12 |
| AGK2 | 80 ± 6 | 30 ± 5 | 5.65 | −4.25 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Li, C.; Chen, W.; Song, C.; Zhang, X.; Yang, F.; Wang, C.; Zhang, Y.; Qian, S.; Wang, Z.; et al. Discovery of (5-Phenylfuran-2-yl)methanamine Derivatives as New Human Sirtuin 2 Inhibitors. Molecules 2019, 24, 2724. https://doi.org/10.3390/molecules24152724
Wang L, Li C, Chen W, Song C, Zhang X, Yang F, Wang C, Zhang Y, Qian S, Wang Z, et al. Discovery of (5-Phenylfuran-2-yl)methanamine Derivatives as New Human Sirtuin 2 Inhibitors. Molecules. 2019; 24(15):2724. https://doi.org/10.3390/molecules24152724
Chicago/Turabian StyleWang, Lijiao, Chao Li, Wei Chen, Chen Song, Xing Zhang, Fan Yang, Chen Wang, Yuanyuan Zhang, Shan Qian, Zhouyu Wang, and et al. 2019. "Discovery of (5-Phenylfuran-2-yl)methanamine Derivatives as New Human Sirtuin 2 Inhibitors" Molecules 24, no. 15: 2724. https://doi.org/10.3390/molecules24152724