
Ion Mobility Spectrometry in Food Analysis: Principles, Current Applications and Future Trends

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
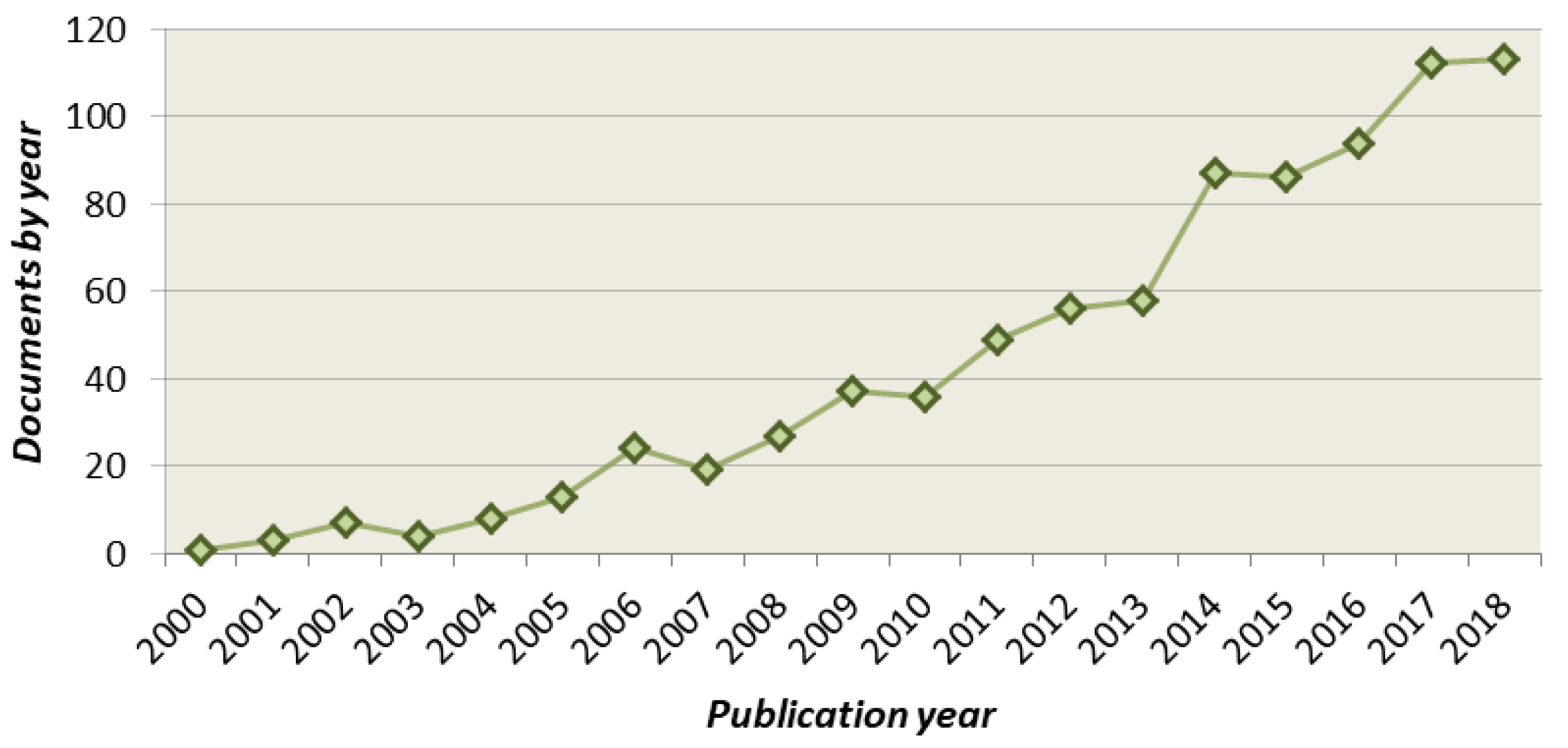
:1. Introduction
2. Overview of Ion Mobility Spectrometry (IMS) Technique and Potential
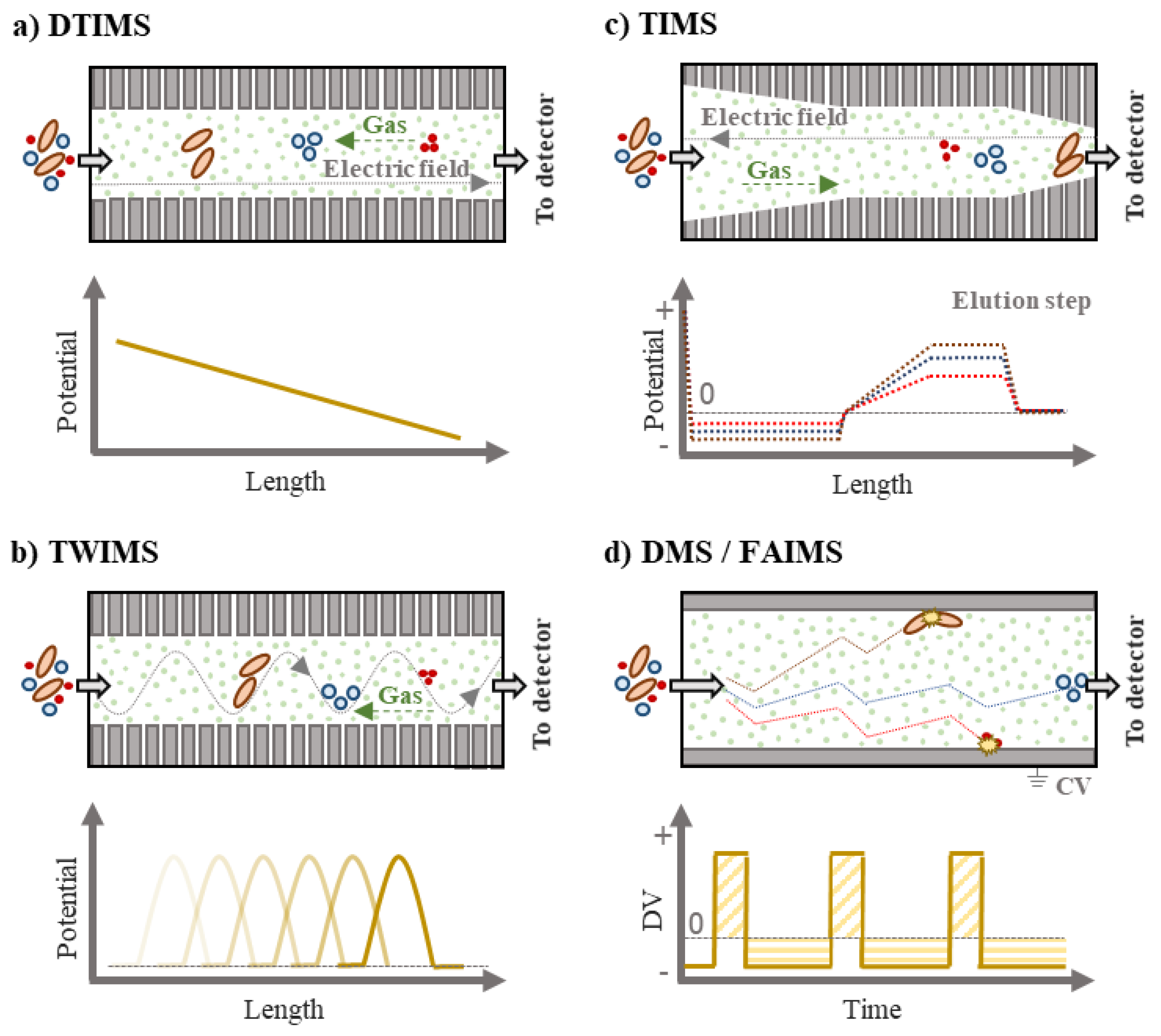
2.1. IMS Instrumentation
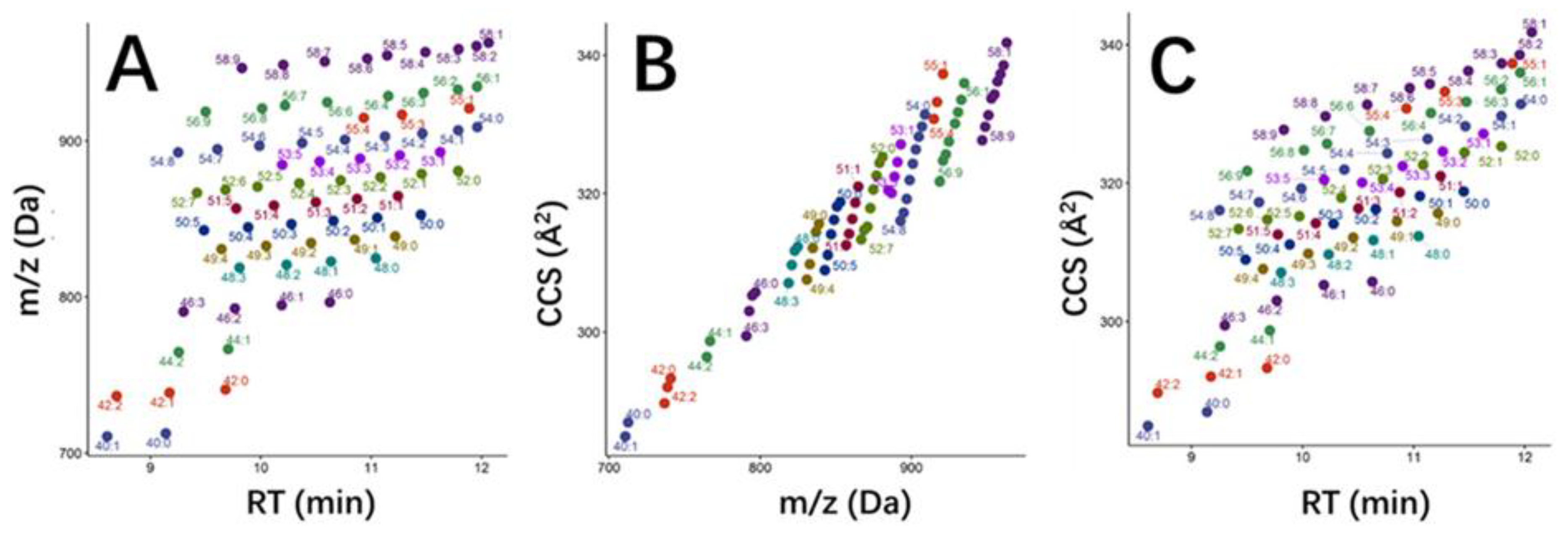
2.2. Collision Cross Section (CCS) for Structural Elucidation
2.3. IMS Hyphenation
3. Applications of IMS in Food Analysis
3.1. Food Composition

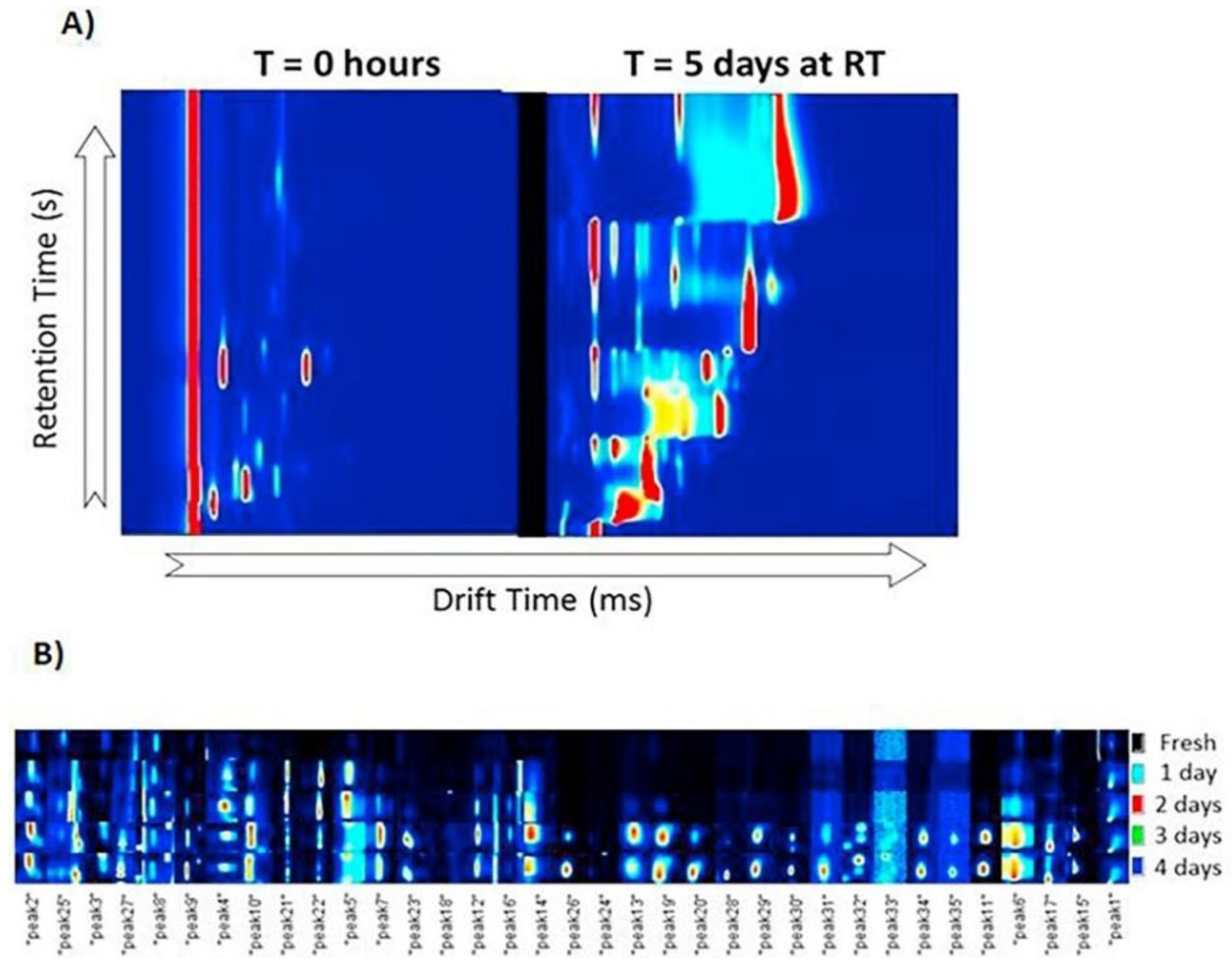
3.2. Food Process Control
3.3. Food Authentication
3.4. Food Adulteration
3.5. Chemical Food Safety
4. Current Perspectives of Ion Mobility Spectrometry
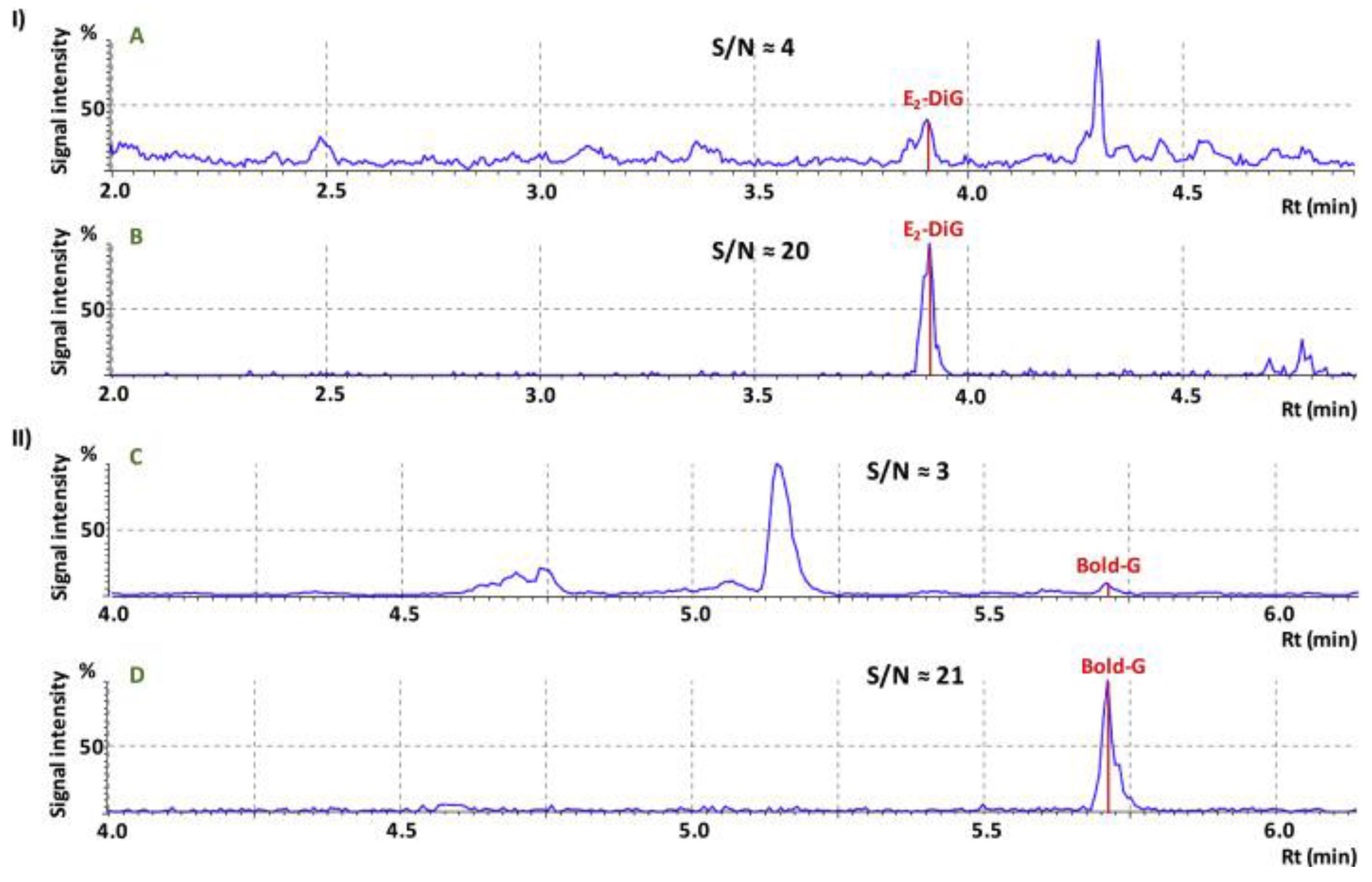
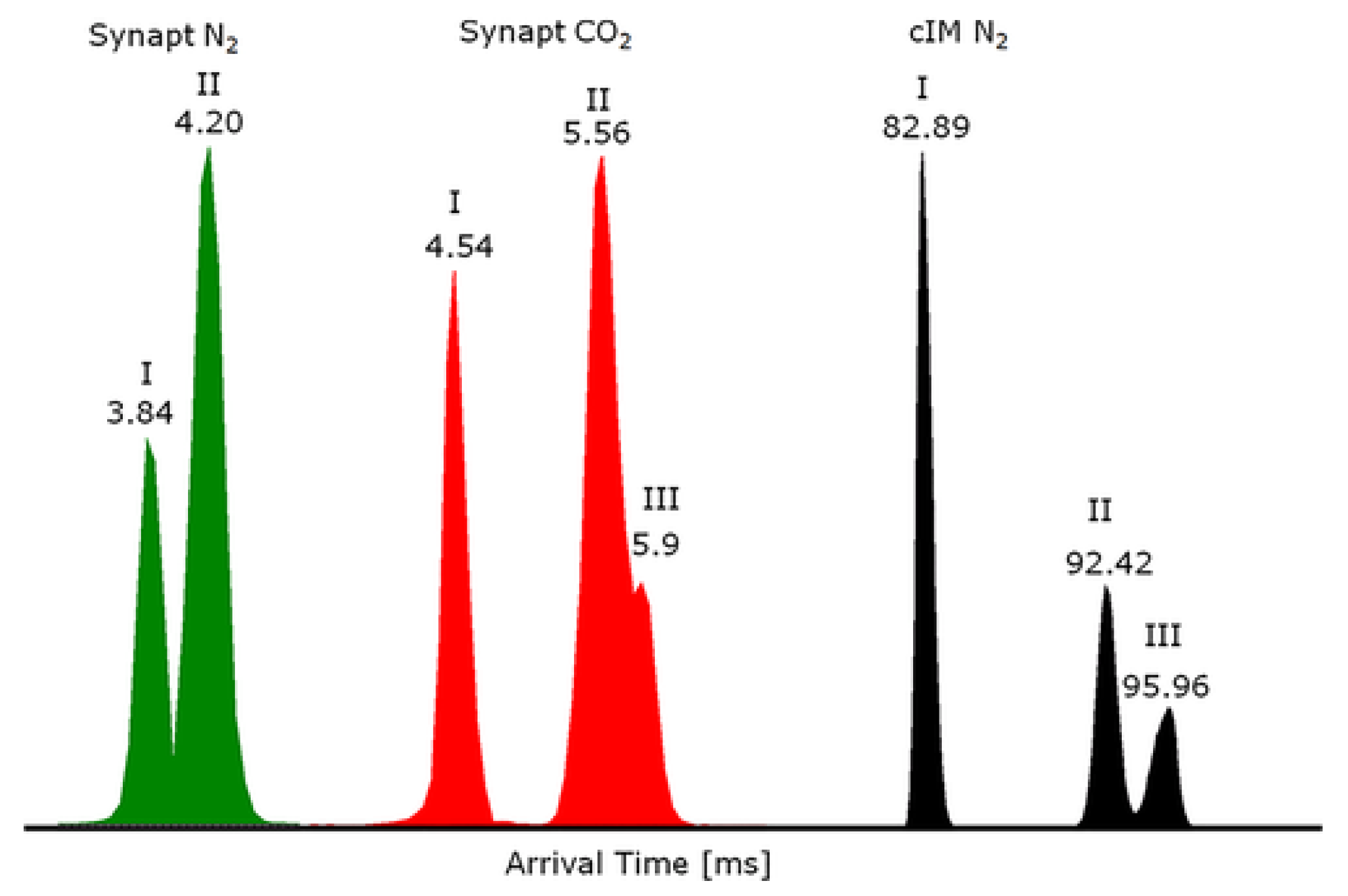
4.1. Improvement in Peak Resolution
4.2. Implementation of CCS in Current Analytical Workflows
5. Conclusions and Perspectives in Food Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Lehotay, S.J.; Chen, Y. Hits and misses in research trends to monitor contaminants in foods. Anal. Bioanal. Chem. 2018, 410, 5331–5351. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.; Pereira, J.A.; Silva, P.; Perestrelo, R.; Câmara, J.S. Food fingerprints—A valuable tool to monitor food authenticity and safety. Food Chem. 2019, 278, 144–162. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wojcik, R.; Zhang, X.; Ibrahim, Y.M.; Burnum-Johnson, K.E.; Orton, D.J.; Monroe, M.E.; Smith, R.D.; Baker, E.S. Coupling front-end separations, ion mobility spectrometry, and mass spectrometry for enhanced multidimensional biological and environmental analyses. Annu. Rev. Anal. Chem. 2017, 10, 71–92. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, V.; Causon, T.; Hernandez-Alba, O.; Mutabazi, A.; Veuthey, J.-L.; Cianferani, S.; Guillarme, D. Adding a new separation dimension to MS and LC-MS: What is the utility of ion mobility spectrometry? J. Sep. Sci. 2018, 41, 20–67. [Google Scholar] [CrossRef] [PubMed]
- Vautz, W.; Zimmermann, D.; Hartmann, M.; Baumbach, J.I.; Nolte, J.; Jung, J. Ion mobility spectrometry for food quality and safety. Food Addit. Contam. 2006, 23, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Arce, L.; Valcárcel, M. The role of ion mobility spectrometry to support the food protected designation of origin. In Comprehensive Analytical Chemistry—Food Protected Designation of Origin: Methodologies and Applications; de la Guardia, M., González, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 60, pp. 221–249. [Google Scholar]
- Karpas, Z. Applications of ion mobility spectrometry (IMS) in the field of foodomics. Food Res. Int. 2013, 54, 1146–1151. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; Escorrou, A.; Monteau, F.; Le Bizec, B.; Dervilly-Pinel, G. Current applications and perspectives of ion mobility spectrometry to answer chemical food safety issues. TrAC Trends Anal. Chem. 2017, 94, 39–53. [Google Scholar] [CrossRef]
- Uetrecht, C.; Rose, R.J.; van Duijn, E.; Lorenzen, K.; Heck, A.J.R. Ion mobility mass spectrometry of proteins and protein assemblies. Chem. Soc. Rev. 2010, 39, 1633–1655. [Google Scholar] [CrossRef]
- Gabelica, V.; Marklund, E. Fundamentals of ion mobility spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 51–59. [Google Scholar] [CrossRef]
- May, J.C.; Morris, C.B.; McLean, J.A. Ion mobility collision cross section compendium. Anal. Chem. 2017, 89, 1032–1044. [Google Scholar] [CrossRef]
- Revercomb, H.E.; Mason, E.A. Theory of plasma chromatography/gaseous electrophoresis—A review. Anal. Chem. 1975, 47, 970–983. [Google Scholar] [CrossRef]
- Creaser, C.S.; Griffiths, J.R.; Bramwell, C.J.; Noreen, S.; Hill, C.A.; Thomas, C.L.P. Ion mobility spectrometry: A review. Part 1. Structural analysis by mobility measurement. Analyst 2004, 129, 984–994. [Google Scholar] [CrossRef]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Picache, J.A.; Rose, B.S.; Balinski, A.; Leaptrot, K.L.; Sherrod, S.D.; May, J.C.; McLean, J.A. Collision cross section compendium to annotate and predict multi-omic compound identities. Chem. Sci. 2019, 10, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Mairinger, T.; Causon, T.J.; Hann, S. The potential of ion mobility-mass spectrometry for non-targeted metabolomics. Curr. Opin. Chem. Biol. 2018, 42, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ewing, M.A.; Glover, M.S.; Clemmer, D.E. Hybrid ion mobility and mass spectrometry as a separation tool. J. Chromatogr. A 2016, 1439, 3–25. [Google Scholar] [CrossRef] [PubMed]
- May, J.C.; McLean, J.A. Ion mobility-mass spectrometry: Time-dispersive instrumentation. Anal. Chem. 2015, 87, 1422–1438. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.B.; Nazarov, E.G.; Londry, F.; Vouros, P.; Covey, T.R. Differential mobility spectrometry/mass spectrometry history, theory, design optimization, simulations, and applications. Mass Spectrom. Rev. 2016, 35, 687–737. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility mass spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef]
- Nichols, C.M.; May, J.C.; Sherrod, S.D.; McLean, J.A. Automated flow injection method for the high precision determination of drift tube ion mobility collision cross sections. Analyst 2018, 143, 1556–1559. [Google Scholar] [CrossRef]
- Stow, S.M.; Causon, T.J.; Zheng, X.; Kurulugama, R.T.; Mairinger, T.; May, J.C.; Rennie, E.E.; Baker, E.S.; Smith, R.D.; McLean, J.A.; et al. An interlaboratory evaluation of drift tube ion mobility-mass spectrometry collision cross section measurements. Anal. Chem. 2017, 89, 9048–9055. [Google Scholar] [CrossRef] [PubMed]
- Kirk, A.T.; Bohnhorst, A.; Raddatz, C.-R.; Allers, M.; Zimmermann, S. Ultra-high-resolution ion mobility spectrometry—Current instrumentation, limitations, and future developments. Anal. Bioanal. Chem. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Ridgeway, M.E.; Lubeck, M.; Jordens, J.; Mann, M.; Park, M.A. Trapped ion mobility spectrometry: A short review. Int. J. Mass Spectrom. 2018, 425, 22–35. [Google Scholar] [CrossRef]
- Michelmann, K.; Silveira, J.A.; Ridgeway, M.E.; Park, M.A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 14–24. [Google Scholar] [CrossRef]
- Cumeras, R.; Figueras, E.; Davis, C.E.; Baumbach, J.I.; Gràcia, I. Review on ion mobility spectrometry. Part 1: Current instrumentation. Analyst 2015, 140, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Eiceman, G.A.; Karpas, Z. Ion. Mobility Spectrometry, 2nd ed.; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2005. [Google Scholar]
- Shvartsburg, A.A.; Smith, R.D. Fundamentals of traveling wave ion mobility spectrometry. Anal. Chem. 2008, 80, 9689–9699. [Google Scholar] [CrossRef]
- Causon, T.J.; Hann, S. Theoretical evaluation of peak capacity improvements by use of liquid chromatography combined with drift tube ion mobility-mass spectrometry. J. Chromatogr. A 2015, 1416, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Mesa, M.; Monteau, F.; Le Bizec, B.; Dervilly-Pinel, G. Potential of ion mobility-mass spectrometry for both targeted and non-targeted analysis of phase II steroid metabolites in urine. Anal. Chim. Acta X 2019, 1, 100006. [Google Scholar] [CrossRef]
- Regueiro, J.; Negreira, N.; Hannisdal, R.; Berntssen, M.H.G. Targeted approach for qualitative screening of pesticides in salmon feed by liquid chromatography coupled to traveling-wave ion mobility/quadrupole time-of-flight mass spectrometry. Food Control. 2017, 78, 116–125. [Google Scholar] [CrossRef]
- Kanu, A.B.; Hill, H.H., Jr. Ion mobility spectrometry detection for gas chromatography. J. Chromatogr. A 2008, 1177, 12–27. [Google Scholar] [CrossRef]
- Lapthorn, C.; Pullen, F.; Chowdhry, B.Z. Ion mobility spectrometry-mass spectrometry (IMS-MS) of small molecules: Separating and assigning structures to ions. Mass Spectrom. Rev. 2013, 32, 43–71. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Polasky, D.A.; Dixit, S.M.; Majmudar, J.D.; Neeson, K.; Ruotolo, B.T.; Martin, B.R. Variable-velocity traveling-wave ion mobility separation enhancing peak capacity for data-independent acquisition proteomics. Anal. Chem. 2017, 89, 5669–5672. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.L.; Turner, M.A.; Reynolds, J.C.; Creaser, C.S. Increasing peak capacity in nontargeted omics applications by combining full scan field asymmetric waveform ion mobility spectrometry with liquid chromatography-mass spectrometry. Anal. Chem. 2017, 89, 3452–3459. [Google Scholar] [CrossRef] [PubMed]
- Hill, H.H.; Simpson, G. Capabilities and limitations of ion mobility spectrometry for field screening applications. Field Anal. Chem. Technol. 1997, 1, 119–134. [Google Scholar] [CrossRef]
- Kaufmann, A.; Walker, S. Comparison of linear intrascan and interscan dynamic ranges of Orbitrap and ion-mobility time-of-flight mass spectrometers. Rapid Commun. Mass Spectrom. 2017, 31, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Widmer, M.; Giles, K.; Uría, D. Are liquid chromatography/ electrospray tandem quadrupole fragmentation rations unequivocal confirmation criteria? Rapid Commun. Mass Spectrom. 2009, 23, 958–998. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Kuballa, J.; Rohn, S.; Jantzen, E.; Luetjohann, J. Evaluation and validation of an ion mobility quadrupole time-of-flight mass spectrometry pesticide screening approach. J. Sep. Sci. 2018, 41, 2178–2187. [Google Scholar] [CrossRef]
- Tu, J.; Zhou, Z.; Li, T.; Zhu, Z.-J. The emerging role of ion mobility-mass spectrometry in lipidomics to facilitate lipid separation and identification. TrAC Trends Anal. Chem. 2019, 116, 332–339. [Google Scholar] [CrossRef]
- Chouinard, C.D.; Nagy, G.; Smith, R.D.; Baker, E.S. Ion mobility-mass spectrometry in metabolomic, lipidomic, and proteomic analyses. Compr. Anal. Chem. 2019, 83, 123–159. [Google Scholar] [CrossRef]
- Manz, C.; Pagel, K. Glycan analysis by ion mobility-mass spectrometry and gas-phase spectroscopy. Curr. Opin. Chem. Biol. 2018, 42, 16–24. [Google Scholar] [CrossRef]
- Kuang, A.; Erlund, I.; Herder, C.; Westerhuis, J.A.; Tuomilehto, J.; Cornelis, M.C. Lipidomic response to coffee consumption. Nutrients 2018, 10, 1851. [Google Scholar] [CrossRef] [PubMed]
- Blaženović, I.; Shen, T.; Mehta, S.S.; Kind, T.; Ji, J.; Piparo, M.; Cacciola, F.; Mondello, L.; Fiehn, O. Increasing compound identification rates in untargeted lipidomics research with liquid chromatography drift time-ion mobility mass spectrometry. Anal. Chem. 2018, 90, 10758–10764. [Google Scholar] [CrossRef] [PubMed]
- López-Morales, C.A.; Vázquez-Leyva, S.; Vallejo-Castillo, L.; Carballo-Uicab, G.; Muñoz-García, L.; Herbet-Pucheta, J.E.; Zepeda-Vallejo, L.G.; Velasco-Velázquez, M.; Pavón, L.; Pérez-Tapia, S.M.; et al. Determination of peptide profile consistency and safety of collagen hydrolysates as quality attributes. J. Food Sci. 2019, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Causon, T.J.; Došen, M.; Reznicek, G.; Hann, S. Workflow development for the analysis of phenolic compounds in wine using liquid chromatography combined with drift-tube ion mobility-mass spectrometry. LC-GC N. Am. 2016, 34, 854–867. [Google Scholar]
- Rodríguez-Maecker, R.; Vyhmeister, E.; Meisen, S.; Martinez Rosales, A.; Kuklya, A.; Telgheder, U. Identification of terpenes and essential oils by means of static headspace gas chromatography-ion mobility spectrometry. Anal. Bioanal. Chem. 2017, 409, 6595–6603. [Google Scholar] [CrossRef] [PubMed]
- Alves, T.O.; D’Almeida, C.T.S.; Victorio, V.C.M.; Souza, G.H.M.F.; Cameron, L.C.; Ferreira, M.S.L. Immunogenic and allergenic profile of wheat flours from different technological qualities revealed by ion mobility mass spectrometry. J. Food Compos. Anal. 2018, 73, 67–75. [Google Scholar] [CrossRef]
- Garrido-Delgado, R.; Dobao-Prieto, M.M.; Arce, L.; Valcárcel, M. Determination of volatile compounds by GC-IMS to assign the quality of virgin olive oil. Food Chem. 2015, 187, 572–579. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, D.; Dong, Y.; Ju, H.; Wu, C.; Lin, S. Characteristic volatiles fingerprints and changes of volatile compounds in fresh and dried Tricholoma matsutake Singer by HS-GC-IMS and HS-SPME-GC-MS. J. Chromatogr. B 2018, 1099, 46–55. [Google Scholar] [CrossRef]
- Browne, C.A.; Forbes, T.P.; Sisco, E. Detection and identification of sugar alcohol sweeteners by ion mobility spectrometry. Anal. Methods 2016, 8, 5611–5618. [Google Scholar] [CrossRef]
- Contreras, M.D.M.; Jurado-Campos, N.; Arce, L.; Arroyo-Manzanares, N. A robustness study of calibration models for olive oil classification: Targeted and non-targeted fingerprint approaches based on GC-IMS. Food Chem. 2019, 288, 315–324. [Google Scholar] [CrossRef]
- Gerhardt, N.; Schwolow, S.; Rohn, S.; Pérez-Cacho, P.R.; Galán-Soldevilla, H.; Arce, L.; Weller, P. Quality assessment of olive oils based on temperature-ramped HS-GC-IMS and sensory evaluation: Comparison of different processing approaches by LDA, kNN, and SVM. Food Chem. 2019, 278, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, S.; Seifert, L.; Ahlmann, N.; Hariharan, C.; Franzke, J.; Vautz, W. Coupling laser desorption with gas chromatography and ion mobility spectrometry for improved olive oil characterization. Food Chem. 2018, 255, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, H.; Yao, Y.; Hua, J.; Yang, Y.; Dong, C.; Deng, Y.; Wang, J.; Li, H.; Jiang, Y.; et al. Rapid volatiles fingerprinting by dopant-assisted positive photoionization ion mobility spectrometry for discrimination and characterization of Green Tea aromas. Talanta 2019, 191, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Guo, J.; Yu, J.; Guo, J.; Jia, X.; Liu, W.; Tian, P. Two-dimensional analysis of phenolic acids in seedling roots by high performance liquid chromatography electrospray ionization-ion mobility spectrometry. Anal. Methods 2019, 11, 610–617. [Google Scholar] [CrossRef]
- Struwe, W.B.; Baldauf, C.; Hofmann, J.; Rudd, P.M.; Pagel, K. Ion mobility separation of deprotonated oligosaccharide isomers—Evidence for gas-phase charge migration. Chem. Commum. 2016, 12353–12356. [Google Scholar] [CrossRef] [PubMed]
- Venter, P.; Muller, M.; Vestner, J.; Stander, M.A.; Tredoux, A.G.J.; Pasch, H.; De Villiers, A. Comprehensive three-dimensional LC × LC × ion mobility spectrometry separation combined with high-resolution MS for the analysis of complex samples. Anal. Chem. 2018, 90, 11643–11650. [Google Scholar] [CrossRef] [PubMed]
- Stephan, S.; Jakob, C.; Hippler, J.; Schmitz, O.J. A novel four-dimensional analytical approach for analysis of complex samples. Anal. Bioanal. Chem. 2016, 408, 3751–3759. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, J.M.; Zheng, D.; Yuan, M.; Wang, Z.Y.; Zhang, H.M.; Zheng, C.W.; Xiao, L.B.; Xu, H.X. A multidimensional analytical approach based on time-decoupled online comprehensive two-dimensional liquid chromatography coupled with ion mobility quadrupole time-of-flight mass spectrometry for the analysis of ginsenosides from white and red ginsengs. J. Pharm. Biomed. Anal. 2019, 163, 24–33. [Google Scholar] [CrossRef]
- Lipok, C.; Hippler, J.; Schmitz, O.J. A four dimensional separation method based on continuous heart-cutting gas chromatography with ion mobility and high resolution mass spectrometry. J. Chromatogr. A 2018, 1536, 50–57. [Google Scholar] [CrossRef]
- McCullagh, M.; Douce, D.; Hoeck, V.; Goscinny, S. Exploring the complexity of steviol glycosides analysis using ion mobility mass spectrometry. Anal. Chem. 2018, 90, 4585–4595. [Google Scholar] [CrossRef]
- Karpas, Z.; Guamán, A.V.; Pardo, A.; Marco, S. Comparison of the performance of three ion mobility spectrometers for measurement of biogenic amines. Anal. Chim. Acta 2013, 758, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Rudnik, D.D.; Laloush, M.; Yakir, D.; Karpas, Z. A novel method for determination of histamine in tuna fish by ion mobility spectrometry. Food Anal. Methods 2015, 8, 2376–2382. [Google Scholar] [CrossRef]
- Cheng, S.; Li, H.; Jiang, D.; Chen, C.; Zhang, T.; Li, Y.; Wang, H.; Zhou, Q.; Li, H.; Tan, M. Sensitive detection of trimethylamine based on dopant-assisted positive photoionization ion mobility spectrometry. Talanta 2017, 162, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Parchami, R.; Kamalabadi, M.; Alizadeh, N. Determination of biogenic amines in canned fish samples using head-space solid phase microextraction based on nanostructured polypyrrole fiber coupled to modified ionization región ion mobility spectrometry. J. Chromatogr. A 2017, 1481, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Zarpas, Z.; Guamán, A.V.; Calvo, D.; Pardo, A.; Marco, S. The potential of ion mobility spectrometry (IMS) for detection of 2,4,6-trichloroanisole (2,4,6-TCA) in wine. Talanta 2012, 93, 200–205. [Google Scholar] [CrossRef]
- Kamalabadi, M.; Ghaemi, E.; Mohammadi, A.; Alizadeh, N. Determination of furfural and hydroxymethylfurfural from baby formula using headspace microextraction base don nanostrutured polypyrrole fiber coupled with ion mobility spectrometry. Food Chem. 2015, 181, 72–77. [Google Scholar] [CrossRef]
- Garrido-Delgado, R.; Dobao-Prieto, M.M.; Arce, L.; Aguilar, J.; Cumplido, J.L.; Valcárcel, M. Ion mobility spectrometry versus classical physico-chemical analysis for assessing the shelf life of extra virgin olive oil according to container type and storage conditions. J. Agric. Food Chem. 2015, 63, 2179–2188. [Google Scholar] [CrossRef]
- Tzschoppe, M.; Haase, H.; Höhnisch, M.; Jaros, D.; Rohm, H. Using ion mobility spectrometry for screening the autoxidation of peanuts. Food Control. 2016, 64, 17–21. [Google Scholar] [CrossRef]
- Raatikainen, O.; Reinikainen, V.; Minkkinen, P.; Ritvanen, T.; Muje, P.; Pursiainen, J.; Hiltunen, T.; Hyvönen, P.; Von Wright, A.; Reinikainen, S.-P. Multivariate modelling of fish freshness index based on ion mobility spectrometry measurements. Anal. Chim. Acta 2005, 544, 128–134. [Google Scholar] [CrossRef]
- Cavanna, D.; Zanardi, S.; Dall’Asta, C.; Suman, M. Ion mobility spectrometry coupled to gas chromatography: A rapid tool to assess eggs freshness. Food Chem. 2019, 271, 691–696. [Google Scholar] [CrossRef]
- Tang, Z.-S.; Zeng, X.-A.; Brennan, M.A.; Han, Z.; Niu, D.; Huo, Y. Characterization of aroma profile and characteristic aromas during lychee wine fermentation. J. Food Process. Preserv. 2019, in press. [Google Scholar] [CrossRef]
- Halbfeld, C.; Ebert, B.E.; Blank, L.M. Multi-capillary column-ion mobility spectrometry of volatile metabolites emitted by Saccharomyces Cerevisiae. Metabolites 2014, 4, 751–774. [Google Scholar] [CrossRef] [PubMed]
- Gloess, A.N.; Yeretzian, C.; Knochenmuss, R.; Groessl, M. On-line analysis of coffee roasting with ion mobility spectrometry-mass spectrometry (IMS-MS). Int. J. Mass Spectrom. 2018, 424, 49–57. [Google Scholar] [CrossRef]
- Danezis, G.P.; Tsagkaris, A.S.; Camin, F.; Brusic, V.; Georgiou, C.A. Food authentication: Techniques, trends & emerging approaches. TrAC Trends Anal. Chem. 2016, 85, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Cubero-Leon, E.; Peñalver, R.; Maquet, A. Review on metabolomics for food authentication. Food Res. Int. 2014, 60, 95–107. [Google Scholar] [CrossRef]
- Causon, T.J.; Ivanova-Petropulos, V.; Petrusheva, D.; Bogeva, E.; Hann, S. Fingerprinting of traditionally produced red wines using liquid chromatography combined with drift tube ion mobility-mass spectrometry. Anal. Chim. Acta 2019, 1052, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Delgado, R.; Mercader-Trejo, F.; Sielemann, S.; de Bruyn, W.; Arce, L.; Valcárcel, M. Direct classification of olive oils by using two types of ion mobility spectrometers. Anal. Chim. Acta 2011, 696, 108–115. [Google Scholar] [CrossRef]
- Gerhardt, N.; Birkenmeier, M.; Sanders, D.; Rohn, S.; Weller, P. Resolution-optimized headspace gas chromatography-ion mobility spectrometry (HS-GC-IMS) for non-targeted olive oil profiling. Anal. Bianal. Chem. 2017, 409, 3933–3942. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Martín-Gómez, A.; Jurado-Campos, N.; Garrido-Delgado, R.; Arce, C.; Arce, L. Target vs spectral fingerprint data analysis of Iberian ham samples for avoiding labelling fraud using headspace– gas chromatography—ion mobility spectrometry. Food Chem. 2018, 246, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Martín-Gómez, A.; Arroyo-Manzanares, N.; Rodríguez-Estévez, V.; Arce, L. Use of a non-destructive sampling method for characterization of Iberian cured ham breed and feeding regime using GC-IMS. Meat Sci. 2019, 146–154. [Google Scholar] [CrossRef]
- Garrido-Delgado, R.; Arce, L.; Guamán, A.V.; Pardo, A.; Marco, S.; Valcárcel, M. Direct coupling of a gas-liquid separator to an ion mobility spectrometer for the classification of different white wines using chemometrics tools. Talanta 2011, 84, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, N.; Birkenmeier, M.; Schwolow, S.; Rohn, S.; Weller, P. Volatile-compound fingerprinting by headspace-gas-chromatography ion-mobility spectrometry (HS-GC-IMS) as a benchtop alternative to 1H NMR profiling for assessment of the authenticity of honey. Anal. Chem. 2018, 90, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, S.; He, J.; Chen, L.; Zhang, J.; Jin, Y.; Zhou, J.; Zhang, Y. A green triple-locked strategy based on volatile-compound imaging, chemometrics, and markers to discriminate winter honey and sapium honey using headspace gas chromatography-ion mobility spectrometry. Food Res. Int. 2019, 119, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Chen, X.; Lu, D.; Chen, B. Detection of adulteration in canola oil by using GC-IMS and chemometric analysis. Int. J. Anal. Chem. 2018, 2018, 3160265. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.; Goggin, K.A.; Tahir, N.I.; Brodrick, E.; Singh, R.; Sambanthamurthi, R.; Parveez, K.A.; Davies, A.N.; Murad, A.J.; Muhammad, N.H.; et al. Use of headspace-gas chromatography-ion mobility spectrometry to detect volatile fingerprints of palm fibre oil and sludge palm oil in samples of crude palm oil. BMC Res. Notes 2019, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Delgado, R.; Muñoz-Pérez, E.; Arce, L. Detection of adulteration in extra virgin olive oils by using UV-IMS and chemometric analysis. Food Control. 2018, 85, 292–299. [Google Scholar] [CrossRef]
- Zhang, L.; Shuai, Q.; Li, P.; Zhang, Q.; Ma, F.; Zhang, W.; Ding, X. Ion mobility spectrometry fingerprints: A rapid detection technology for adulteration of sesame oil. Food Chem. 2016, 192, 60–66. [Google Scholar] [CrossRef]
- Shuai, Q.; Zhang, L.; Li, P.; Zhang, Q.; Wang, X.; Ding, X.; Zhang, W. Rapid adulteration detection for flaxseed oil using ion mobility spectrometry and chemometric methods. Anal. Methods 2014, 6, 9575–9580. [Google Scholar] [CrossRef]
- Khademi, S.M.S.; Telgheder, U.; Valadbeigi, Y.; Ilbeigi, V.; Tabrizchi, M. Direct detection of glyphosate in drinking water using corona-discharge ion mobility spectrometry: A theoretical and experimental study. Int. J. Mass Spectrom. 2019, 442, 29–34. [Google Scholar] [CrossRef]
- European Commission. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed; SANTE/11813/2017; European Commission: Brussels, Belgium, 2017.
- European Commission. Commission Decision (EEC) 2002/657/EC. Off. J. Eur. Commun. 2002, L221, 8. [Google Scholar]
- European Commission. Guidance Document on Identification of Mycotoxins in Food and Feed; SANTE/12089 /2016; European Commission: Brussels, Belgium, 2016.
- Wageningen University & Research. Available online: https://www.wur.nl/upload_mm/e/6/e/0d3c53a2-28b8-4e4d-a436-b84cf471f20e_20181015-9%20Revision%202002-657%20confirmation%20criteria.pdf (accessed on 24 July 2019).
- Weickhardt, C.; Kaiser, N.; Borsdorf, H. Ion mobility spectrometry of laser desorbed pesticides from fruit surfaces. Int. J. Ion. Mobil. Spectrom. 2012, 15, 55–62. [Google Scholar] [CrossRef]
- Armenta, S.; de la Guardia, M.; Abad-Fuentes, A.; Abad-Somovilla, A.; Esteve-Turrillas, F.A. Off-line coupling of multidimensional immunoaffinity chromatography and ion mobility spectrometry: A promising partnership. J. Chromatogr. A 2015, 1426, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, A.; Tabrizchi, M.; Ghaziaskar, H.S. Determination of aflatoxins B1 and B2 using ion mobility spectrometry. Talanta 2008, 75, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Aria, A.A.; Sorribes-Soriano, A.; Jafari, M.T.; Nourbakhsh, F.; Esteve-Turrilas, F.A.; Armenta, S.; Herrero-Martínez, J.M.; de la Guardia, M. Uptake and translocation monitoring of imidacloprid to chili and tomato plants by molecularly imprinting extraction—Ion mobility spectrometry. Microchem. J. 2019, 144, 195–202. [Google Scholar] [CrossRef]
- Saraji, M.; Jafari, M.T.; Mossaddegh, M. Carbon nanotubes@silicon dioxide nanohybrids coating for solid-phase microextraction of organophosphorus pesticides followed by gas chromatography-corona discharge ion mobility spectrometric detection. J. Chromatogr. A 2016, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kermani, M.; Jafari, M.; Saraji, M. Porous magnetized carbon sheet nanocomposites for dispersive solid-phase microextraction of organophosphorus pesticides prior to analysis by gas chromatography-ion mobility spectrometry. Microchim. Acta 2019, 186, 88. [Google Scholar] [CrossRef] [PubMed]
- McCooeye, M.; Kolakowski, B.; Boison, J.; Mester, Z. Evaluation of high-field asymmetric waveform ion mobility spectrometry mass spectrometry for the analysis of the mycotoxin zearalenone. Anal. Chim. Acta 2008, 627, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, J.; Chen, A.; Ma, X.; Yang, S. A new retrospective, multi-evidence veterinary drug screening method using drift tube ion mobility mass spectrometry. Rapid Commun. Mass Spectrom. 2018, 32, 1141–1148. [Google Scholar] [CrossRef]
- Ahmed, E.; Kabir, K.M.M.; Wang, H.; Xiao, D.; Fletcher, J.; Donald, W.A. Rapid separation of isomeric perfluoroalkyl substances by high-resolution differential ion mobility mass spectrometry. Anal. Chim. Acta 2019, 1058, 127–135. [Google Scholar] [CrossRef]
- Beach, D.G.; Melanson, J.E.; Purves, R.W. Analyis of paralytic shellfish toxins using high-field asymmetric waveform ion mobility spectrometry with liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2473–2484. [Google Scholar] [CrossRef]
- Poyer, S.; Loutelier-Bourhis, C.; Coadou, G.; Mondeguer, F.; Enche, J.; Bossée, A.; Hess, P.; Afonso, C. Identification and separation of saxitoxins using hydrophilic interaction liquid chromatography coupled to travelling wave ion mobility-mass spectrometry. J. Mass Spectrom. 2015, 50, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Beucher, L.; Dervilly-Pinel, G.; Prévost, S.; Monteau, F.; Le Bizec, B. Determination of a large set of β-adrenergic agonists in animal matrices based on ion mobility and mass separations. Anal. Chem. 2015, 87, 9234–9242. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Dupuis, K.T.; Aly, N.A.; Zhou, Y.; Smith, F.B.; Tang, K.; Smith, R.D.; Baker, E.S. Utilizing ion mobility spectrometry and mass spectrometry for the analysis of polyciyclic aromatic hydrocarbons, polychlorinated biphenyls, polybrominated diphenyl ethers and their metabolites. Anal. Chim. Acta 2018, 1037, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Varesio, E.; Le Blanc, J.C.Y.; Hopfgartner, G. Real-time 2D separation by LC x differential ion mobility hyphenated to mass spectrometry. Anal. Bioanal. Chem. 2012, 402, 2555–2564. [Google Scholar] [CrossRef] [PubMed]
- Beach, D.G.; Kerrin, E.S.; Quilliam, M.A. Selective quantitation of the neurotoxin BMAA by use of hydrophilic-interaction liquid chromatography-differential mobility spectrometry-tandem mass spectrometry (HILIC-DMS-MS/MS). Anal. Bioanal. Chem. 2015, 407, 8397–8409. [Google Scholar] [CrossRef] [PubMed]
- Goscinny, S.; Joly, L.; De Pauw, E.; Hanot, V.; Eppe, G. Travelling-wave ion mobility time-of-flight mass spectrometry as an alternative strategy for screening of multi-class pesticides in fruits and vegetables. J. Chromatogr. A 2015, 1405, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Goscinny, S.; McCullagh, M.; Far, J.; De Pauw, E.; Eppe, G. Towards the use of ion mobility mass spectrometry derived collision cross section as a screening approach for unambiguous identification of targeted pesticides in food. Rapid Commun. Mass Spectrom. 2019, 33, 34–48. [Google Scholar] [CrossRef] [Green Version]
- Regueiro, J.; Negreira, N.; Berntssen, M.H.G. Ion-mobility-derived collision cross section as an additional identification point for multiresidue screening of pesticides in fish feed. Anal. Chem. 2016, 88, 11169–11177. [Google Scholar] [CrossRef]
- Hines, K.M.; Ross, D.H.; Davidson, K.L.; Bush, M.F.; Xu, L. Large-scale structural characterization of drug and drug-like compounds by high-throughput ion mobility-mass spectrometry. Anal. Chem. 2017, 89, 9023–9030. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; Le Bizec, B.; Monteau, F.; García-Campaña, A.M.; Dervilly-Pinel, G. Collision Cross Section (CCS) database: An additional measure to characterize steroids. Anal. Chem. 2018, 90, 4616–4625. [Google Scholar] [CrossRef]
- Tejada-Casado, C.; Hernández-Mesa, M.; Monteau, F.; Lara, F.J.; del Olmo-Iruela, M.; García-Campaña, A.M.; Le Bizec, B.; Dervilly-Pinel, G. Collision cross section (CCS) as a complementary parameter to characterize human and veterinary drugs. Anal. Chim. Acta 2018, 1043, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Righetti, L.; Bergmann, A.; Galaverna, G.; Rolfsson, O.; Paglia, G.; Dall’Asta, C. Ion mobility-derived collision cross section database: Application to mycotoxin analysis. Anal. Chim. Acta 2018, 1014, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Righetti, L.; Fenclova, M.; Dellafiora, L.; Hajslova, J.; Stranska-Zachariasova, M.; Dall’Asta, C. High resolution-ion mobility mass spectrometry as an additional powerful tool for structural characterization of mycotoxin metabolites. Food Chem. 2018, 245, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Luetjohann, J.; Hanschen, F.S.; Schreiner, M.; Kuballa, J.; Jantzen, E.; Rohn, S. Identification and characterization of pesticide metabolites in Brassica species by liquid chromatography travelling wave ion mobility quadrupole time-of-flight mass spectrometry (UPLC-TWIMS-QTOF-MS). Food Chem. 2018, 244, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Dodds, J.N.; May, J.C.; McLean, J.A. Correlating resolving power, resolution, and collision cross section: Unifying cross-platform assessment of separation efficiency in ion mobility spectrometry. Anal. Chem. 2019, 89, 12176–12184. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Webb, I.K.; Garimella, S.V.B.; Hamid, A.M.; Zheng, X.; Norheim, R.V.; Prost, S.A.; Anderson, G.A.; Sandoval, J.A.; Baker, E.S.; et al. Serpentine ultralong path with extended routing (SUPER) high resolution traveling wave ion mobility-MS using structures for lossless ion manipulations. Anal. Chem. 2017, 89, 4628–4634. [Google Scholar] [CrossRef] [PubMed]
- Merenbloom, S.I.; Glaskin, R.S.; Henson, Z.B.; Clemmer, D.E. High-resolution ion cyclotron mobility spectrometry. Anal. Chem. 2009, 81, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Glaskin, R.S.; Ewing, M.A.; Clemmer, D.E. Ion trapping for ion mobility spectrometry measurements in a cyclical drift tube. Anal. Chem. 2013, 85, 7003–7008. [Google Scholar] [CrossRef]
- Giles, K.; Wildgoose, J.; Pringle, S.; Garside, J.; Carney, P.; Nixon, P.; Langridge, D. Design and utility of a multi-pass cyclic ion mobility separator. In Annual Conference Proceedings; ASMS: Baltimore, MD, USA, 2014. [Google Scholar]
- Ujma, J.; Ropartz, D.; Giles, K.; Richardson, K.; Langridge, D.; Wildgoose, J.; Green, M. Cyclic ion mobility mass spectrometry distinguishes anomers and open-ring forms of pentasaccharides. J. Am. Soc. Mass Spectrom. 2019, 30, 1028–1037. [Google Scholar] [CrossRef]
- McCullagh, M.; Giles, K.; Richardson, K.; Stead, S.; Palmer, M. Investigations into the performance of travelling wave enabled conventional and cyclic ion mobility systems to characterize protomers of fluoroquinolone antibiotic residues. Rapid Commun. Mass Spectrom. 2019, 33, 11–21. [Google Scholar] [CrossRef]
- Hamid, A.M.; Ibrahim, Y.M.; Garimella, S.V.B.; Webb, I.K.; Deng, L.; Chen, T.-C.; Anderson, G.A.; Prost, S.A.; Norheim, R.V.; Tolmachev, A.V.; et al. Characterization of traveling wave ion mobility separations in structures for lossless ion manipulations. Anal. Chem. 2015, 87, 11301–11308. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ibrahim, Y.M.; Baker, E.S.; Aly, N.A.; Hamid, A.M.; Zhang, X.; Zheng, X.; Garimella, S.V.B.; Webb, I.K.; Prost, S.A.; et al. Ion mobility separations of isomers based upon long path length structures for lossless ion manipulations combined with mass spectrometry. ChemistrySelect 2016, 1, 2396–2399. [Google Scholar] [CrossRef] [PubMed]
- Faleh, A.B.; Warnke, S.; Rizzo, T.R. Combining ultrahigh-resolution ion-mobility spectrometry with cryogenic infrared spectroscopy for the analysis of glycan mixtures. Anal. Chem. 2019, 91, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tu, J.; Zhu, Z.-J. Advancing the large-scale CCS database for metabolomics and lipidomics at the machine-learning era. Curr. Opin. Chem. Biol. 2018, 42, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, L.; Bade, R.; Celma, A.; Mullin, L.; Cleland, G.; Stead, S.; Hernandez, F.; Sancho, J.V. Prediction of collision cross-section values for small molecules: Application to pesticide residue analysis. Anal. Chem. 2017, 89, 6583–6589. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.B.; May, J.C.; Leaptrot, K.L.; McLean, J.A. Evaluating separation selectivity and collision cross section measurement reproducibility in helium, nitrogen, argon, and carbon dioxide drift gases for drift tube ion mobility-mass spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Kurulugama, R.T.; Darland, E.; Kuhlmann, F.; Stafford, G.; Fjeldsted, J. Evaluation of drift gas selection in complex sample analysis using a high performance drift tube ion mobility-QTOF mass spectrometer. Analyst 2015, 140, 6834–6844. [Google Scholar] [CrossRef]
- Oranzi, N.R.; Kemperman, R.H.J.; Wei, M.S.; Petkovska, V.I.; Granato, S.W.; Rochon, B.; Kaszycki, J.; La Rotta, A.; Jeanne Dit Fouque, K.; Fernandez-Lima, F.; et al. Measuring the integrity of gas-phase conformers of sodiated 25-hydroxyvitamin d3 by drift tube, traveling wave, trapped, and high-field asymmetric ion mobility. Anal. Chem. 2019, 91, 4092–4099. [Google Scholar] [CrossRef]
- Hinnenkamp, V.; Klein, J.; Mecklmann, S.W.; Balsaa, P.; Schmidt, T.C.; Schmitz, O.J. Comparison of CCS values determined by traveling wave ion mobility mass spectrometry and drift tube ion mobility mass spectrometry. Anal. Chem. 2018, 90, 12042–12050. [Google Scholar] [CrossRef]
- Paglia, G.; Williams, J.P.; Menikarachchi, L.; Thompson, J.W.; Tyldesley-Worster, R.; Halldórson, S.; Rolfsson, O.; Moseley, A.; Grant, D.; Langridge, J.; et al. Ion mobility derived collision cross sections to support metabolomics applications. Anal. Chem. 2014, 86, 3985–3993. [Google Scholar] [CrossRef]
- Paglia, G.; Astarita, G. Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nat. Protoc. 2017, 12, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Struwe, W.B.; Scarff, C.A.; Scrivens, J.H.; Harvey, D.J.; Pagel, K. Estimating collision cross sections of negatively charged n-glycans using traveling wave ion mobility-mass spectrometry. Anal. Chem. 2014, 86, 10789–10795. [Google Scholar] [CrossRef] [PubMed]
- Hines, K.M.; May, J.C.; McLean, J.A.; Xu, L. Evaluation of collision cross section calibrants for structural analysis of lipids by travelling wave ion mobility-mass spectrometry. Anal. Chem. 2016, 88, 7329–7336. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Mesa, M.; Ropartz, D.; García-Campaña, A.M.; Rogniaux, H.; Dervilly-Pinel, G.; Le Bizec, B. Ion Mobility Spectrometry in Food Analysis: Principles, Current Applications and Future Trends. Molecules 2019, 24, 2706. https://doi.org/10.3390/molecules24152706
Hernández-Mesa M, Ropartz D, García-Campaña AM, Rogniaux H, Dervilly-Pinel G, Le Bizec B. Ion Mobility Spectrometry in Food Analysis: Principles, Current Applications and Future Trends. Molecules. 2019; 24(15):2706. https://doi.org/10.3390/molecules24152706
Chicago/Turabian StyleHernández-Mesa, Maykel, David Ropartz, Ana M. García-Campaña, Hélène Rogniaux, Gaud Dervilly-Pinel, and Bruno Le Bizec. 2019. "Ion Mobility Spectrometry in Food Analysis: Principles, Current Applications and Future Trends" Molecules 24, no. 15: 2706. https://doi.org/10.3390/molecules24152706