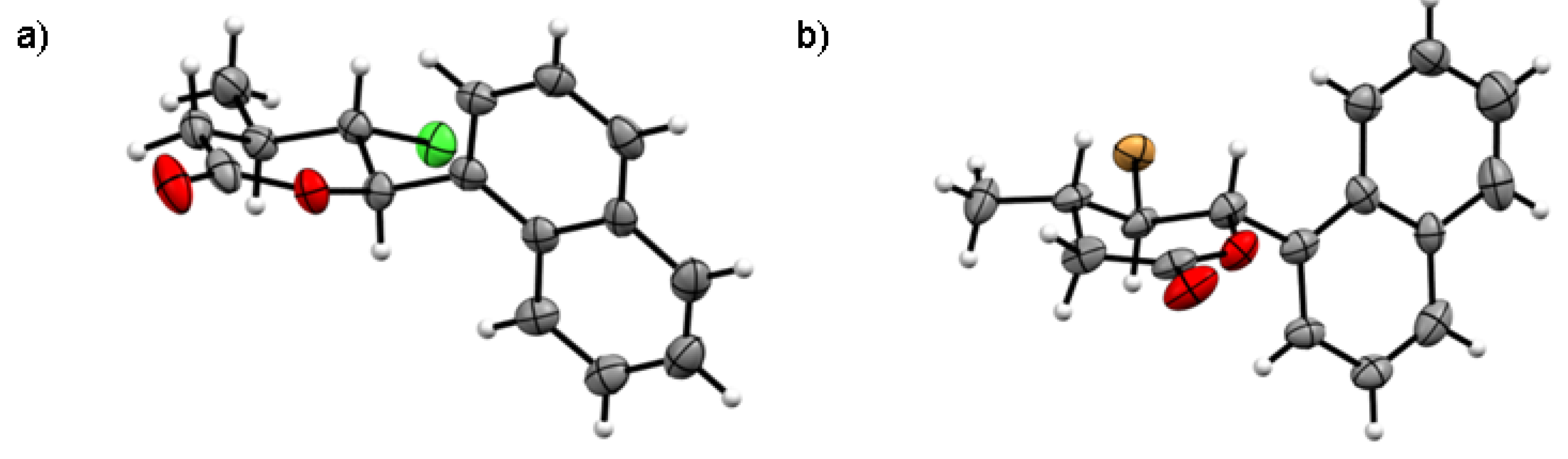
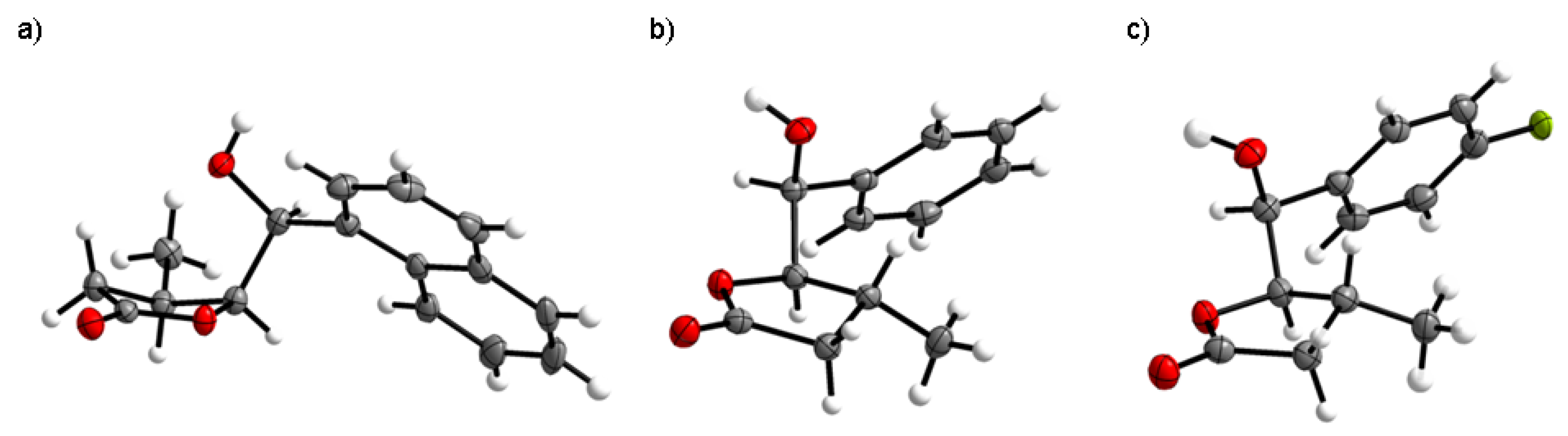
All used chemicals were purchased from Sigma-Aldrich and Fluka and used without further purification. The NMR spectra were obtained in CDCl on a Bruker Avance DRX 500 MHz. FTIR spectra were recorded using an attenuated total reflection technique on a Perkin Elmer Spectrum 400 spectrometer. High-resolution electrospray ionization mass spectra (HR-ESI-MS) were acquired on a Bruker micrOTOF-Q II. Gas chromatography was performed using a Thermo Scientific-Trace 1310 chromatograph equipped with a TG-5HT column (30 m × 0.25 mm). Melting points were determined with a Boetius micro melting point apparatus and are uncorrected. The refractive index was determined with an Abbe refractometer (Atago RX-7000 CX). TLC analysis was performed on silica gel F254 plates (Merck). Column chromatography was performed on silica gel 60 (230–400 mesh, Merck) using mixtures of hexane, ethyl acetate and acetone as eluents. The crystal structure was determined by single-crystal X-ray diffraction (Xcalibur, Sapphire 2 CCD detector).
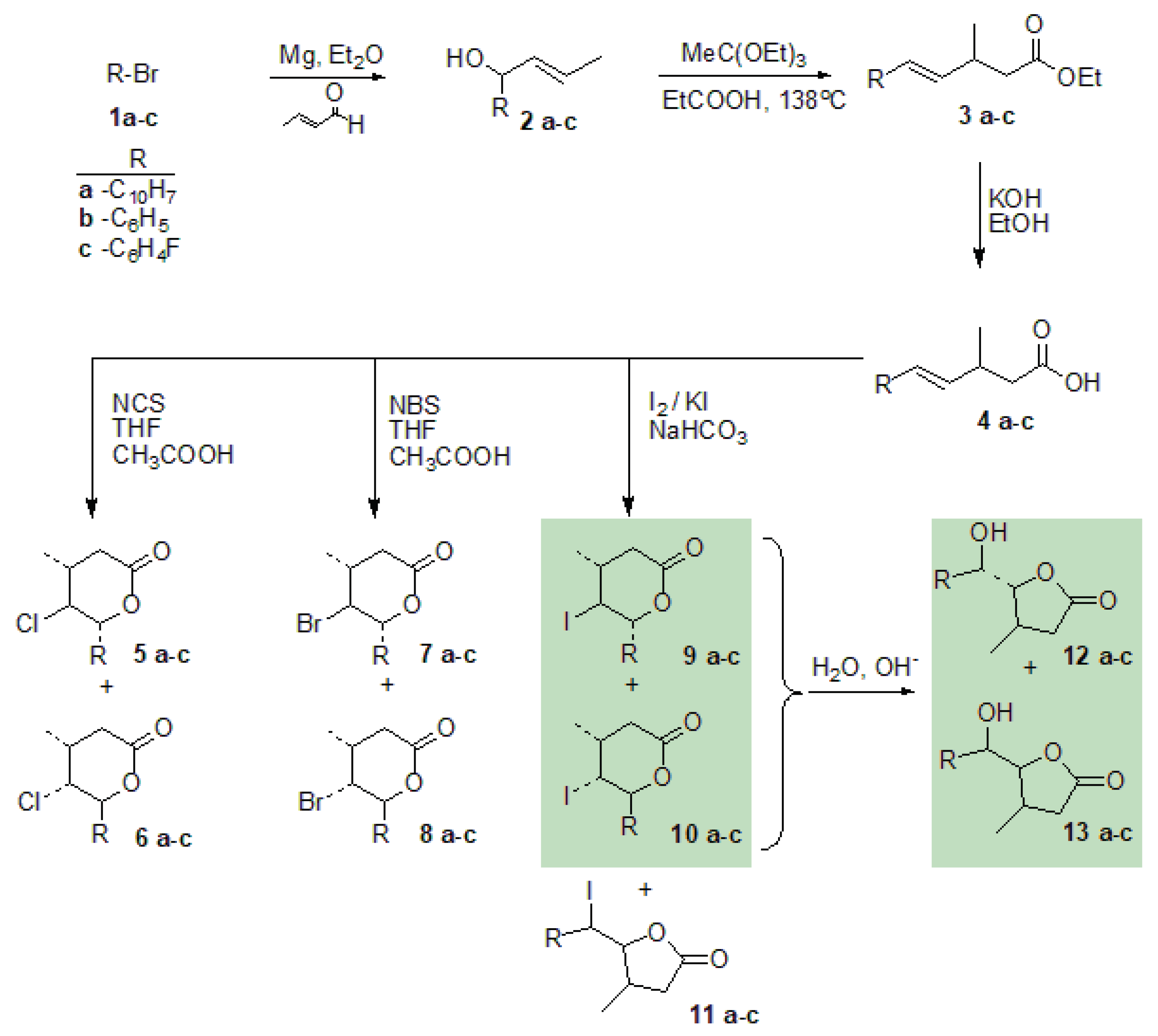
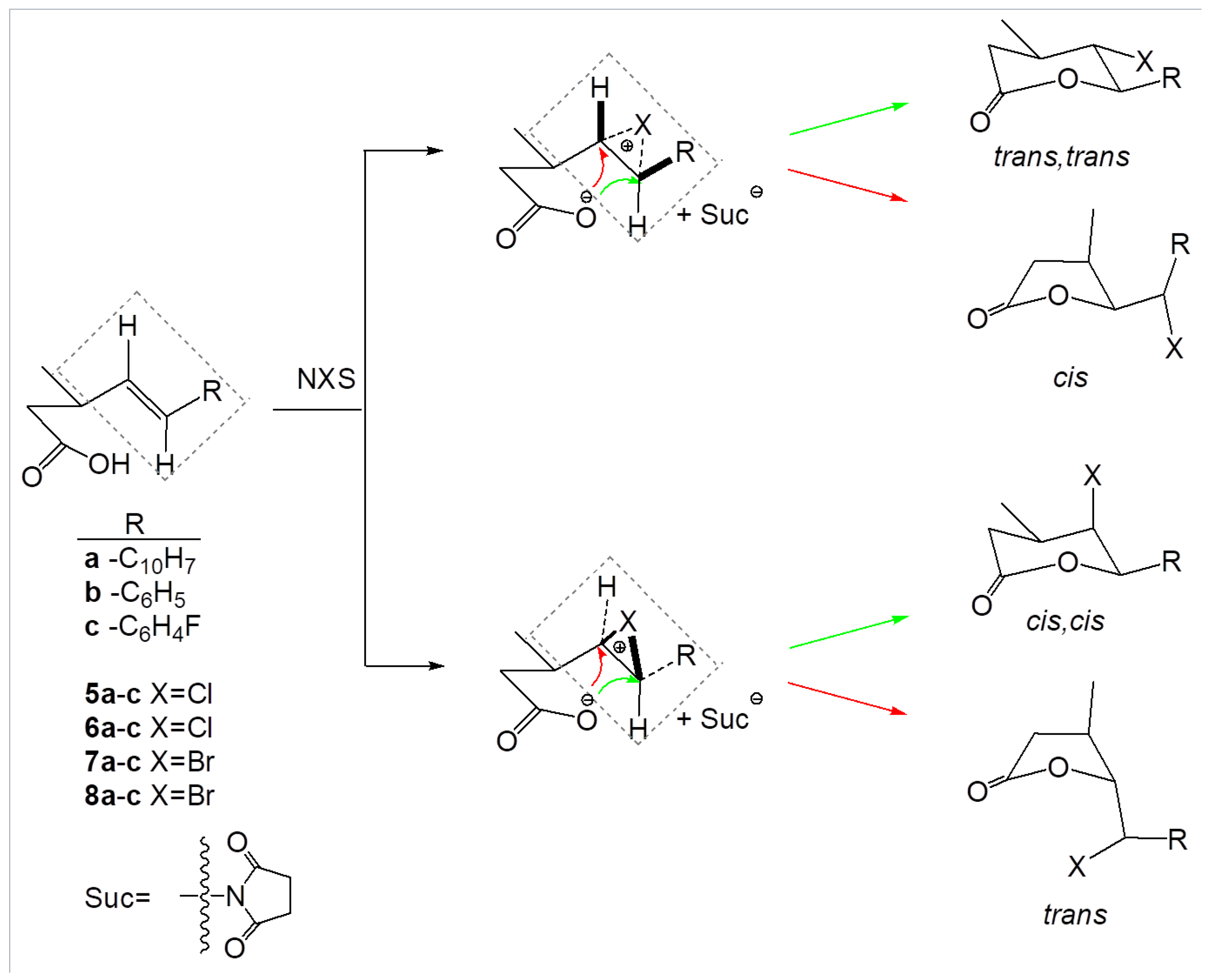
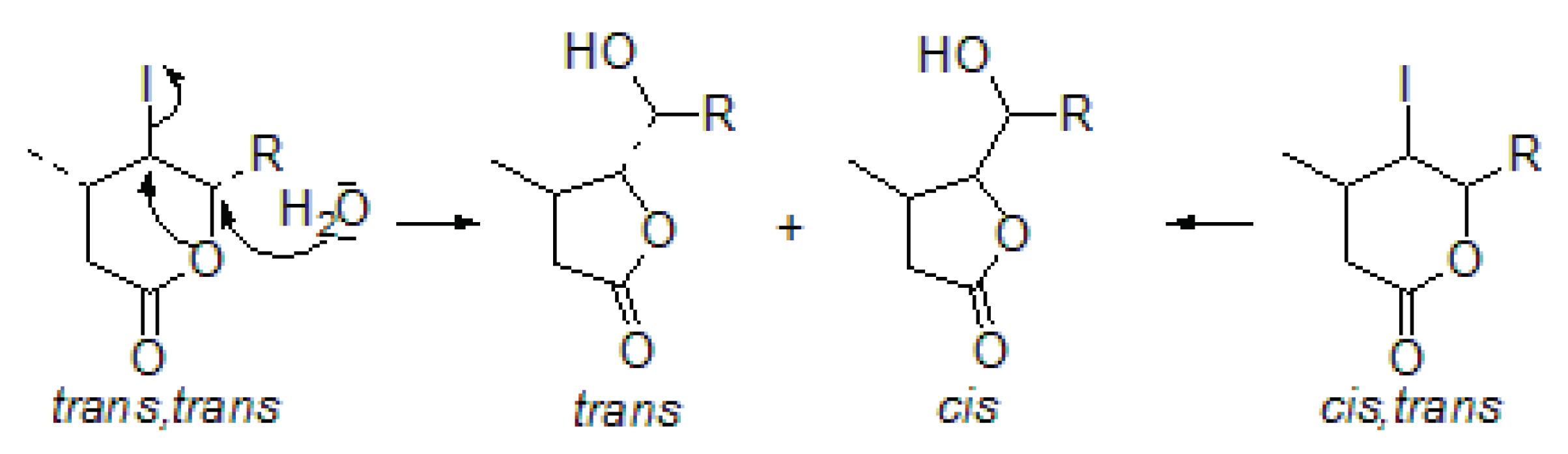
3.4. The Chlorolactonization of Unsaturated Carboxylic Acids 4a–c
The mixture of an unsaturated carboxylic acid (0.005 mol) and
N-chlorosuccinimide (0.009 mol) was dissolved in 60 mL of THF. Acetic acid was added dropwise and the reaction mixture was stirred at room temperature for 48 h. When the reaction was completed, the mixture was dissolved in diethyl ether, washed with saturated
and dried over anhydrous magnesium sulfate. The crude product was purified by column chromatography on silica gel using a mixture of acetone and hexane (1:10) [
28]. Spectral data are given below. The chlorolactonization of
4a gave chlorolactones
5a and
6a with a total yield of 51.19% (
5a:
6a 81.55:18.45). The chlorolactonization of
4b gave chlorolactones
5b and
6b with a total yield of 82.21% (
5b:
6b 84.42:15.58). The chlorolactonization of
4c gave chlorolactones
5c and
6c with a total yield of 61.67% (
5c:
6c 81.02:18.98).
Trans,trans-5-chloro-tetrahydro-4-methyl-6-(naphthalen-1-yl)pyran-2-one (5a): The product was obtained as a colorless solid; mp = 157–158 C; = 0.19 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.29 (d, 3H, J = 6.4 Hz, CH(CH)), 2.47–2.61 (m, 2H, CH(CH), CHaHb), 3.07 (dd, 1H, = 18.5 Hz, = 10.0 Hz, CHaHb), 4.18 (t, 1H, J = 9.7 Hz, CHCl), 5.99 (d, 1H, J = 10.0 Hz, CHAr), 7.47–7.61 (m, 4H, HAr), 7.90 (d, 2H, J = 8.3 Hz, HAr), 8.09 (d, 1H, J = 8.5 Hz, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.70 (CH(CH)), 35.98 (CH(CH)), 37.15 (CH), 62.72 (CHCl), 81.97 (CHAr), 169.03 (C = O), CAr: 123.01, 125.00, 125.85, 125.97, 126.62, 129.09, 130.10, 131.18, 131.85, 133.87; IR (cm): 1727, 1596, 1512, 1453, 1408, 1375, 1350, 1324, 1286, 1263, 1239, 1216, 1175, 1105, 1085, 1062, 1022, 997, 950, 900, 860, 834, 805, 775, 732, 674, 640; HR-MS (ESI-TOF) calculated for , m/z [M + K]: 313.039760; experimental value: 313.041132.
Cis,trans-5-chloro-tetrahydro-4-methyl-6-(naphthalen-1-yl)pyran-2-one (6a): The product was obtained as a colorless solid; mp = 98–99.5 C; = 0.28 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 0.98 (d, 3H, J = 6.5 Hz, CH(CH)), 2.03–2.11 (m, 1H, CH(CH)), 2.67 (dd, 1H, = 18.7 Hz, = 11.4 Hz, CHaHb), 2.75 (dd, 1H, = 18.7 Hz, = 6.3 Hz, CHaHb), 4.51 (t, 1H, J = 1.9 Hz, CHCl), 6.55 (d, 1H, J = 1.9 Hz, CHAr), 7.44–7.59 (m, 4H, HAr), 7.89 (d, 2H, J = 8.2 Hz, HAr), 8.07 (d, 1H, J = 8.4 Hz, HAr); C NMR (125 MHz, CDCl), [ppm]: 17.81 (CH(CH)), 27.12 (CH(CH)), 33.06 (CH), 62.19 (CHCl), 84.33 (CHAr), 169.24 (C = O), CAr: 121.76, 123.56, 125.22, 126.30, 127.35, 129.46, 129.51, 130.12, 132.99, 133.83; IR (cm): 1733, 1598, 1507, 1455, 1393, 1342, 1281, 1218, 1169, 1112, 1074, 1038, 997, 945, 909, 886, 863, 802, 777, 737, 703, 615; HR-MS (ESI-TOF) calculated for , m/z [M + Na]: 297.065822; experimental value: 297.067366.
Trans,trans-5-chloro-tetrahydro-4-methyl-6-phenylpyran-2-one (5b): The product was obtained as a colorless solid; mp = 105–106.5 C, = 0.18 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.25 (d, 3H, J = 6.4 Hz, CH(CH)), 2.35–2.47 (m, 2H, J = 17.2 Hz, CHaHb), 2.92–3.01 (m, 1H, CHaHb), 3.76 (t, 1H, J = 9.8Hz, CHCl), 5.18 (d, 1H, J = 10.0 Hz, CHAr), 7.33–7.43 (m, 5H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.62 (CH(CH)), 35.22 (CH(CH), 37.10 (CH), 63.27 (CHCl), 85.05 (CHAr), 168.95 (C = O), CAr: 127.51, 128.52, 129.29, 136.44; IR (cm): 1730, 1496, 1460, 1401, 1376, 1345, 1288, 1240, 1218, 1184, 1107, 1074, 1055, 1031, 1020, 995, 930, 906, 893, 850, 800, 757, 670, 651, 618; HR-MS (ESI-TOF) calculated for , m/z [M + Na]: 247.050173; experimental value: 247.051037.
Cis,trans-5-chloro-tetrahydro-4-methyl-6-phenylpyran-2-one (6b): The product was obtained as a colorless oil; = 1.5453; = 0.27 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.05 (d, 3H, J = 6.6 Hz, CH(CH)), 2.24–2.36 (m, 1H, CH(CH)), 2.59 (dd, 1H, = 18.4 Hz, = 10.3 Hz, CHaHb), 2.64 (dd, 1H, = 18.4 Hz, = 6.9 Hz, CHaHb), 4.33 (t, 1H, J = 2.5 Hz, CHCl), 5.79 (d, 1H, J = 2.5 Hz, CHAr), 7.25–7.28 (m, 2H, HAr), 7.32–7.47 (m, 3H, HAr); C NMR (125 MHz, CDCl), [ppm]: 17.65 (CH(CH)), 27.07 (CH(CH), 33.54 (CH), 63.32 (CHCl), 85.20 (CHAr), 168.97 (C = O), CAr: 125.30, 128.67, 129.00, 137.80; IR (cm): 1736, 1949, 1453, 1415, 1370, 1333, 1286, 1243, 1220, 1167, 1112, 1071, 1031, 1004, 949, 909, 881, 860, 830, 802, 757, 710, 699, 606; HR-MS (ESI-TOF) calculated for , m/z [M + Na]: 247.050173; experimental value: 247.050609.
Trans,trans-5-chloro-6-(4-fluorophenyl)-tetrahydro-4-methylpyran-2-one (5c): The product was obtained as a colorless solid; mp = 80–81 C; = 0.16 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.25 (d, 3H, J = 6.3 Hz, CH(CH)), 2.34–2.49 (m, 2H, CHaHb, CH(CH)), 2.94–3.05 (m, 1H, CHaHb), 3.71 (t, 1H, J = 9.8 Hz, CHCl), 5.17 (d, 1H, J = 10.0 Hz, CHAr), 7.03–7.13 (m, 2H, HAr), 7.32–7.42 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.62 (CH(CH)), 35.51 (CH(CH), 37.04 (CH), 63.25 (CHCl), 84.32 (CHAr), 168.71 (C = O), CAr: 115.53 (d, = 21.9 Hz), 129.35 (d, = 8.4 Hz), 132.33 (d, = 3.2 Hz), 163.10 (d, = 248.4 Hz); IR (cm): 1731, 1609, 1514, 1453, 1435, 1427, 1381, 1352, 1334, 1260, 1221, 1192, 1160, 1105, 1083, 1065, 1035, 1010, 960, 919, 883, 844, 825, 810, 786, 719, 687; HR-MS (ESI-TOF) calculated for , m/z [M + Na]: 265.040751; experimental value: 265.041057.
Cis,trans-5-chloro-6-(4-fluorophenyl)-tetrahydro-4-methylpyran-2-one (6c): The product was obtained as a colorless oil; = 1.5453, = 0.27 (acetone:heksane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.10 (d, 3H, J = 6.6 Hz, CH(CH)), 2.27–2.37 (m, 1H, CH(CH)), 2.62 (dd, 1H, = 18.4 Hz, = 9.7 Hz, CHaHb), 2.66 (dd, 1H, = 18.4 Hz, = 7.0 Hz, CHaHb), 4.30 (t, 1H, J = 2.8 Hz, CHCl), 5.75 (d, 1H, J = 2.7 Hz, CHAr), 7.09–7.15 (m, 2H, HAr), 7.23–7.33 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 17.42 (CH(CH)), 27.41 (CH(CH), 33.71 (CH), 63.02 (CHCl), 84.46 (CHAr), 168.69 (C = O), CAr: 116.02 (d, = 21.9 Hz), 127.29 (d, = 8.3 Hz), 133.56 (d, = 3.1 Hz), 163.65 (d, = 248.4 Hz); IR (cm): 1737, 1605, 1510, 1453, 1414, 1372, 1358, 1340, 1286, 1272, 1218, 1176, 1155, 1110, 1096, 1078, 1060, 1040, 1000, 950, 911, 888, 860, 834, 796, 745, 723, 692, 631; HR-MS (ESI-TOF) calculated for , m/z [M + Na]: 265.040751; experimental value: 265.041871.
The bromolactonization of unsaturated carboxylic acids
4a–
c: The mixture of an unsaturated carboxylic acid (0.005 mol) and
N-bromosuccinimide (0.01 mol) was dissolved in 60 mL of THF. Acetic acid was added dropwise and reaction mixture was stirred at room temperature for 48 h. When the reaction was completed, the mixture was dissolved in diethyl ether, washed with saturated NaHCO
and dried over anhydrous magnesium sulfate. The crude product was purified by column chromatography on silica gel using a mixture of acetone and hexane (1:10) [
28]. Spectral data are given below. The bromolactonization of
4a gave bromolactones
7a and
8a with a total yield of 72.23% (
7a:
8a 82.30:17.70). The bromolactonization of
4b gave bromolactones
7b and
8b with a total yield of 71.94% (
7b:
8b 78.15:21.85). The bromolactonization of
4c gave bromolactones
7c and
8c with a total yield of 77.72% (
7c:
8c 76.27:23.73).
Trans,trans-5-bromo-tetrahydro-4-methyl-6-(naphthalen-1-yl)pyran-2-one (7a): The product was obtained as a colorless solid; mp = 197–198 C; = 0.13 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.33 (d, 3H, J = 6.6 Hz, CH(CH)), 2.55 (dd, 1H, = 17.4 Hz, = 10.1 Hz, CHaHb), 2.59–2.72 (m, 1H, CH(CH)), 3.08 (dd, 1H, = 17.4 Hz, = 5.7 Hz, CHaHb), 4.28 (dd, 1H, = 10.3 Hz, = 9.6 Hz, ), 6.12 (d, 1H, J = 10.3 Hz, CHAr), 7.49–7.53 (m, 2H, HAr), 7.53–7.59 (m, 4H, HAr), 7.88–7.93 (m, 2H, HAr), 8.10 (d, 1H, J = 8.6 Hz, HAr); C NMR (125 MHz, CDCl), [ppm]: 21.13 (CH(CH)), 36.34 (CH(CH)), 37.49 (CH), 54.75 (), 82.12 (CHAr), 169.22 (C = O), CAr: 123.06, 125.01, 125.89, 126.08, 126.65, 129.14, 130.19, 131.14, 132.24, 133.90; IR (cm): 1724, 1596, 1514, 1453, 1413, 1400, 1376, 1345, 1324, 1285, 1236, 1187, 1170, 1105, 1062, 1013, 994, 947, 906, 893, 860, 834, 800, 773, 735, 660, 631; HR-MS (ESI-TOF) calculated for CHBrO, m/z [M + Na]: 341.015305; experimental value: 341.016902.
Cis,trans-5-bromo-tetrahydro-4-methyl-6-(naphthalen-1-yl)pyran-2-one (8a): The product was obtained as a yellow oil, = 1.5140, = 0.22 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 0.96 (d, 3H, J = 6.4 Hz, CH(CH)), 2.04–2.12 (m, 1H, CH(CH)), 2.66 (dd, 1H, = 18.5 Hz, = 11.4 Hz, CHaHb), 2.72 (ddd, 1H, = 18.5 Hz, = 6.3 Hz, = 1.0 Hz, CHaHb), 4.62 (m, 1H, ), 6.65 (d, 1H, J = 1.7 Hz, CHAr), 7.33–7.71 (m, 4H, HAr), 7.83 (d, 1H, J = 8.2 Hz, HAr), 7.88 (d, 1H, J = 8.2 Hz, HAr), 7.92–7.96 (m, 1H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.26 (CH(CH)), 27.13 (CH(CH)), 34.27 (CH), 56.19 (), 84.74 (CHAr), 169.10 (C = O), CAr: 121.81, 123.52, 125.22, 126.32, 127.41, 129.15, 129.47, 129.52, 133.60, 133.85; IR (cm): 1740, 1596, 1507, 1453, 1390, 1340, 1270, 1236, 1200, 1153, 1107, 1076, 1037, 994, 947, 904, 886, 861, 800, 780, 746, 683, 606; HR-MS (ESI-TOF) calculated for CHBrO, m/z [M + K]: 356.989243; experimental value: 356.990538.
Trans,trans-5-bromo-tetrahydro-4-methyl-6-phenylpyran-2-one (7b): The product was obtained as a colorless solid; mp = 111–112 C; = 0.16 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.28 (d, 3H, J = 6.5 Hz, CH(CH)), 2.44 (dd, 1H, = 17.2 Hz, = 10.0 Hz, CHaHb), 2.47–2.59 (m, 1H, CH(CH)), 2.99 (dd, 1H, = 17.2 Hz, = 5.6 Hz, CHaHb), 3.86 (dd, 1H, = 10.2 Hz, = 9.7 Hz, ), 5.32 (d, 1H, J = 10.3 Hz, CHAr), 7.34–7.44 (m, 5H, HAr); C NMR (125 MHz, CDCl), [ppm]: 21.04 (CH(CH)), 35.84 (CH(CH), 37.39 (CH), 55.44 (), 85.36 (CHAr), 169.19 (C = O); CAr: 127.59, 128.55, 129.36, 136.70; IR (cm): 1720, 1497, 1458, 1372, 1345, 1284, 1248, 1184, 1170, 1103, 1061, 1006, 956, 906, 889, 845, 775, 753, 696, 631, 617; HR-MS (ESI-TOF) calculated for CHBrO, m/z [M + Na]: 290.999656; experimental value: 291.000717.
Cis,trans-5-bromo-tetrahydro-4-methyl-6-phenylpyran-2-one (8b): The product was obtained as a yellow oil; = 1.5140, = 0.23 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.04 (d, 3H, J = 6.5 Hz, CH(CH)), 2.05–2.14 (m, 1H, CH(CH)), 2.59 (dd, 1H, = 18.4 Hz, = 10.8 Hz, CHaHb), 2.66 (dd, 1H, = 18.4 Hz, = 6.0 Hz, CHaHb), 4.46 (t, 1H, J = 2.5 Hz, ), 5.89 (d, 1H, J = 2.5 Hz, CHAr), 7.27–7.45 (m, 5H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.07 (CH(CH)), 27.18 (CH(CH), 34.69 (CH), 57.01 (), 85.53 (CHAr), 168.80 (C = O), CAr: 125.35, 128.67, 129.01, 138.31; IR (cm): 1742, 1598, 1451, 1370, 1338, 1281, 1270, 1239, 1162, 1105, 1067, 1035, 1001, 954, 920, 850, 800, 764, 740, 701, 683; HR-MS (ESI-TOF) calculated for CHBrO, m/z [M + K]: 306.973594; experimental value: 306.974724.
Trans,trans-5-bromo-6-(4-fluorophenyl)-tetrahydro-4-methylpyran-2-one (7c): The product was obtained as a colorless solid; mp = 123–124 C; = 0.12 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.28 (d, 3H, J = 6.5 Hz, CH(CH)), 2.44 (dd, 1H, = 17.2 Hz, = 10.0 Hz, CHaHb), 2.47–2.61 (m, 1H, CH(CH)), 2.99 (dd, 1H, = 17.2 Hz, = 5.6 Hz, CHaHb), 3.80 (dd, 1H, = 10.4 Hz, = 9.7 Hz, ), 5.31 (d, 1H, J = 10.4 Hz, CHAr), 7.04–7.16 (m, 2H, HAr), 7.32–7.42 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 21.07 (CH(CH)), 35.88 (CH(CH), 37.33 (CH), 55.53 (), 84.64 (CHAr), 168.93 (C = O), CAr: 115.56 (d, = 21.8 Hz), 129.48 (d, = 8.5 Hz), 132.77 (d, = 3.3 Hz), 163.14 (d, = 248.6 Hz); IR (cm): 1729, 1607, 1512, 1454, 1424, 1372, 1331, 1261, 1220, 1190, 1160, 1035, 1004, 850, 918, 877, 840, 823, 790, 773, 719, 660; HR-MS (ESI-TOF) calculated for CHBrO, m/z [M + Na]: 308.990234; experimental value: 308.991651.
Cis,trans-5-bromo-6-(4-fluorophenyl)-tetrahydro-4-methylpyran-2-one (8c): The product was obtained as a colorless oil; = 1.5140, = 0.21 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.07 (d, 3H, J = 6.5 Hz, CH(CH)), 2.08–2.12 (m, 1H, CH(CH)), 2.60 (dd, 1H, = 18.4 Hz, = 10.3 Hz, CHaHb), 2.67 (ddd, 1H, = 18.4 Hz, = 6.1 Hz, = 0.9 Hz, CHaHb), 4.41 (td, 1H, = 3.1 Hz, = 0.8 Hz, ), 5.84 (dd, 1H, = 3.1 Hz, = 0.5 Hz CHAr), 7.08–7.13 (m, 2H, HAr), 7.24–7.29 (m, 2H, HAr); C NMR (125Hz, CDCl), [ppm]: 18.84 (CH(CH)), 27.54 (CH(CH), 34.84 (CH), 56.58 (), 84.80 (CHAr), 168.63 (C = O), CAr: 116.06 (d, = 21.9 Hz), 127.38 (d, = 8.3 Hz), 134.02 (d, = 3.2 Hz), 162.67 (d, = 248.6 Hz); IR (cm): 1740, 1602, 1507, 1453, 1413, 1370, 1359, 1338, 1273, 1227, 1190, 1160, 1115, 1096, 1074, 1062, 1033, 1004, 951, 911, 855, 832, 802, 790, 750, 716, 670; HR-MS (ESI-TOF) calculated for CHBrO, m/z [M + Na]: 308.990234; experimental value: 308.991301.
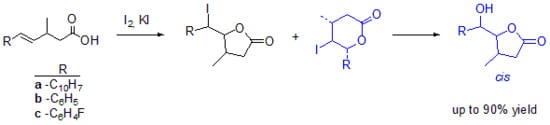
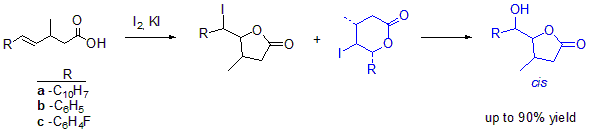
The iodolactonization of unsaturated carboxylic acids
4a–c: The mixture of an unsaturated carboxylic acid (0.005 mol), diethyl ether and saturated aqueous
was stirred for 30 min and a solution of iodine in potassium iodide was dropwise added. The reaction mixture was refluxed for 24 h. When the reaction was completed, the mixture was dissolved in diethyl ether and washed with saturated
solution. The organic layer was washed with saturated
and dried over anhydrous magnesium sulfate. The crude product was purified by column chromatography on silica gel using a mixture of acetone and hexane (1:15) [
29]. Spectral data are given below. The iodolactonization of
4a gave
-hydroxylactones
12a and
13a with a total yield of 84.59% (
12a:
13a 10.24:89.76). The iodolactonization of
4b gave
-iodolactones
9b,
10b,
-iodoactone
11b and
-hydroxylactone
12b,
13b with total yield of 75.28% (
9b:
10b:
11b:
12b:
13b 41.71:33.19:7.00:13.79:4.65). The iodolactonization of
4c gave
-iodolactones
9c,
10c,
-iodoactone
11c and
-hydroxylactone
12c,
13c with total yield of 68.55% (
9c:
10c:
11c:
12c:
13c 30.26:37.42:8.00:7.23:18.08).
Trans-dihydro-5-((S)-hydroxy(naphthalen-1-yl)methyl)-4-methylfuran-2(3H)-one (12a): The product was obtained as a yellow oil; = 1.427; = 0.08 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 0.56 (d, 3H, J = 6.8 Hz, CH), 2.11 (dd, 1H, = 17.5 Hz, = 6.6 Hz, CHaHb), 2.72–2.79 (m, 1H, CH(CH)), 2.85 (dd, 1H, = 17.5 Hz, = 9.3 Hz, CHaHb), 4.53 (dd, 1H, = 5.5 Hz, = 2.8 Hz, COCH), 5.92 (d, 1H, J = 2.3 Hz, CHOH), 7.50–7.59 (m, 3H, HAr), 7.80–7.86 (m, 2H, HAr), 7.90 (d, 1H, J = 8.1 Hz, HAr), 7.96 (d, 1H, J = 8.1 Hz, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.74 (CH), 28.38 (CH(CH)), 37.34 (CH), 69.97 (CHOH), 88.20 (COCH), 175.91 (C = O), CAr: 122.11, 123.65, 125.49, 125.82, 126.64, 128.69, 129.18, 129.88, 133.56, 133.69; IR (cm): 3422, 1774, 1593, 1510, 1458, 1417, 1379, 1359, 1327, 1291, 1254, 1209, 1166, 1112, 1092, 1078, 1037, 1008, 975, 936, 920, 881, 854, 823, 802, 782, 741, 694, 660, 628, 615; HR-MS (ESI-TOF) calculated for CHO, m/z [M + Na]: 279.099709; experimental value: 279.100786.
Cis-dihydro-5-((S)-hydroxy(naphthalen-1-yl)methyl)-4-methylfuran-2(3H)-one (13a): The product was obtained as a brown solid; mp = 123–124 C; = 0.06 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.31 (d, 3H, J = 7.0 Hz, CH), 1.57 (s, 1H, ), 2.57 (dd, 1H, = 17.0 Hz, = 8.8 Hz, CHaHb), 2.64 (dd, 1H, = 17.0 Hz, = 8.6 Hz, CHaHb), 2.72–2.83 (m, 1H, CH(CH)), 4.86 (dd, 1H, = 7.2 Hz, = 3.4 Hz, COCH), 5.72 (t, 1H, J = 3.5 Hz, CHOH), 7.47–7.58 (m, 3H, HAr), 7.71 (d, 1H, J = 6.9 Hz, HAr), 7.84 (d, 1H, J = 8.2 Hz, HAr), 7.90 (d, 1H, J = 8.0 Hz, HAr), 8.02 (d, 1H, J = 7.9 Hz, HAr); C NMR (125 MHz, CDCl), [ppm]: 14.30 (CH), 33.02 (CH(CH)), 36.87 (CH), 68.97 (CHOH), 83.96 (COCH), 177.11 (C = O), CAr: 122.28, 122.95, 125.49, 125.76, 126.58, 129.02, 129.24, 130.36, 133.88, 134.90; IR (cm): 3492, 1747, 1593, 1507, 1453, 1415, 1374, 1350, 1327, 1286, 1275, 1257, 1214, 1189, 1173, 1153, 1110, 1096, 1037, 1026, 1000, 942, 918, 852, 834, 787, 773, 755, 730, 655, 644, 622; HR-MS (ESI-TOF) calculated for CHO, m/z [M + K]: 295.073647; experimental value: 295.074508.
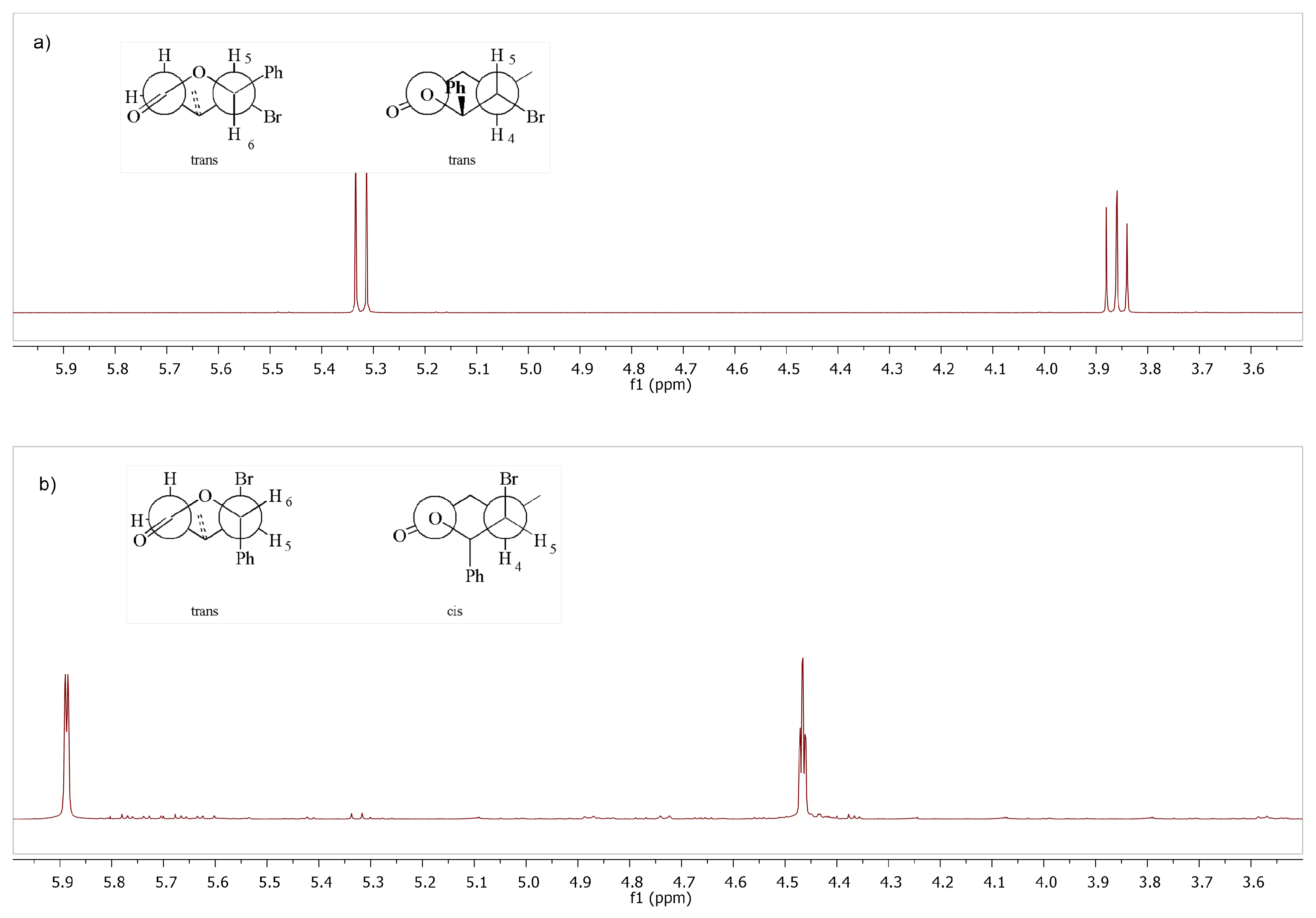
Trans,trans-tetrahydro-5-iodo-4-methyl-6-phenylpyran-2-one (9b): The product was obtained as a colorless solid; mp = 107–108 C; = 0.15 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.29 (d, 3H, J = 6.6 Hz, CH(CH)), 2.41 (dd, 1H, = 17.4 Hz, = 9.8 Hz, CHaHb), 2.48–2.61 (m, 1H, CH(CH)), 2.93 (dd, 1H, = 17.4 Hz, = 5.9 Hz, CHaHb), 3.97 (dd, 1H, = 10.9 Hz, = 10.0 Hz, CHI), 5.45 (d, 1H, J = 10.9 Hz, CHAr), 7.32–7.44 (m, 5H, HAr); C NMR (125 MHz, CDCl), [ppm]: 23.57 (CH(CH)), 36.00 (CH(CH), 37.12 (CH), 38.88 (CHI), 86.82 (CHAr), 169.49 (C = O), CAr: 127.74, 128.50, 129.43, 137.53; IR (cm): 1719, 1499, 1453, 1413, 1372, 1340, 1290, 1245, 1211, 1140, 1100, 1070, 996, 950, 933, 907, 887, 850, 769, 743, 695; HR-MS (ESI-TOF) calculated for CHIO, m/z [M + Na]: 338.985797 experimental value: 338.986432.
Cis,trans-tetrahydro-5-iodo-4-methyl-6-phenylpyran-2-one (10b): The product was obtained as a colorless oil, = 1.5227, = 0.19 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 0.97 (d, 3H, J = 6.4 Hz, CH(CH)), 1.27–1.31 (m, 1H, CH(CH)), 2.49 (dd, 1H, = 18.5 Hz, = 10.5 Hz, CHaHb), 2.67 (ddd, 1H, = 18.5 Hz, = 5.6 Hz, = 1.1 Hz, CHaHb), 4.60 (td, 1H, = 3.1 Hz, = 1.1 Hz, CHI), 5.91 (d, 1H, J = 3.1 Hz, CHAr), 7.25–7.43 (m, 5H, HAr); C NMR (125 MHz, CDCl), [ppm]: 21.03 (CH(CH)), 27.34 (CH(CH), 36.36 (CH), 38.08 (), 86.32 (CHAr), 168.32 (C = O), CAr: 125.06, 128.18, 128.51, 138.42; IR (cm): 1735, 1495, 1451, 1407, 1380, 1368, 1354, 1334, 1322, 1285, 1265, 1233, 1190, 1132, 1103, 1061, 1030, 998, 950, 903, 883, 852, 796, 763, 735, 695, 664; HR-MS (ESI-TOF) calculated for CHIO, m/z [M + K]: 354.959735 experimental value: 354.958974.
Cis-dihydro-5-(iodo(phenyl)methyl)-4-methylfuran-2(3H)-one (11b): The product was obtained as a colorless solid, mp = 86–87 C; = 0.15 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.17 (d, 3H, J = 7.0 Hz, CH), 2.32 (dd, 1H, = 17.0 Hz, = 0.5 Hz, CHaHb), 2.87 (dd, 1H, = 17.0 Hz, = 7.4 Hz, CHaHb), 2.96–3.03 (m, 1H, CH(CH)), 4.96 (d, 1H, J = 11.2 Hz, CHI), 5.11 (dd, 1H, = 11.2 Hz, = 4.5 Hz, COCH), 7.34–7.39 (m, 3H, HAr), 7.39–7.44 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 12.82 (CH), 28.38 (CHI), 34.00 (CH(CH)), 38.88 (CH), 84.45 (COCH), 176.00 (C = O), CAr: 127.79, 128.61, 128.85, 140.09; IR (cm): 1765, 1500, 1452, 1414, 1372, 1341, 1290, 1247, 1212, 1141, 1100, 1071, 997, 950, 932, 908, 886, 851, 769, 742, 695; HR-MS (ESI-TOF) calculated for CHIO, m/z [M + Na]: 338.985797 experimental value: 338.986737.
Trans-dihydro-5-(hydroxy(phenyl)methyl)-4-methylfuran-2(3H)-one (12b): The product was obtained as a colorless solid, mp = 86–87 C; = 0.11 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 0.81 (d, 3H, J = 6.9 Hz, CH), 2.10 (dd, 1H, = 17.6 Hz, = 6.7 Hz, CHaHb), 2.57–2.76 (m, 1H, CH(CH)), 2.73 (dd, 1H, = 17.6 Hz, = 9.2 Hz, CHaHb), 4.30 (dd, 1H, = 5.5 Hz, = 3.3 Hz, COCH), 5.09 (t, 1H, J = 3.6 Hz, CHOH), 7.29–7.34 (m, 3H, HAr), 7.36–7.42 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.77 (CH), 28.80 (CH(CH)), 37.06 (CH), 72.29 (CHOH), 89.74 (COCH), 176.99 (C = O), CAr: 126.00, 128.12, 128.63, 138.34; IR (cm): 3442, 1767, 1600, 1496, 1451, 1415, 1379, 1356, 1324, 1275, 1225, 1214, 1202, 1162, 1103, 1085, 1060, 1008, 983, 936, 915, 879, 859, 818, 762, 707, 694, 676, 617; HR-MS (ESI-TOF) calculated for CHO, m/z [M + Na]: 229.084059 experimental value: 229.084576.
Cis-dihydro-5-(hydroxy(phenyl)methyl)-4-methylfuran-2(3H)-one (13b): The product was obtained as a colorless solid, mp = 89–91 C; = 0.11 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.32 (d, 3H, J = 6.9 Hz, CH), 1.58 (s, 1H, ), 2.20 (dd, 1H, = 17.2 Hz, = 4.8 Hz, CHaHb), 2.77 (dd, 1H, = 17.3 Hz, = 8.4 Hz, CHaHb), 2.80–2.89 (m, 1H, CH(CH)), 4.85 (dd, 1H, = 7.2 Hz, = 4.0 Hz, COCH), 5.11 (d, 1H, J = 3.9 Hz, CHOH), 7.27–7.35 (m, 2H, HAr), 7.37–7.48 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.20 (CH), 32.85 (CH(CH)), 35.03 (CH), 69.70 (CHOH), 84.59 (COCH), 175.49 (C = O), CAr: 125.67, 128.29, 128.78, 138.37; IR (cm): 3419, 1751, 1587, 1497, 1434, 1403, 1387, 1361, 1301, 1255, 1243, 1202, 1167, 1075, 1067, 1054, 1011, 983, 933, 887, 828, 785, 752, 732, 697; HR-MS (ESI-TOF) calculated for CHO, m/z [M + Na]: 229.084059 experimental value: 229.084781.
Trans,trans-6-(4-fluorophenyl)-tetrahydro-5-iodo-4-methylpyran-2-one (9c): The product was obtained as a colorless solid, mp = 136–138 C, = 0.16 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.28 (d, 3H, J = 6.5 Hz, CH(CH)), 2.36 (dd, 1H, = 17.2 Hz, = 10.0 Hz, CHaHb), 2.51–2.57 (m, 1H, CH(CH)), 2.92 (dd, 1H, = 17.5 Hz, = 6.0 Hz, CHaHb), 3.90 (dd, 1H, = 10.8 Hz, = 10.1 Hz, CHI), 5.42 (d, 1H, J = 10.8 Hz, CHAr), 7.05–7.09 (m, 2H, HAr), 7.32–7.36 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 23.6 (CH(CH)), 27.03 (CH(CH), 31.66 (CH), 37.69 (CHI), 86.43 (CHAr), 169.68 (C = O), CAr: 115.53 (d, = 21.7 Hz), 129.58 (d, = 8.4 Hz), 133.78 (d, = 3.3 Hz), 163.09 (d, = 248.60 Hz); IR (cm): 1720, 1601, 1515, 1459, 1421, 1411, 1366, 1334, 1300, 1285, 1223, 1207, 1176, 1162, 1110, 1090, 1071, 1053, 1004, 952, 909, 896, 858, 844, 822, 778, 775, 717; HR-MS (ESI-TOF) calculated for CHFIO, m/z [M + Na]: 356.976375 experimental value: 356.977702.
Cis,trans-6-(4-fluorophenyl)-tetrahydro-5-iodo-4-methylpyran-2-one (10c): The product was obtained as a yellow oil; = 1.5227, = 0.24 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.04 (d, 3H, J = 6.5 Hz, CH(CH)), 1.39–1.48 (m, 1H, CH(CH)), 2.52 (dd, 1H, = 18.4 Hz, = 9.8 Hz, CHaHb), 2.71 (ddd, 1H, = 18.4 Hz, = 5.5 Hz, = 1.0 Hz, CHaHb), 4.55 (td, 1H, = 3.9 Hz, = 0.9 Hz, CHI), 5.86 (d, 1H, J = 3.9 Hz, CHAr), 7.04–7.13 (m, 2H, HAr), 7.22–7.29 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 21.13 (CH(CH)), 28.37 (CH(CH), 30.90 (CH), 36.88 (CHI), 85.92 (CHAr), 168.53 (C = O), CAr: 115.96 (d, = 21.8 Hz), 127.56 (d, = 8.3 Hz), 134.62 (d, = 3.0 Hz), 162.62 (d, = 248.8 Hz); IR (cm): 1735, 1605, 1508, 1453, 1411, 1384, 1366, 1354, 1340, 1328, 1283, 1263, 1225, 1184, 1157, 1133, 1095, 1073, 1056, 998, 946, 907, 890, 851, 829, 802, 782, 750, 714; HR-MS (ESI-TOF) calculated for CHFIO, m/z [M + Na]: 356.976375 experimental value: 356.977461.
Cis-5-((4-fluorophenyl)iodomethyl)-dihydro-4-methylfuran-2(3H)-one (11c): The product was obtained as a colorless solid, mp = 118–119 C; = 0.16 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.16 (d, 3H, J = 7.1 Hz, CH), 2.33 (dd, 1H, = 17.1 Hz, = 0.5 Hz, CHaHb), 2.87 (dd, 1H, = 17.1 Hz, = 7.3 Hz, CHaHb), 2.95–3.00 (m, 1H, CH(CH)), 4.94 (d, 1H, = 11.2 Hz, CHI), 5.04 (dd, 1H, = 11.2 Hz, = 4.5 Hz, COCH), 6.97–7.10 (m, 2H, HAr), 7.32–7.43 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 12.85 (CH), 28.45 (CHI), 34.05 (CH(CH)), 38.91 (CH), 84.30 (COCH), 176.11 (C = O), CAr: 116.24 (d, = 22.1 Hz), 127.64 (d, = 8.0 Hz), 133.62 (d, = 3.0 Hz), 162.38 (d, = 246.3 Hz). IR (cm): 1760, 1600, 1512, 1460, 1420, 1412, 1361, 1335, 1301, 1284, 1226, 1203, 1174, 1161, 1108, 1091, 1070, 1052, 1001, 950, 906, 894, 857, 842, 821, 779, 776, 714; HR-MS (ESI-TOF) calculated for CHFIO, m/z [M + Na]: 356.976375 experimental value: 356.976932.
Trans-5-((4-fluorophenyl)(hydroxy)methyl)-dihydro-4-methylfuran-2(3H)-one (12c): The product was obtained as a colorless solid, mp = 118–119 C, = 0.10 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 0.83 (d, 3H, J = 6.9 Hz, CH), 2.11 (dd, 1H, = 17.7 Hz, = 6.9 Hz, CHaHb), 2.53–2.64 (m, 1H, CH(CH)), 2.74 (dd, 1H, = 17.7 Hz, = 9.2 Hz, CHaHb), 4.26 (dd, 1H, = 5.3 Hz, = 3.4 Hz, COCH), 5.07 (d, 1H, J = 3.2 Hz, CHOH), 6.98–7.12 (m, 2H, HAr), 7.33–7.43 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.74 (CH), 28.84 (CH(CH)), 37.03 (CH), 72.70 (CHOH), 89.65 (COCH), 176.93 (C = O), CAr: 115.57 (d, = 21.5 Hz), 127.72 (d, = 8.1 Hz), 134.17 (d, = 3.1 Hz), 163.46 (d, = 246.6 Hz); IR (cm): 3431, 1772, 1593, 1509, 1456, 1412, 1378, 1361, 1329, 1287, 1257, 1215, 1169, 1114, 1093, 1075, 1041, 1008, 975, 936, 920, 881, 854, 826, 802, 782, 749, 715; HR-MS (ESI-TOF) calculated for CHFO, m/z [M + Na]: 247.074638 experimental value: 247.075208.
Cis-5-((4-fluorophenyl)(hydroxy)methyl)-dihydro-4-methylfuran-2(3H)-one (13c): The product was obtained as a colorless solid, mp = 118–119 C; = 0.08 (acetone:hexane 1:7); H NMR (CDCl, 500 MHz), [ppm]: 1.34 (d, 3H, J = 6.8 Hz, CH), 1.59 (s, 1H, ), 2.24 (dd, 1H, = 17.3 Hz, = 4.8 Hz, CHaHb), 2.78 (dd, 1H, = 17.3 Hz, = 8.5 Hz, CHaHb), 2.81–2.89 (m, 1H, CH(CH)), 4.81 (dd, 1H, = 7.3 Hz, = 4.1 Hz, COCH), 5.09 (d, 1H, J = 4.1 Hz, CHOH), 7.14–7.24 (m, 2H, HAr), 7.29–7.38 (m, 2H, HAr); C NMR (125 MHz, CDCl), [ppm]: 19.22 (CH), 32.86 (CH(CH)), 35.05 (CH), 69.72 (CHOH), 84.57 (COCH), 175.47 (C = O), CAr: 116.26 (d, = 22.1 Hz), 127.69 (d, = 8.0 Hz), 133.63 (d, = 3.0 Hz), 162.34 (d, = 246.4 Hz); IR (cm): 3439, 1751, 1592, 1507, 1459, 1410, 1380, 1365, 1327, 1285, 1259, 1216, 1169, 1115, 1094, 1077, 1044, 1010, 973, 937, 920, 882, 853, 827, 801, 782, 748, 716; HR-MS. HR-MS (ESI-TOF) calculated for CHFO, m/z [M + Na]: 247.074638 experimental value: 247.075284.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}