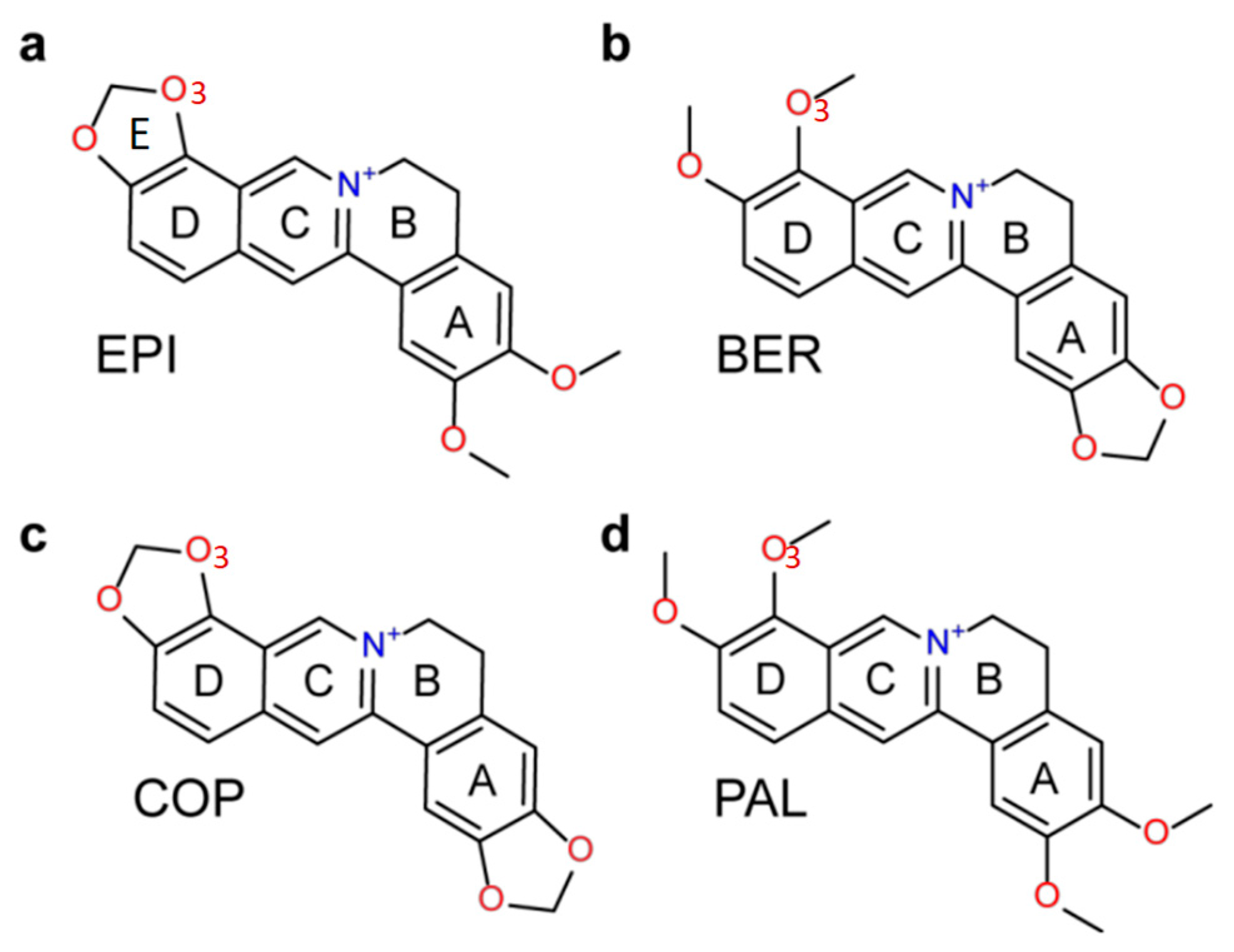
Ligand Selectivity in the Recognition of Protoberberine Alkaloids by Hybrid-2 Human Telomeric G-Quadruplex: Binding Free Energy Calculation, Fluorescence Binding, and NMR Experiments

 ,
,
Abstract
:1. Introduction
2. Results and Discussions
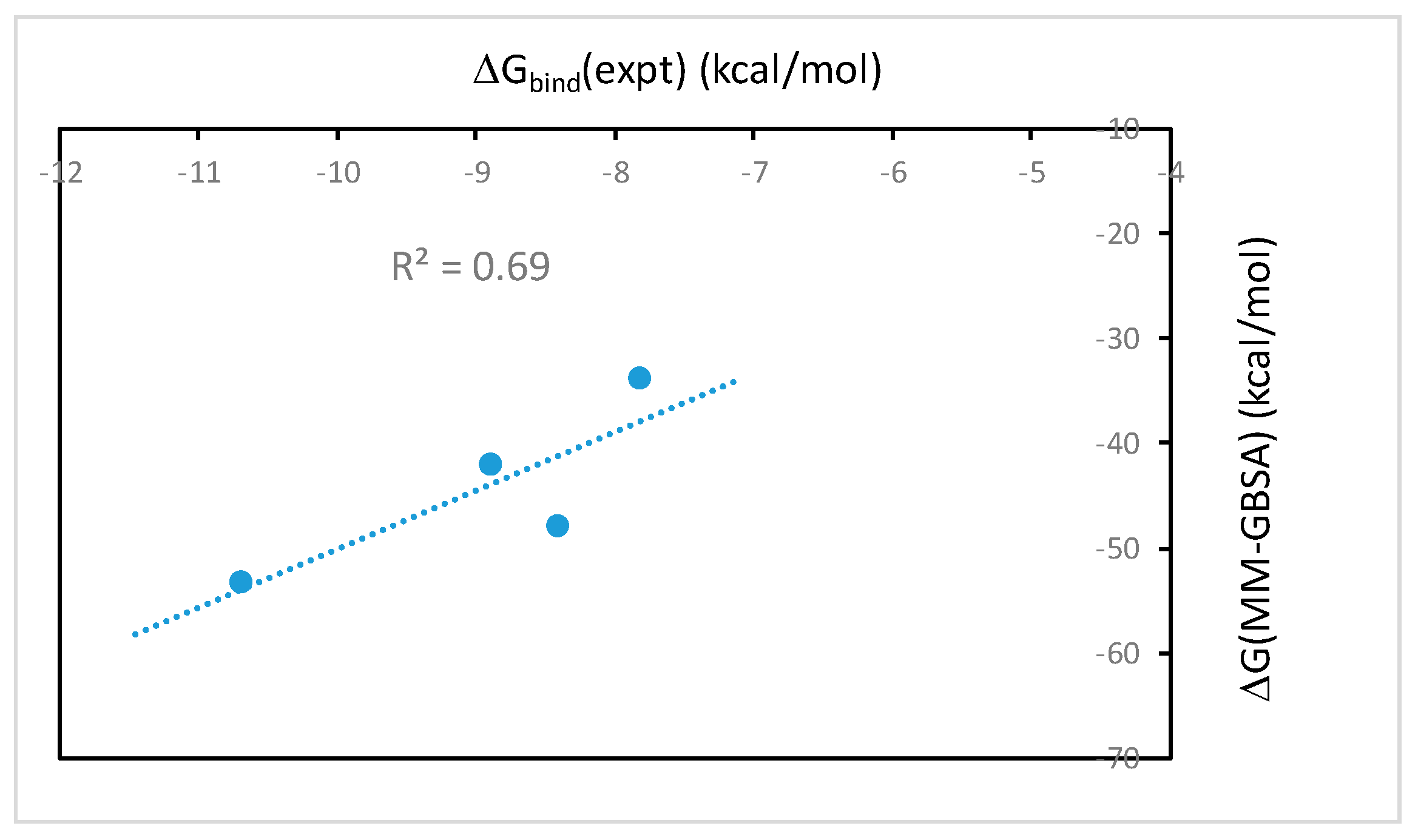
2.1. The MM-PB(GB)SA Binding Free Energies Calculated from Molecular Dynamics Simulations Correlate Well with Experimental Binding Affinities
2.2. The MM-PB(GB)SA Binding Free Energies Calculated Using Single Energy-Minimized Structures Show No Correlation with the Experimental Data
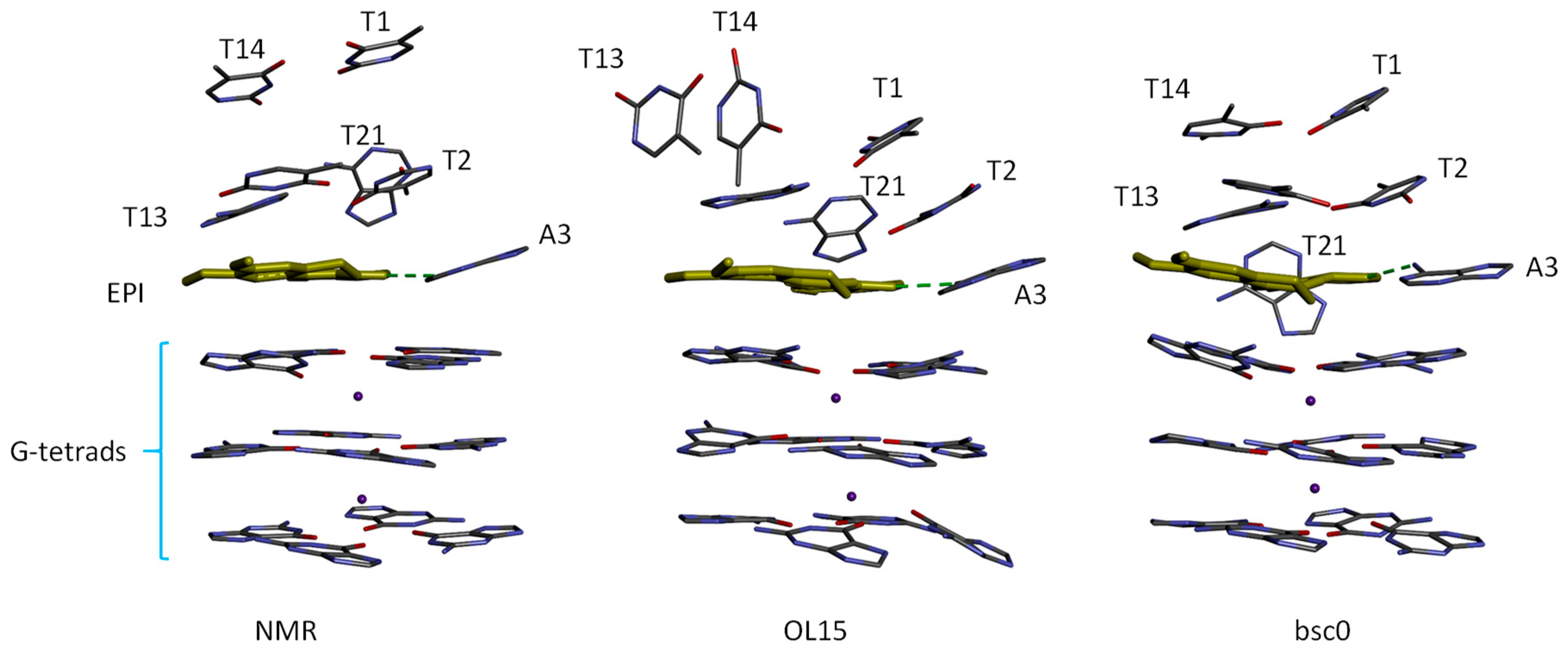
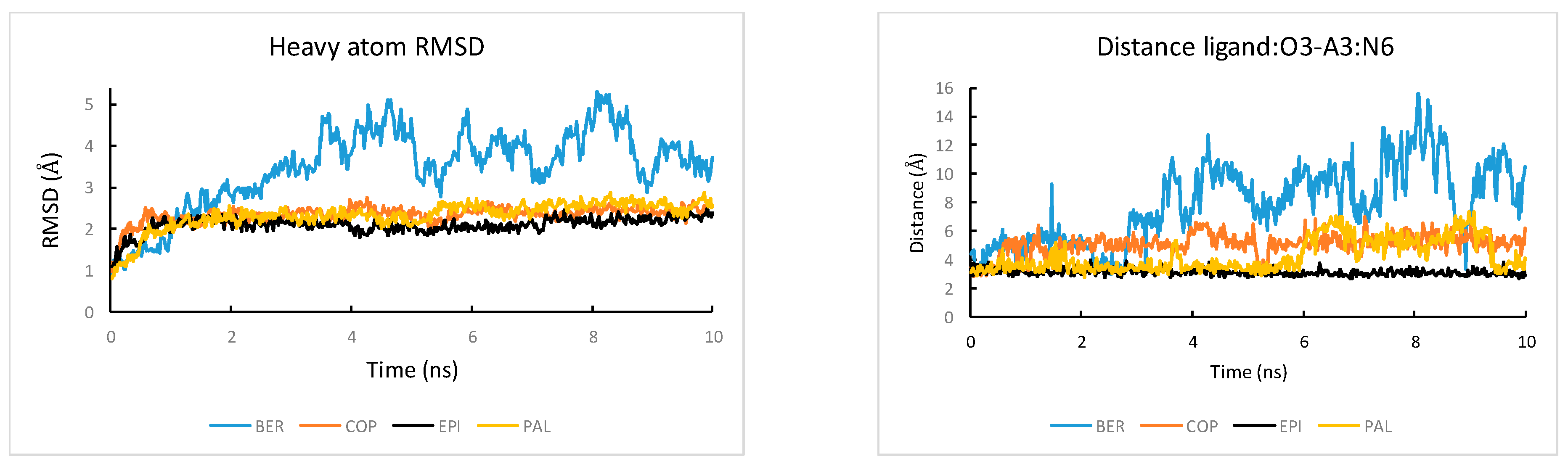
2.3. MM-PB(GB)SA Free Energy Calculated Using MD with OL15 Force Field also Reproduced Experimental Trend Well but the Simulations Show Structural Distortion in the Protoberberine Binding Site
2.4. Thermodynamic Driving Force for the Protoberberine-wtTel26 Binding
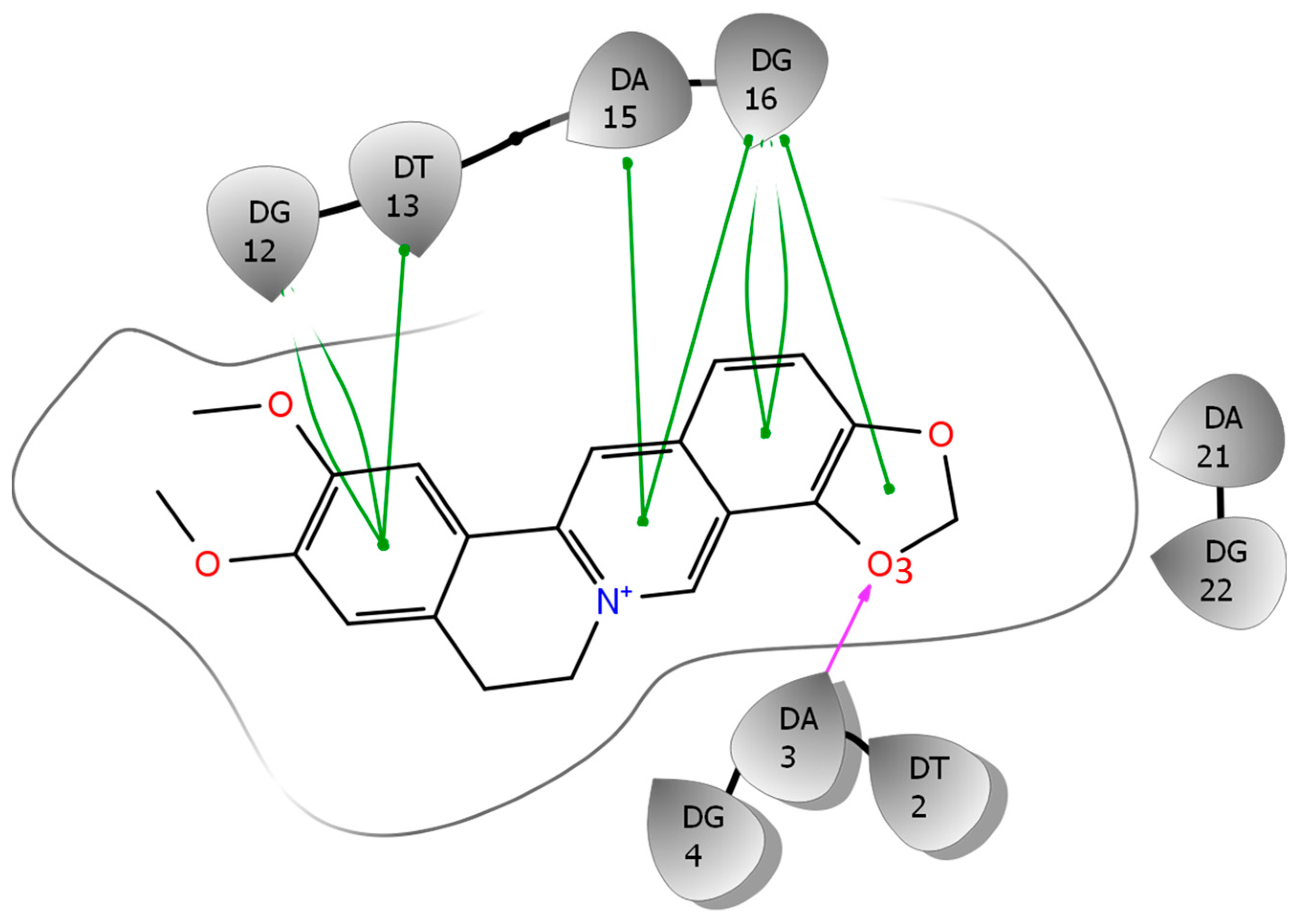
2.5. Binding Free Energy Components and MD Trajectory Analysis Help Rationalize the Ligand Selectivity
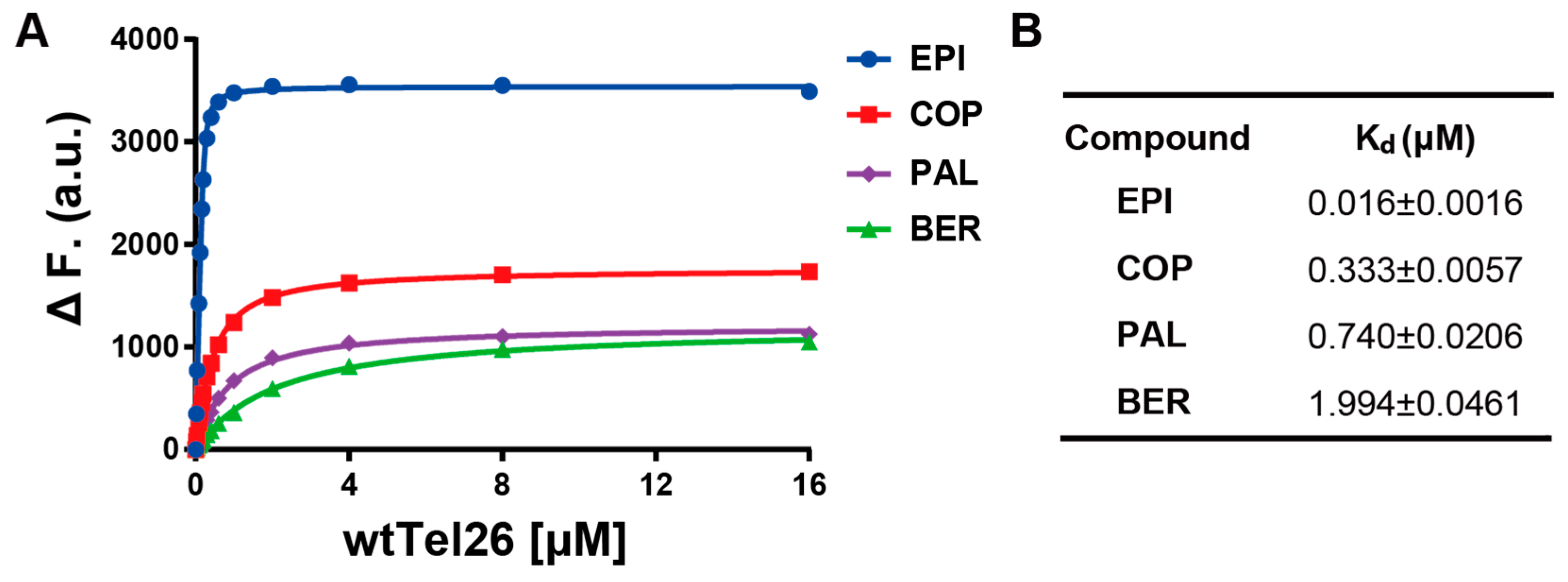
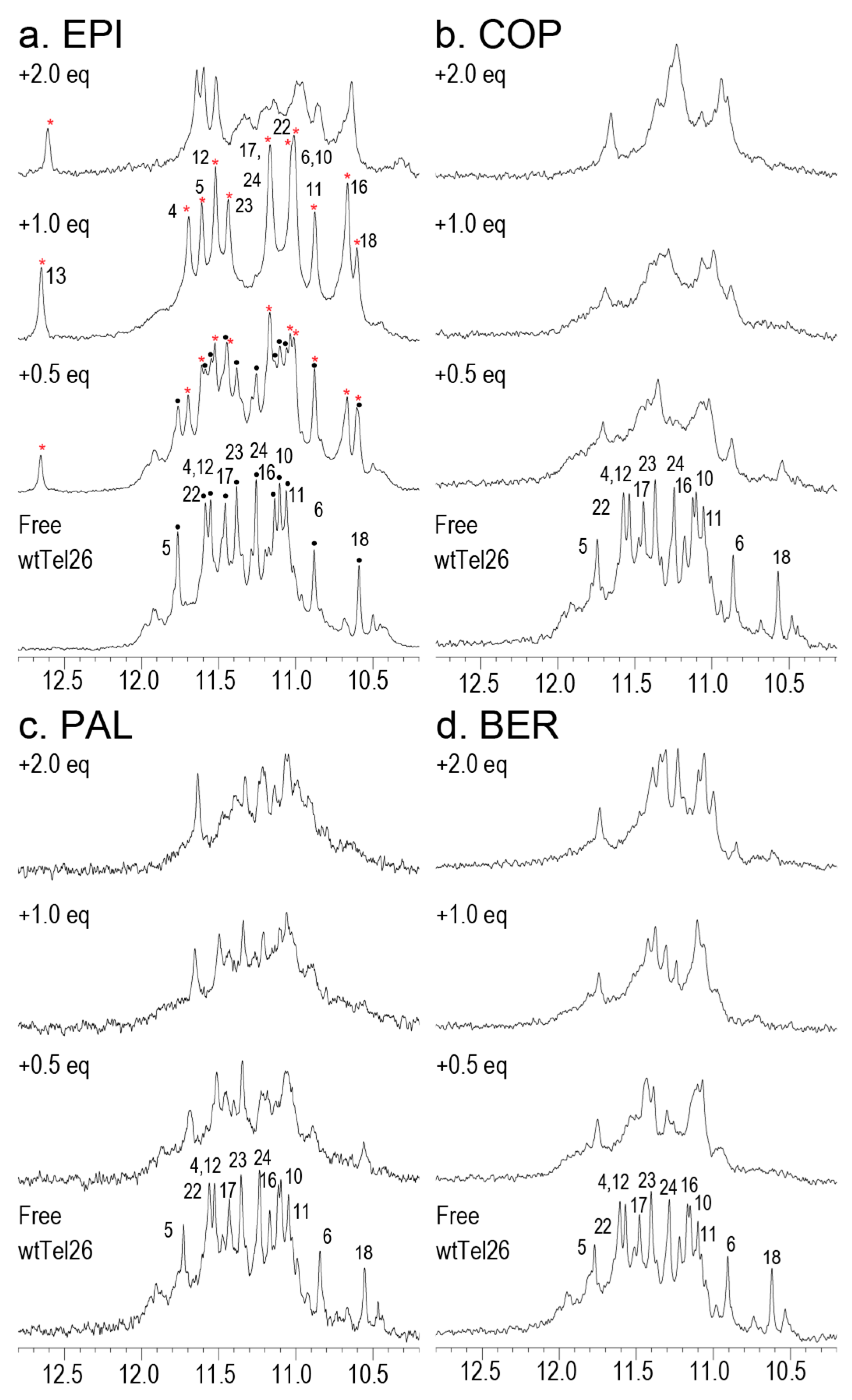
2.6. NMR Titration Shows that Among the Protoberberines Studied Here Only EPI Binds Specifically to the Hybrid-2 Human Telomeric G4
2.7. The More Rigorous DDM Calculations Make Incorrect Predictions for the Top Binder of the Hybrid-2 Human Telomeric G4
3. Materials and Methods
3.1. Fluorescence Measurements of the Binding Dissociation Constant
3.2. NMR Titration
3.3. MD Setup and MM-PB(GB)SA Calculation
3.4. DDM Calculation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Okamoto, K. Structural insights into G-quadruplexes: Towards new anticancer drugs. Future Med. Chem. 2010, 2, 619–646. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomere states and cell fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Piatyszek, M.; Prowse, K.; Harley, C.; West, M.; Ho, P.; Coviello, G.; Wright, W.; Weinrich, S.; Shay, J. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, D. Human Telomeric G-Quadruplex Structures and G-Quadruplex-Interactive Compounds. Methods Mol. Biol. 2017, 1587, 171–196. [Google Scholar] [PubMed] [Green Version]
- Lin, C.; Wu, G.; Wang, K.; Onel, B.; Sakai, S.; Shao, Y.; Yang, D. Molecular Recognition of the Hybrid-2 Human Telomeric G-Quadruplex by Epiberberine: Insights into Conversion of Telomeric G-Quadruplex Structures. Angew. Chem. Int. Ed. Engl. 2018, 57, 10888–10893. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, L.M.; Lombardi, P.; Tillhon, M.; Scovassi, A.I. Berberine, an epiphany against cancer. Molecules 2014, 19, 12349–12367. [Google Scholar] [CrossRef] [PubMed]
- Franceschin, M.; Rossetti, L.; D’Ambrosio, A.; Schirripa, S.; Bianco, A.; Ortaggi, G.; Savino, M.; Schultes, C.; Neidle, S. Natural and synthetic G-quadruplex interactive berberine derivatives. Bioorg. Med. Chem. Lett. 2006, 16, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, H.; Shao, Y.; Lin, C.; Jia, H.; Chen, G.; Yang, D.; Wang, Y. Selective lighting up of epiberberine alkaloid fluorescence by fluorophore-switching aptamer and stoichiometric targeting of human telomeric DNA G-quadruplex multimer. Anal. Chem. 2015, 87, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.-J.; Cieplak, P. Insights into affinity and specificity in the complexes of α-lytic protease and its inhibitor proteins: Binding free energy from molecular dynamics simulation. Phys. Chem. Chem. Phys. 2009, 11, 4968. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.-J.; Zhang, P.; Cieplak, P.; Lai, L. Elucidating the Energetics of Entropically Driven Protein-Ligand Association: Calculations of Absolute Binding Free Energy and Entropy. J. Phys. Chem. B 2011, 115, 11902–11910. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Wickstrom, L.; Cieplak, P.; Lin, C.; Yang, D. Resolving the Ligand-Binding Specificity in c-MYC G-Quadruplex DNA: Absolute Binding Free Energy Calculations and SPR Experiment. J. Phys. Chem. B 2017, 121, 10484–10497. [Google Scholar] [CrossRef]
- Gilson, M.; Given, J.; Bush, B.; McCammon, J. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997, 72, 1047–1069. [Google Scholar] [CrossRef]
- Deng, Y.; Roux, B. Calculation of Standard Binding Free Energies: Aromatic Molecules in the T4 Lysozyme L99A Mutant. J. Chem. Theory Comput. 2006, 2, 1255–1273. [Google Scholar] [CrossRef] [PubMed]
- Lybrand, T.P.; McCammon, J.A.; Wipff, G. Theoretical calculation of relative binding affinity in host-guest systems. Proc. Natl. Acad. Sci. USA 1986, 83, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Thomas, L.L. Perspective on Free-Energy Perturbation Calculations for Chemical Equilibria. J. Chem. Theory Comput. 2008, 4, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wang, L.; Mobley, D.L. Is ring breaking feasible in relative binding free energy calculations? J. Chem. Inf. Model. 2015, 55, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Deng, Y.; Wu, Y.; Kim, B.; LeBard, D.N.; Wandschneider, D.; Beachy, M.; Friesner, R.A.; Abel, R. Accurate Modeling of Scaffold Hopping Transformations in Drug Discovery. J. Chem. Theory Comput. 2017, 13, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Islam, B.; Stadlbauer, P.; Gil-Ley, A.; Perez-Hernandez, G.; Haider, S.; Neidle, S.; Bussi, G.; Banas, P.; Otyepka, M.; Sponer, J. Exploring the Dynamics of Propeller Loops in Human Telomeric DNA Quadruplexes Using Atomistic Simulations. J. Chem. Theory Comput. 2017, 13, 2458–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.; Piana, S.; Dirks, R.M.; Shaw, D.E. RNA force field with accuracy comparable to state-of-the-art protein force fields. Proc. Natl. Acad. Sci. USA 2018, 115, E1346–E1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α/γ Conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [Green Version]
- Zgarbova, M.; Sponer, J.; Otyepka, M.; Cheatham, T.E., 3rd; Galindo-Murillo, R.; Jurecka, P. Refinement of the Sugar-Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Machireddy, B.; Kalra, G.; Jonnalagadda, S.; Ramanujachary, K.; Wu, C. Probing the Binding Pathway of BRACO19 to a Parallel-Stranded Human Telomeric G-Quadruplex Using Molecular Dynamics Binding Simulation with AMBER DNA OL15 and Ligand GAFF2 Force Fields. J. Chem. Inf. Model. 2017, 57, 2846–2864. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Punchihewa, C.; Jones, R.A.; Yang, D. Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: Insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007, 35, 4927–4940. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, D.S.C.T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Greene, D.; Homeyer, N.; Izadi, S.; et al. AMBER 2017; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [Green Version]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Boresch, S.; Tettinger, F.; Leitgeb, M.; Karplus, M. Absolute Binding Free Energies: A Quantitative Approach for Their Calculation. J. Phys. Chem. B 2003, 107, 9535–9551. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | ΔE(vdw) | ΔE(elec) | ΔG(solv_elec) | ΔG(solv_np) | ΔG(MM-GBSA) a | ΔG(total_elec) a | ΔG(total_np) a |
|---|---|---|---|---|---|---|---|
| BER | −46.8 ± 0.1 | −539.1 ± 10.3 | 554.9 ± 11.5 | −3.0 ± 0.1 | −33.9 ± 1.2 | 15.9 ± 1.1 | −49.8 ± 0.0 |
| COP | −59.4 ± 0.4 | −561.1 ± 0.2 | 582.0 ± 0.8 | −3.6 ± 0.2 | −42.1 ± 0.5 | 20.9 ± 1.0 | −63.0 ± 0.5 |
| EPI | −68.1 ± 0.2 | −579.2 ± 0.5 | 598.0 ± 0.6 | −4.0 ± 0.0 | −53.3 ± 0.4 | 18.8 ± 0.1 | −72.1 ± 0.2 |
| PAL | −65.2 ± 2.4 | −573.8 ± 5.0 | 595.6 ± 5.2 | −4.5 ± 0.1 | −47.9 ± 2.4 | 21.8 ± 0.2 | −69.7 ± 2.5 |
| Ligand | ΔE(vdw) | ΔE(elec) | ΔG(solv_elec) | ΔG(solv_np) | ΔG(MM-GBSA) | ΔG(total_elec) | ΔG(total_np) |
|---|---|---|---|---|---|---|---|
| BER | −60.7 ± 2.6 | −557.2 ± 5.9 | 577.4 ± 6.2 | −4.1 ± 0.1 | −44.6 ± 2.2 | 20.2 ± 0.3 | −64.8 ± 2.5 |
| COP | −60.9 ± 1.7 | −566.5 ± 2.9 | 585.4 ± 2.6 | −3.4 ± 0.0 | −45.3 ± 2.1 | 18.9 ± 0.3 | −64.3 ± 1.7 |
| EPI | −64.7 ± 0.9 | −573.4 ± 3.2 | 592.4 ± 3.9 | −4.1 ± 0.0 | −49.9 ± 0.2 | 19.0 ± 0.7 | −68.8 ± 0.9 |
| PAL | −67.2 ± 0.4 | −581.3 ± 1.3 | 605.4 ± 5.2 | −4.4 ± 0.1 | −47.5 ± 0.8 | 24.1 ± 0.4 | −71.6 ± 0.5 |
| Ligand | a | a | |||
|---|---|---|---|---|---|
| BER | −42.8 ± 0.5 | 27.9 ± 0.0 | 7.3 | −7.6 ± 0.5 | −7.82 ± 0.06 |
| COP | −45.9 ± 0.8 | 28.5 ± 0.1 | 7.3 | −10.1 ± 0.8 | −8.89 ± 0.1 |
| EPI | −43.3 ± 0.4 | 27.1 ± 0.0 | 7.3 | −8.9 ± 0.4 | −10.70 ± 0.02 |
| PAL | −40.3 ± 0.3 | 24.6 ± 0.1 | 7.3 | −8.4 ± 0.3 | −8.41 ± 0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, N.; Xia, J.; Wickstrom, L.; Lin, C.; Wang, K.; He, P.; Yin, Y.; Yang, D. Ligand Selectivity in the Recognition of Protoberberine Alkaloids by Hybrid-2 Human Telomeric G-Quadruplex: Binding Free Energy Calculation, Fluorescence Binding, and NMR Experiments. Molecules 2019, 24, 1574. https://doi.org/10.3390/molecules24081574
Deng N, Xia J, Wickstrom L, Lin C, Wang K, He P, Yin Y, Yang D. Ligand Selectivity in the Recognition of Protoberberine Alkaloids by Hybrid-2 Human Telomeric G-Quadruplex: Binding Free Energy Calculation, Fluorescence Binding, and NMR Experiments. Molecules. 2019; 24(8):1574. https://doi.org/10.3390/molecules24081574
Chicago/Turabian StyleDeng, Nanjie, Junchao Xia, Lauren Wickstrom, Clement Lin, Kaibo Wang, Peng He, Yunting Yin, and Danzhou Yang. 2019. "Ligand Selectivity in the Recognition of Protoberberine Alkaloids by Hybrid-2 Human Telomeric G-Quadruplex: Binding Free Energy Calculation, Fluorescence Binding, and NMR Experiments" Molecules 24, no. 8: 1574. https://doi.org/10.3390/molecules24081574