Advances in the Understanding of Skin Cancer: Ultraviolet Radiation, Mutations, and Antisense Oligonucleotides as Anticancer Drugs


 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
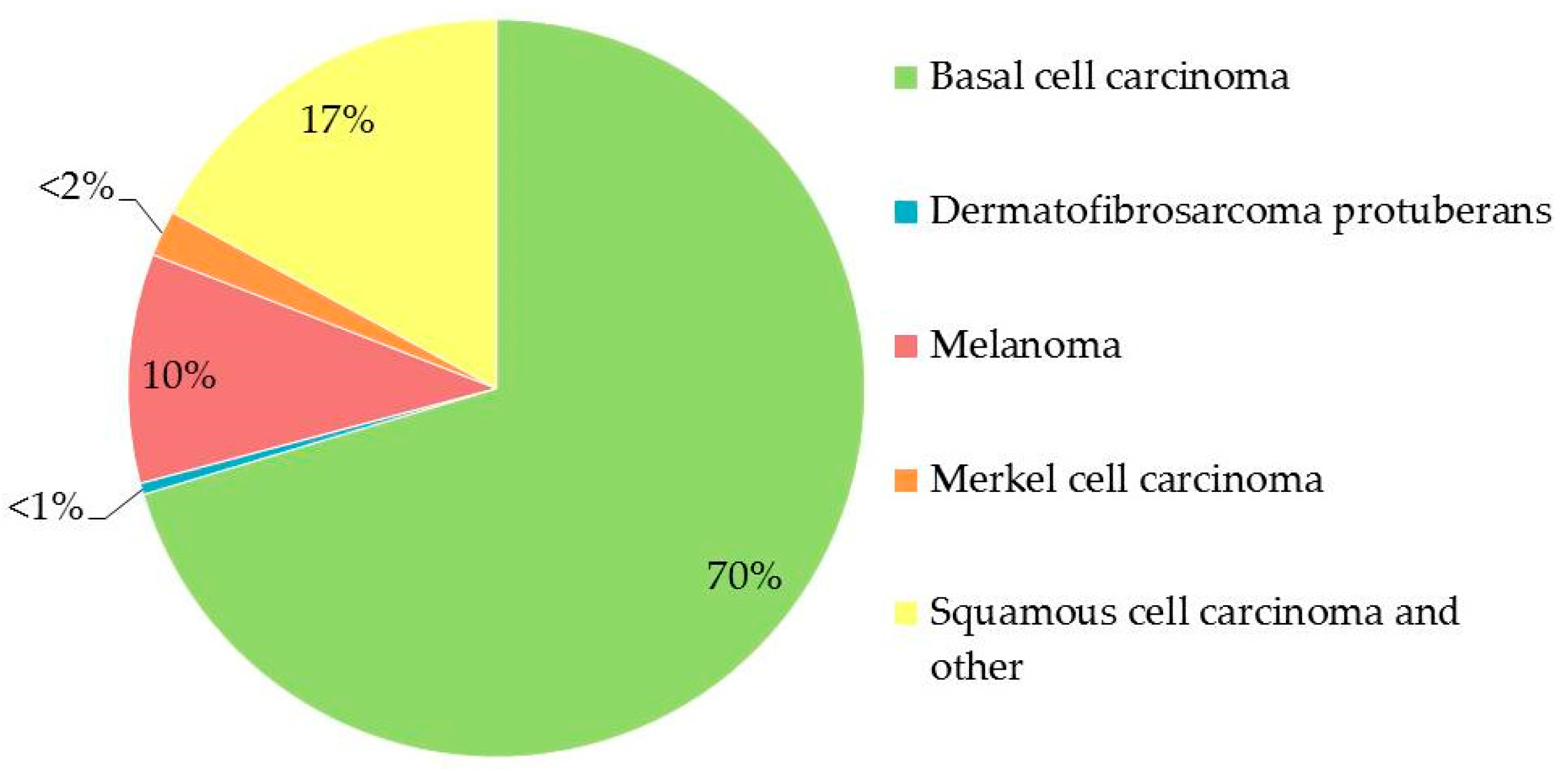
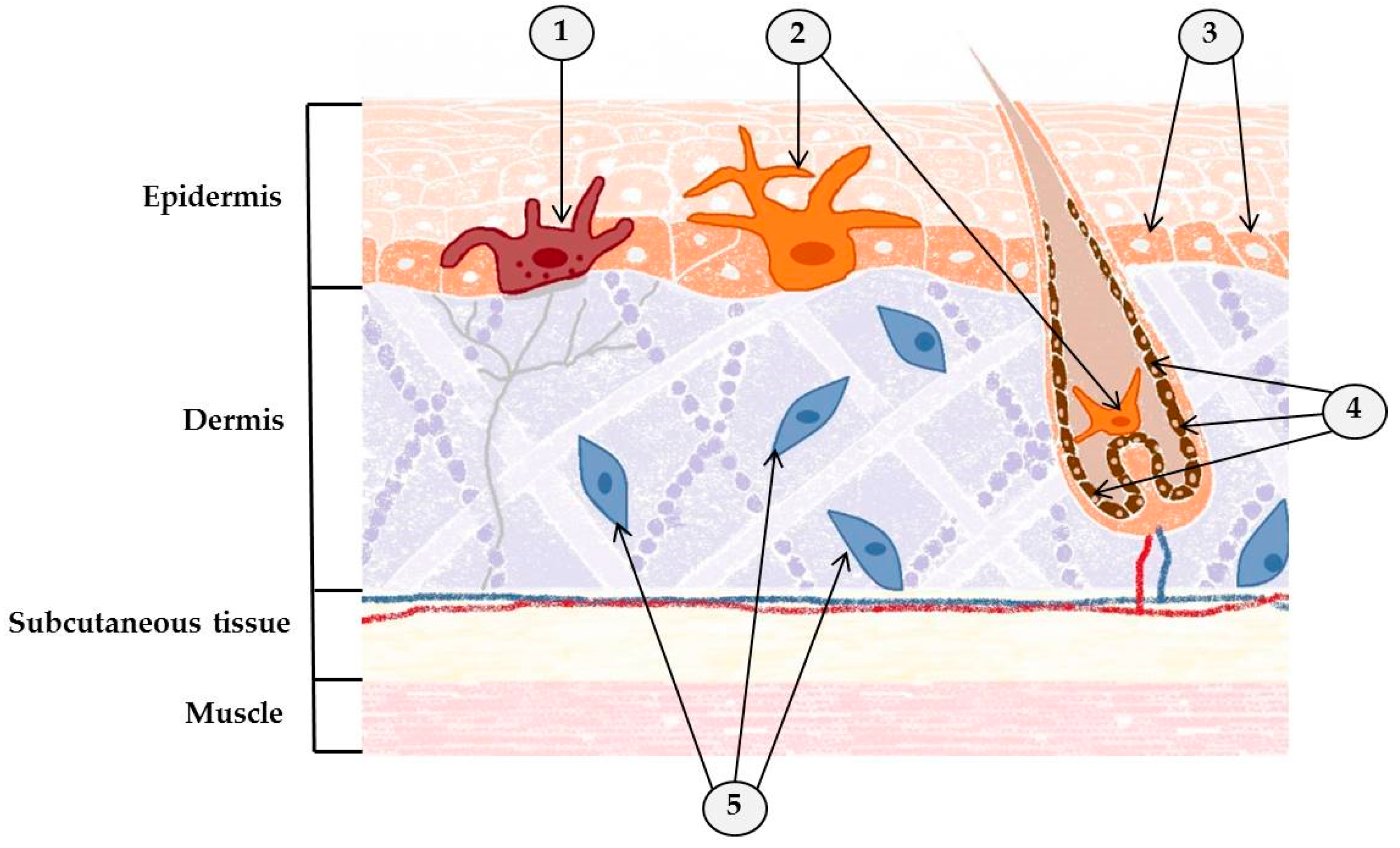
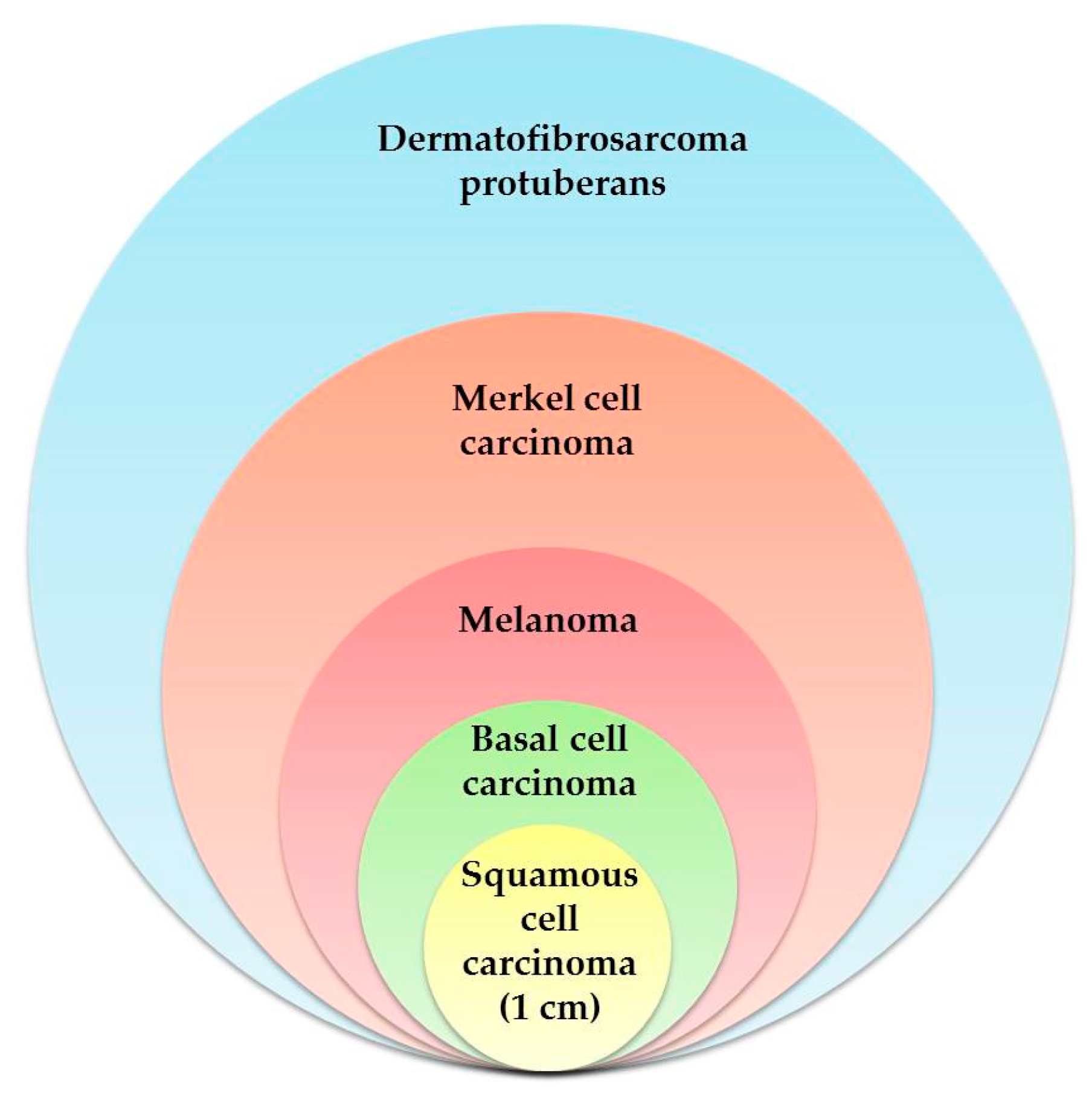
2. Skin: Where One-Third of New Cancers Are Born
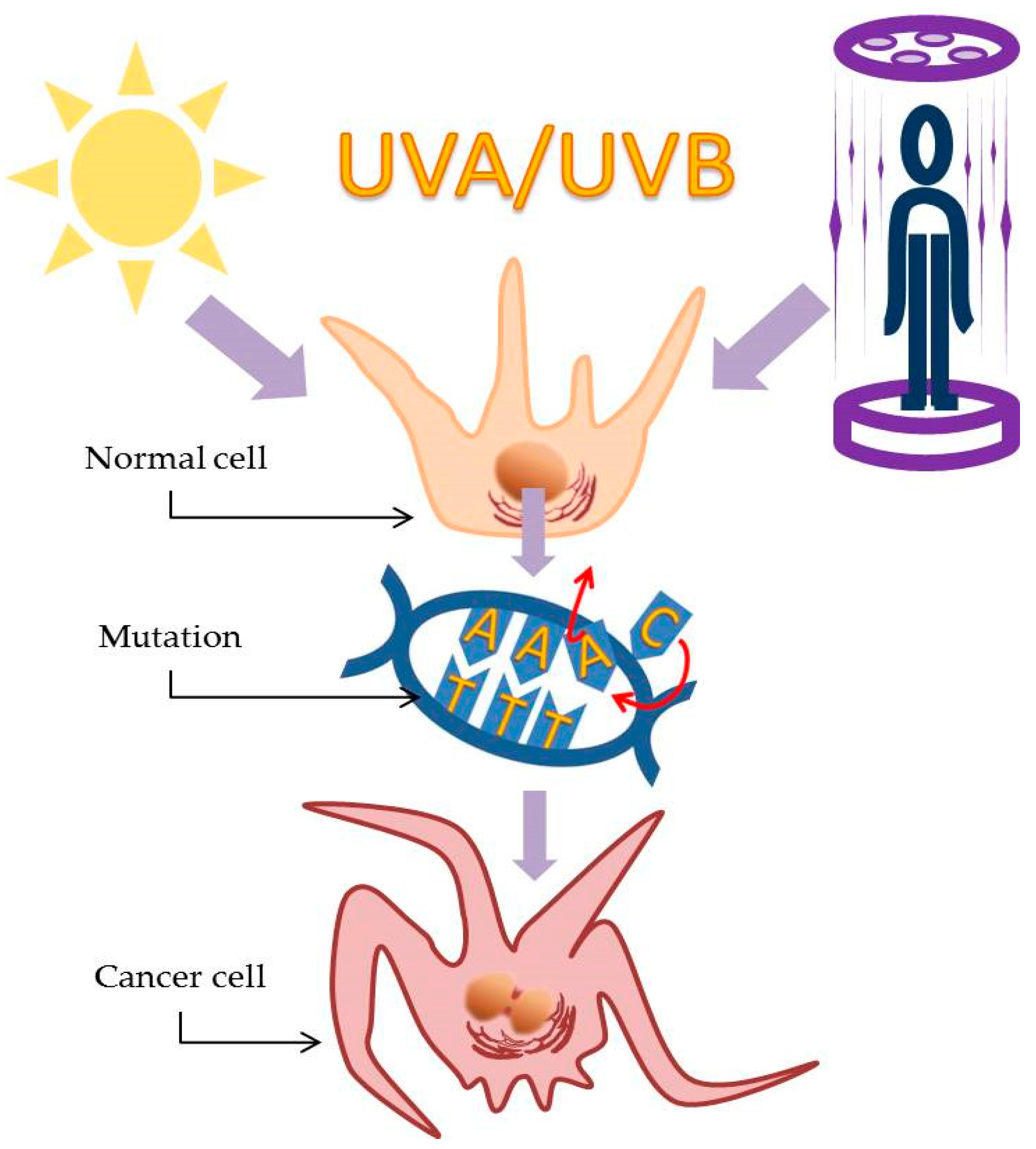
3. Ultraviolet Light Illuminates the Path of Skin Cancer
4. Help the Skin by Cutting the Cancer Out in Time
5. What Happens if the Surgeon Does Not Succeed?
5.1. Botanicals as a Hope
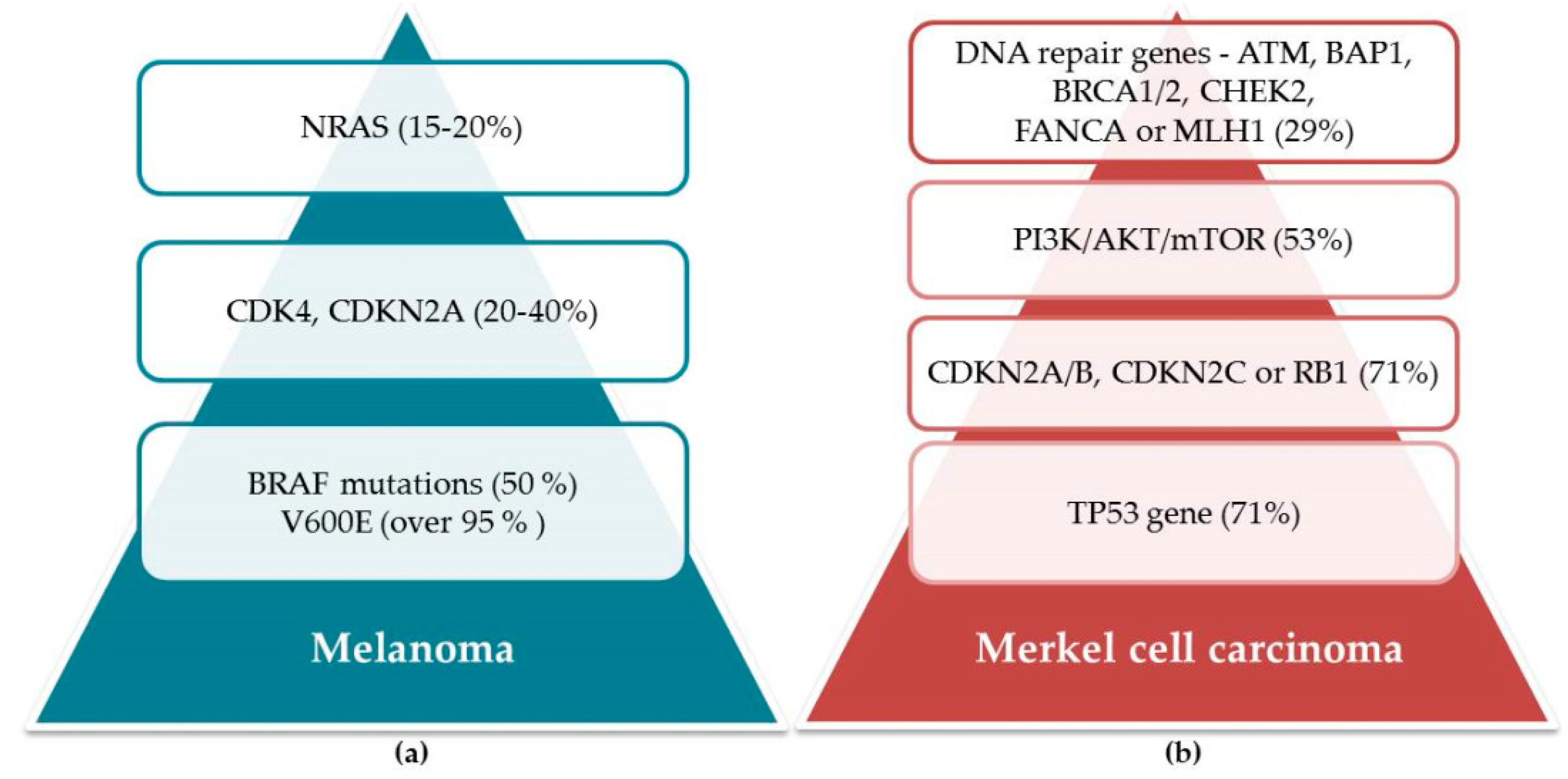
5.2. Mutations as a Grounding for Efficient Treatment
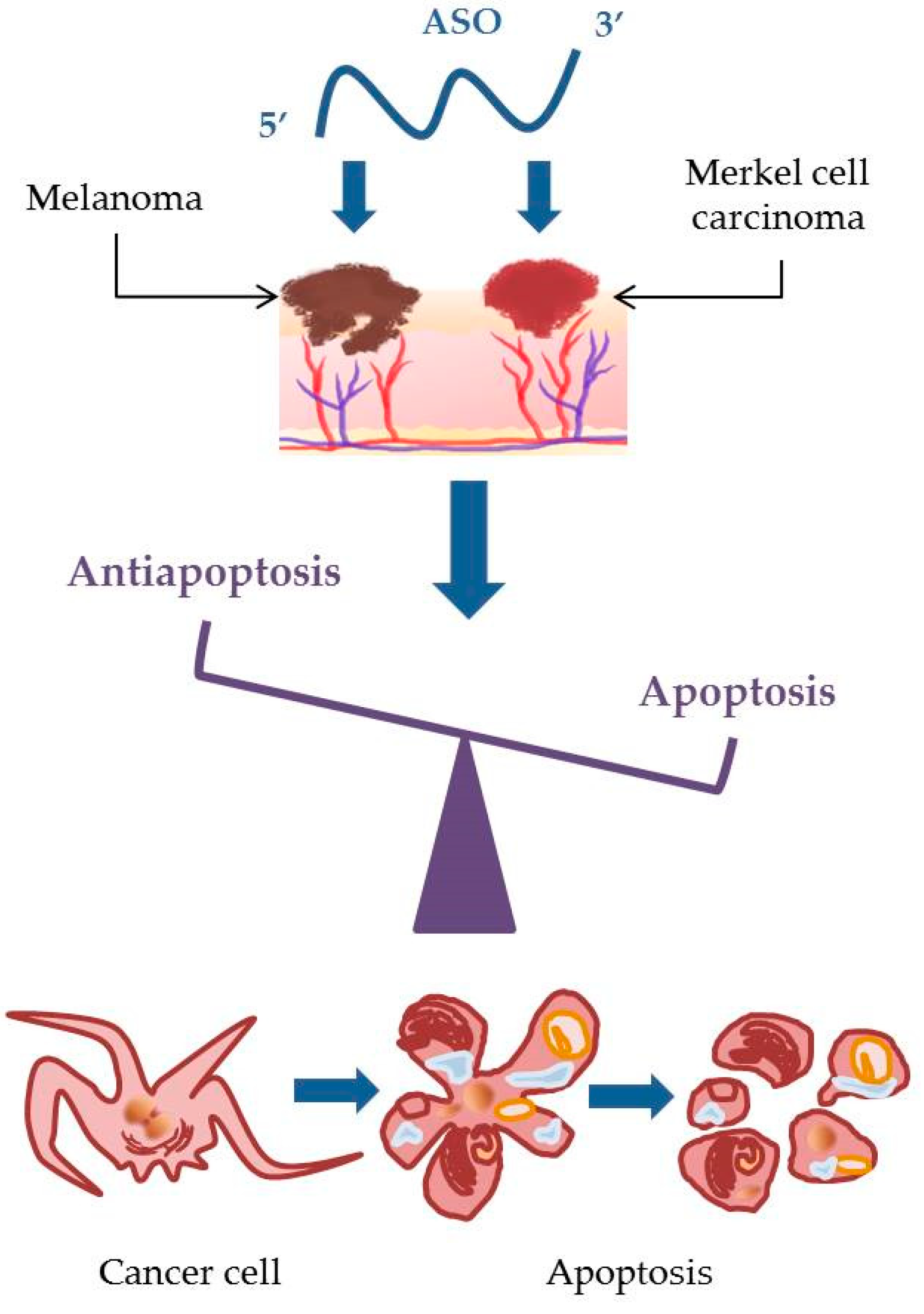
6. Keep Silencing: Approaches for the Treatment of Melanoma and Merkel Cell Carcinoma Using Antisense Oligonucleotides
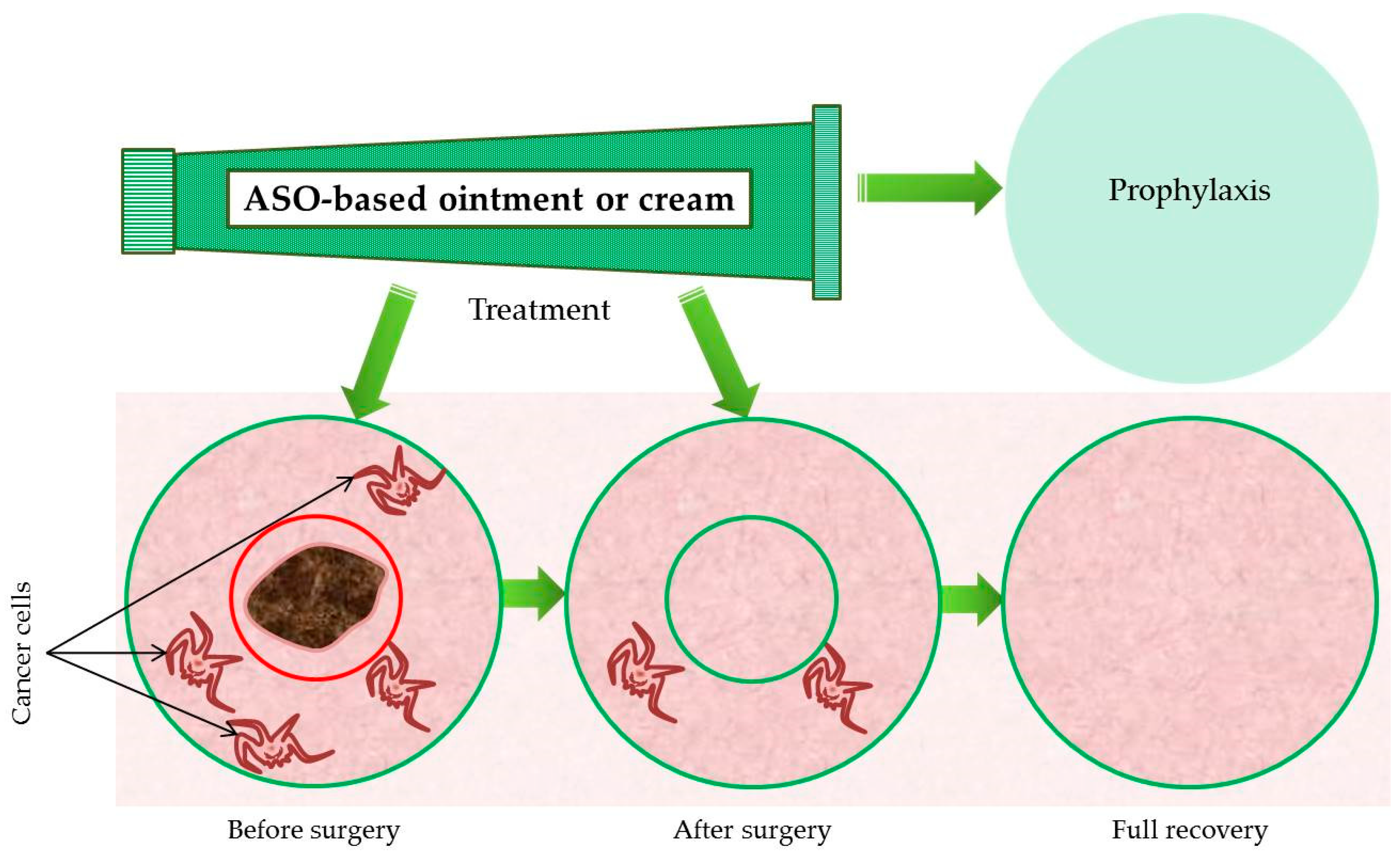
7. Future Perspectives
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Tsao, H. Genetics of non-melanoma skin cancer. Arch. Dermatol. 2001, 137, 1486–1492. [Google Scholar] [CrossRef]
- Diepgen, T.L.; Mahler, V. The epidemiology of skin cancer. Br. J. Dermatol. 2002, 146, 1–6. [Google Scholar] [CrossRef]
- Leiter, U.; Garbe, C. Epidemiology of melanoma and non-melanoma skin cancer—The role of sunlight. Adv. Exp. Med. Biol. 2008, 624, 89–103. [Google Scholar] [PubMed]
- World Health Organization. Skin Cancers /online/. Available online: http://www.who.int/uv/faq/skincancer/en/print.html (accessed on 3 February 2019).
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer. Dermatol. Pract. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of non-melanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Connolly, S.M.; Baker, D.R.; Coldiron, B.M.; Coldiron, B.M.; Fazio, M.J.; Storrs, P.A.; Vidimos, A.T.; Zalla, M.J.; Brewer, J.D.; Smith Begolka, W.; et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: A report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J. Am. Acad. Dermatol. 2012, 67, 531–550. [Google Scholar] [CrossRef]
- Karimkhani, C.; Green, A.C.; Nijsten, T.; Weinstock, M.A.; Dellavalle, R.P.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Lemos, B.D.; Storer, B.E.; Iyer, J.G.; Phillips, J.L.; Bichakjian, C.K.; Fang, L.C.; Johnson, T.M.; Liegeois-Kwon, N.J.; Otley, C.C.; Paulson, K.G.; et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: Analysis of 5823 cases as the basis of the first consensus staging system. J. Am. Acad. Dermatol. 2010, 63, 751–761. [Google Scholar] [CrossRef]
- Fitzgerald, T.L.; Dennis, S.; Kachare, S.D.; Vohra, N.A.; Wong, J.H.; Zervos, E.E. Dramatic increase in the incidence and mortality from Merkel cell carcinoma in the United States. Am. Surg. 2015, 81, 802–806. [Google Scholar] [PubMed]
- Miller, S.J.; Alam, M.; Andersen, J.S.; Berg, D.; Bichakjian, C.K.; Bowen, G.M.; Cheney, R.T.; Glass, L.F.; Grekin, R.C.; Ho, A.L.; et al. Dermatofibrosarcoma protuberans. J. Nat. Compr. Cancer Netw. 2012, 10, 312–318. [Google Scholar] [CrossRef]
- Strassburg, M.A. The global eradication of smallpox. Am. J. Infect. Control 1982, 10, 53–59. [Google Scholar] [CrossRef]
- Elwood, J.M.; Jopson, J. Melanoma and sun exposure: An overview of published studies. Int. J. Cancer 1997, 73, 198–203. [Google Scholar] [CrossRef]
- Lowe, N.J. An overview of ultraviolet radiation, sunscreens, and photo-induced dermatoses. Dermatol. Clin. 2006, 24, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the skin. Endocr. Rev. 2000, 21, 457–487. [Google Scholar] [CrossRef]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell Biol. 2012, 212, 1–115. [Google Scholar]
- Setlow, R.B. The Wavelengths in Sunlight Effective in Producing Skin Cancer: A Theoretical Analysis. Proc. Natl. Acad. Sci. USA 1974, 71, 3363–3366. [Google Scholar] [CrossRef]
- Berwick, M.; Wiggins, C. The current epidemiology of cutaneous malignant melanoma. Front. BioSci. 2006, 11, 1244–1254. [Google Scholar] [CrossRef]
- Brash, D.E.; Heffernan, T.; Ngheim, P. Carcinogenesis: Ultraviolet radiation. In Fitzpatrick’s Dermatology in General Medicine; Wolff, K., Goldsmith, L.A., Katz, S.I., Gilchrest, B.A., Paller, A.S., Leffell, D.J., Eds.; McGraw-Hill: New York, NY, USA, 2008; pp. 999–1006. [Google Scholar]
- Donovan, J. Review of the hair follicle origin hypothesis for basal cell carcinoma. Dermatol. Surg. 2009, 35, 1311–1323. [Google Scholar] [CrossRef]
- Youssef, K.K.; Van Keymeulen, A.; Lapouge, G.; Beck, B.; Michaux, C.; Achouri, Y.; Sotiropoulou, P.A.; Blanpain, C. Identification of the cell lineage at the origin of basal cell carcinoma. Nat. Cell Biol. 2010, 12, 299–305. [Google Scholar] [CrossRef]
- Tlholoe, M.M.; Khammissa, R.A.; Bouckaert, M.; Altini, M.; Lemmer, J.; Feller, L. Oral Mucosal Melanoma: Some Pathobiological Considerations and an Illustrative Report of a Case. Head Neck Pathol. 2015, 9, 127–134. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell. 2015, 6, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Kim, H.; Jin, U.S.; Minn, K.W.; Chang, H. Wide local excision for dermatofibrosarcoma protuberans: A single-center series of 90 patients. BioMed Res. Int. 2015, 2015, 642549. [Google Scholar] [CrossRef] [PubMed]
- Noujaim, J.; Thway, K.; Fisher, C.; Jones, R.L. Dermatofibrosarcoma protuberans: From translocation to targeted therapy. Cancer Biol. Med. 2015, 12, 375–384. [Google Scholar]
- Becker, J.C.; zur Hausen, A. Cells of origin in skin cancer. J. Investig. Dermatol. 2014, 134, 2491–2493. [Google Scholar] [CrossRef]
- Schadendorf, D.; Lebbé, C.; Hausen zur, A.; Avril, M.F.; Hariharan, S.; Bharmal, M.; Becker, J.C. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur. J. Cancer 2017, 71, 53–69. [Google Scholar] [CrossRef]
- Arora, R.; Chang, Y.; Moore, P.S. MCV and Merkel cell carcinoma: A molecular success story. Curr. Opin. Virol. 2012, 2, 489–498. [Google Scholar] [CrossRef]
- Tilling, T.; Moll, I. Which are the cells of origin in merkel cell carcinoma? J. Skin Cancer 2012, 2012, 680410. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.S.; Byrne, P.J.; Jacobs, L.K.; Taube, J.M. Merkel cell carcinoma: Update and review. Semin. Cutan. Med. Surg. 2011, 30, 49–56. [Google Scholar] [CrossRef]
- Young, C. Solar ultraviolet radiation and skin cancer. Occup. Med. (Lond.) 2009, 59, 82–88. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, J.; Jarrett, S.; Maro-Ortiz, A.; Scott, T. UV Radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef]
- Meeran, S.M.; Punathil, T.; Katiyar, S.K. Interleukin-12-deficiency exacerbates inflammatory responses in UVirradiated skin and skin tumors. J. Investig. Dermatol. 2008, 128, 2716–2727. [Google Scholar] [CrossRef]
- Rangwala, S.; Tsai, K.Y. Roles of the immune system in skin cancer. Br. J. Dermatol. 2011, 165, 953–965. [Google Scholar] [CrossRef]
- Benjamin, C.L.; Ananthaswamy, H.N. p53 and the pathogenesis of skin cancer. Toxicol. Appl. Pharmacol. 2007, 224, 241–248. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Liley, J.B.; McKenzie, R.L. Where on Earth has the highest UV? UV Radiation and Its Effects: An Update. Natl. Inst. Water Atmos. Res. 2006, 68, 36–37. [Google Scholar]
- World Health Organization. Ultraviolet Radiation (UV) /online/. Available online: www.who.int/uv/intersunprogramme/activities/uv_index/en/index3.html (accessed on 4 February 2019).
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordóñez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef]
- Elmets, C.A.; Ledet, J.J.; Athar, M. Cyclooxygenases: Mediators of UV-induced skin cancer and potential targets for prevention. J. Investig. Dermatol. 2014, 134, 2497–2502. [Google Scholar] [CrossRef]
- Timerman, D.; McEnery-Stonelake, M.; Joyce, C.J.; Nambudiri, V.E.; Hodi, F.S.; Claus, E.B.; Ibrahim, N.; Lin, J.Y. Vitamin D deficiency is associated with a worse prognosis in metastatic melanoma. Oncotarget. 2017, 8, 6873–6882. [Google Scholar] [CrossRef]
- Juzeniene, A.; Moan, J. Beneficial effects of UV radiation other than via vitamin D production. Dermatoendocrinology 2012, 4, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Feller, L.; Khammissa, R.A.; Kramer, B.; Altini, M.; Lemmer, J. Basal cell carcinoma, squamous cell carcinoma and melanoma of the head and face. Head Face Med. 2016, 12, 11. [Google Scholar] [CrossRef]
- Grossman, D.; Leffell, D.J. Squamous cell carcinoma. In Fitzpatrick’s Dermatology in General Medicine; Wolff, K., Goldsmith, L.A., Katz, S.I., Gilchrest, B.A., Paller, A.S., Leffell, D.J., Eds.; McGraw-Hill: New York, NY, USA, 2008; pp. 1028–1036. [Google Scholar]
- Bakos, L.; Masiero, N.C.; Bakos, R.M.; Burttet, R.M.; Wagner, M.B.; Benzano, D. European ancestry and cutaneous melanoma in Southern Brazil; I.K. Crombie Racial differences in melanoma incidence. Br. J. Cancer 1979, 40, 185–193. [Google Scholar]
- Scherer, D.; Kumar, R. Genetics of pigmentation in skin cancer–A review. Mutat. Res. 2010, 705, 141–153. [Google Scholar] [CrossRef]
- Horrell, E.M.W.; Boulanger, M.; D’Orazio, J.A. Melanocortin 1 receptor: Structure, function and regulation. Front. Genet. 2016, 7, 95. [Google Scholar] [CrossRef]
- Grabowski, J.; Saltzstein, S.L.; Sadler, G.R.; Tahir, Z.; Blair, S. A comparison of Merkel cell carcinoma and melanoma: Results from the california cancer registry. Clin. Med. Oncol. 2008, 2, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Mogha, A.; Fautrel, A.; Mouchet, N.; Guo, N.; Corre, S.; Adamski, H.; Watier, E.; Misery, L.; Galibert, M.D. Merkel cell polyomavirus small T antigen mRNA level is increased following in vivo UV-radiation. PLoS ONE 2010, 5, e11423. [Google Scholar] [CrossRef]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 170–177. [Google Scholar] [CrossRef]
- Girschik, J.; Thorn, K.; Beer, T.W.; Heenan, P.J.; Fritschi, L. Merkel cell carcinoma in Western Australia: A population-based study of incidence and survival. Br. J. Dermatol. 2011, 165, 1051–1057. [Google Scholar] [CrossRef]
- Dabner, M.; McClure, R.J.; Harvey, N.T.; Budgeon, C.A.; Beer, T.W.; Amanuel, B.; Wood, B.A. Merkel cell polyomavirus and p63 status in Merkel cell carcinoma by immunohistochemistry: Merkel cell polyomavirus positivity is inversely correlated with sun damage, but neither is correlated with outcome. Pathology 2014, 46, 205–210. [Google Scholar] [CrossRef]
- Chockalingam, R.; Downing, C.; Tyring, S.K. Cutaneous squamous cell carcinomas in organ transplant recipients. J. Clin. Med. 2015, 4, 1229–1239. [Google Scholar] [CrossRef]
- Heratizadeh, A.; Völker, B.; Kupsch, E.; Wichmann, K.; Kapp, A.; Werfel, T. Successful symptomatic treatment of epidermodysplasia verruciformis with imiquimod 5% cream. Hautarzt 2010, 61, 1052–1055. [Google Scholar] [CrossRef]
- Moncrieff, M. Excision margins for melanomas: How wide is enough? Lancet Oncol. 2016, 17, 127–128. [Google Scholar] [CrossRef]
- Di Trolio, R.; Simeone, E.; Di Lorenzo, G.; Buonerba, C.; Ascierto, P.A. The use of interferon in melanoma patients: A systematic review. Cytokine Growth Factor Rev. 2015, 26, 203–212. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Eng. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Eng. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Nikolaou, V.A.; Stratigos, A.J.; Flaherty, K.T.; Tsao, H. Melanoma: New insights and new therapies. J. Investig. Dermatol. 2012, 132, 854–863. [Google Scholar] [CrossRef]
- Ji, Z.; Flaherty, K.T.; Tsao, H. Targeting the RAS pathway in melanoma. Trends Mol. Med. 2012, 18, 27–35. [Google Scholar] [CrossRef]
- Flaherty, K.T. Targeting metastatic melanoma. Annu. Rev. Med. 2012, 63, 171–183. [Google Scholar] [CrossRef]
- Hodi, F.S.; Oble, D.A.; Drappatz, J.; Velazquez, E.F.; Ramaiya, N.; Ramakrishna, N.; Day, A.L.; Kruse, A.; Mac Rae, S.; Hoos, A.; et al. CTLA-4 blockade with ipilimumab induces significant clinical benefit in a female with melanoma metastases to the CNS. Nat. Clin. Pract. Oncol. 2008, 5, 557–561. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Eng. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Serrone, L.; Hersey, P. The chemoresistance of human malignant melanoma: An update. Melanoma Res. 1999, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Grossman, D.; Altieri, D.C. Drug resistance in melanoma: Mechanisms, apoptosis, and new potential therapeutic targets. Cancer Metastasis Rev. 2001, 20, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Helmbach, H.; Rossmann, E.; Kern, M.A.; Schadendorf, D. Drug-resistance in human melanoma. Int. J. Cancer 2001, 93, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Glazer, A.M.; Winkelmann, R.R.; Farberg, A.S.; Rigel, D.S. Analysis of trends in US melanoma incidence and mortality. JAMA Dermatol. 2016, 153, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Buzaid, A.C.; Soong, S.J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A., Jr.; Kirkwood, J.M.; et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. Clin. Oncol. 2001, 19, 3635–3648. [Google Scholar] [CrossRef] [PubMed]
- Jansen, B.; Wacheck, V.; Heere-Ress, E.; Schlagbauer-Wadl, H.; Hoeller, C.; Lucas, T.; Hoermann, M.; Hollenstein, U.; Wolff, K.; Pehamberger, H. Chemosensitisation of malignant melanoma by BCL2 antisense therapy. Lancet 2000, 356, 1728–1733. [Google Scholar] [CrossRef]
- Megahed, M.; Schon, M.; Selimovic, D.; Schon, M.P. Reliability of diagnosis of melanoma in situ. Lancet 2002, 359, 1921–1922. [Google Scholar] [CrossRef]
- Zalaudek, I.; Hofmann-Wellenhof, R.; Cerroni, L.; Kerl, H. “White” dysplastic melanocytic naevi. Lancet 2002, 359, 1999–2000. [Google Scholar] [CrossRef]
- Hudson, L.E.; Maithel, S.K.; Carlson, G.W.; Rizzo, M.; Murray, D.R.; Hestley, A.C.; Delman, K.A. 1 or 2 cm margins of excision for T2 melanomas: Do they impact recurrence or survival? Ann. Surg. Oncol. 2013, 20, 346–351. [Google Scholar] [CrossRef]
- Ross, M.I.; Gershenwald, J.E. Evidence-based treatment of early-stage melanoma. J. Surg. Oncol. 2011, 104, 341–353. [Google Scholar] [CrossRef]
- Ross, M.I.; Balch, C.M. Excision margins of melanoma make a difference: New data support an old paradigm. Ann. Surg. Oncol. 2016, 23, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.L. The genetics of sun sensitivity in humans. Am. J. Hum. Genet. 2004, 75, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Lv, R.; Sun, Q. A Network Meta-Analysis of Non-Melanoma Skin Cancer (NMSC) Treatments: Efficacy and Safety Assessment. J. Cell. Biochem. 2017, 118, 3686–3695. [Google Scholar] [CrossRef] [PubMed]
- Griffin, L.; Lear, J. Photodynamic Therapy and Non-Melanoma Skin Cancer. Cancers 2016, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Lee, P. Photodynamic Therapy for Non-Melanoma Skin Cancers. Cancers 2016, 8, 90. [Google Scholar] [CrossRef]
- Hamdan, I.; Donnelly, R. Microneedle-assisted photodynamic therapy: Delivery of a NIR photosensitiser for the treatment of skin cancers. Photodiagnosis Photodyn. Ther. 2017, 17, A63. [Google Scholar] [CrossRef]
- Cheraghi, N.; Cognetta, A.; Goldberg, D. Radiation Therapy in Dermatology: Non-Melanoma Skin Cancer. J. Drugs Dermatol. 2017, 16, 464–469. [Google Scholar]
- Kumar, R.; Deep, G.; Agarwal, R. An overview of ultraviolet B radiation-induced skin cancer chemoprevention by Silibinin. Curr. Pharmacol. Rep. 2015, 1, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Nahhas, A.F.; Scarbrough, C.A.; Trotter, S.A. Review of the global guidelines on surgical margins for nonmelanoma skin cancers. J. Clin. Aesthetic Dermatol. 2017, 10, 37–46. [Google Scholar]
- Duprat, J.P.; Landman, G.; Salvajoli, J.V.; Brechtbühl, E.R. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics 2011, 66, 1817–1823. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.J.; Zhang, Z.F.; Coit, D.G. Surgical management of Merkel cell carcinoma. Ann. Surg. 1999, 229, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Lemos, B.; Nghiem, P. Merkel cell carcinoma: More deaths but still no pathway to blame. J. Investig. Dermatol. 2007, 127, 2100–2103. [Google Scholar] [CrossRef]
- Mohs, F. Chemosurgical treatment of cancer of the lip: A microscopically controlled method of excision. Arch. Surg. 1944, 48, 478–488. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology; Dermatofibrosarcoma Protuberans. Available online: http://www.nccn.org (accessed on 5 February 2019).
- McArthur, G.; Demetri, G.D.; Oosterom, A.; Heinrich, M.C.; Debiec-Rychter, M.; Corless, C.L.; Nikolova, Z.; Dimitrijevic, S.; Fletcher, J.A. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib target exploration consortium study B2225. J. Clin. Oncol. 2005, 23, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Bogucki, B.; Neuhaus, I.; Hurst, E.A. Dermatofibrosarcoma protuberans: A review of the literature. Dermatol. Surg. 2012, 38, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.C.; Hameed, M.; Qin, L.X.; Moraco, N.; Jia, X.; Maki, R.G.; Singer, S.; Brennan, M.F. Dermatofibrosarcoma protuberans (DFSP): Predictors of recurrence and the use of systemic therapy. Ann. Surg. Oncol. 2011, 18, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Leigheb, M.; Zavattaro, E.; Bellinzona, F.; Furlan, G.; Leigheb, G. Micrographic surgery (Tubingen torte technique) for the treatment of an invasive dermatofibrosarcoma protuberans with muscular involvement. G. Ital. Dermatol. Venereol. 2010, 145, 309–311. [Google Scholar]
- Madhunapantula, S.V.; Robertson, G.P. Chemoprevention of melanoma. Adv. Pharmacol. 2012, 65, 361–398. [Google Scholar]
- Keith, R.L. Chemoprevention of lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, S. Technology Motivating Industry. The Scientist, 1996; 10, 1–7. [Google Scholar]
- Jiang, G.; Li, R.H.; Sun, C.; Liu, Y.Q.; Zheng, J.N. Dacarbazine combined targeted therapy versus dacarbazine alone in patients with malignant melanoma: a meta-analysis. PLoS ONE 2014, 9, e111920. [Google Scholar] [CrossRef] [PubMed]
- Bajetta, E.; Del Vecchio, M.; Bernard-Marty, C.; Vitali, M.; Buzzoni, R.; Rixe, O.; Nova, P.; Aglione, S.; Taillibert, S.; Khayat, D. Metastatic melanoma: Chemotherapy. Semin. Oncol. 2002, 29, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Serrone, L.; Zeuli, M.; Sega, F.M.; Cognetti, F. Dacarbazine-based chemotherapy for metastatic melanoma: Thirty-year experience overview. J. Exp. Clin. Cancer Res. 2000, 19, 21–34. [Google Scholar]
- Avril, M.F.; Aamdal, S.; Grob, J.J.; Hauschild, A.; Mohr, P.; Bonerandi, J.J.; Weichenthal, M.; Neuber, K.; Bieber, T.; Gilde, K.; et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: A Phase III Study. J. Clin. Oncol. 2004, 22, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Chiarion Sileni, V.; Nortilli, R.; Aversa, S.M.; Paccagnella, A.; Medici, M.; Corti, L.; Favaretto, A.G.; Cetto, G.L.; Monfardini, S. Phase II randomized study of dacarbazine, carmustine, cisplatin, and tamoxifen versus dacarbazine alone in advanced melanoma patients. Melanoma Res. 2001, 11, 189–196. [Google Scholar] [CrossRef]
- Strickland, L.R.; Pal, H.C.; Elmets, C.A.; Afaq, F. Targeting drivers of melanoma with synthetic small molecules and phytochemicals. Cancer Lett. 2015, 359, 20–35. [Google Scholar] [CrossRef]
- Richtig, G.; Hoeller, C.; Kashofer, K.; Aigelsreiter, A.; Heinemann, A.; Kwong, L.N.; Pichler, M.; Richtig, E. Beyond the BRAFV600E hotspot-biology and clinical implications of rare BRAF gene mutations in melanoma patients. Br. J. Dermatol. 2017, 177, 936–944. [Google Scholar] [CrossRef]
- Edwards, R.H.; Ward, M.R.; Wu, H.; Medina, C.A.; Brose, M.S.; Volpe, P.; Nussen-Lee, S.; Haupt, H.M.; Martin, A.M.; Herlyn, M.; et al. Absence of BRAF mutations in UV-protected mucosal melanomas. J. Med. Genet. 2004, 41, 270–272. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]
- Mukherjee, N.; Schwan, J.V.; Fujita, M.; Norris, D.A.; Shellman, Y.G. Alternative treatments for melanoma: Targeting BCL-2 family members to de-bulk and kill cancer stem cells. J. Investig. Dermatol. 2015, 135, 2155–2161. [Google Scholar] [CrossRef]
- Liu, F.; Cao, J.; Wu, J.; Sullivan, K.; Shen, J.; Ryu, B.; Xu, Z.; Wei, W.; Cui, R. Stat3-targeted therapies overcome the acquired resistance to vemurafenib in melanomas. J. Investig. Dermatol. 2013, 133, 2041–2049. [Google Scholar] [CrossRef]
- Becker, T.M.; Boyd, S.C.; Mijatov, B.; Gowrishankar, K.; Snoyman, S.; Pupo, G.M.; Scolyer, R.A.; Mann, G.J.; Kefford, R.F.; Zhang, X.D.; et al. Mutant B-RAF-Mcl-1 survival signaling depends on the STAT3 transcription factor. Oncogene 2014, 33, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Marconcini, R.; Spagnolo, F.; Stucci, L.S.; Ribero, S.; Marra, E.; Rosa, F.; Picasso, V.; Di Guardo, L.; Cimminiello, C.; Cavalieri, S.; et al. Current status and perspectives in immunotherapy for metastatic melanoma. Oncotarget 2018, 9, 12452–12470. [Google Scholar] [CrossRef]
- Munoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; Garcia, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef]
- Jakob, J.A.; Bassett, R.L., Jr.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, J.W.; Kim, Y.S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Erstad, D.J.; Cusack, J.C., Jr. Mutational analysis of Merkel cell carcinoma. Cancers 2014, 6, 2116–2136. [Google Scholar] [CrossRef] [PubMed]
- Larramendy, M.L.; Koljonen, V.; Bohling, T.; Tukiainen, E.; Knuutila, S. Recurrent DNA copy number changes revealed by comparative genomic hybridization in primary Merkel cell carcinomas. Mod. Pathol. 2004, 17, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Lee, L.; Caramuta, S.; Hoog, A.; Browaldh, N.; Bjornhagen, V.; Larsson, C.; Lui, W.O. MicroRNA expression patterns related to Merkel cell polyomavirus infection in human Merkel cell carcinoma. J. Investig. Dermatol. 2014, 134, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef]
- Busam, K.J.; Jungbluth, A.A.; Rekthman, N.; Coit, D.; Pulitzer, M.; Bini, J.; Arora, R.; Hanson, N.C.; Tassello, J.A.; Frosina, D.; et al. Merkel cell polyomavirus expression in Merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am. J. Surg. Pathol. 2009, 33, 1378–1385. [Google Scholar] [CrossRef]
- Tadmor, T.; Aviv, A.; Polliack, A. Merkel cell carcinoma, chronic lymphocytic leukemia and other lymphoproliferative disorders: An old bond with possible new viral ties. Ann. Oncol. 2011, 22, 250–256. [Google Scholar] [CrossRef]
- Laude, H.C.; Jonchere, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.F.; Dupin, N.; et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar] [CrossRef]
- Sahi, H.; Savola, S.; Sihto, H.; Koljonen, V.; Bohling, T.; Knuutila, S. Rb1 gene in merkel cell carcinoma: Hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. APMIS 2014, 122, 1157–1166. [Google Scholar] [CrossRef]
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. Uv-b-type mutations and chromosomal imbalances indicate common pathways for the development of merkel and skin squamous cell carcinomas. Int. J. Cancer 2002, 9, 352–360. [Google Scholar] [CrossRef]
- Cohen, P.R.; Tomson, B.N.; Elkin, S.K.; Marchlik, E.; Carter, J.L.; Kurzrock, R. Genomic portfolio of Merkel cell carcinoma as determined by comprehensive genomic profiling: Implications for targeted therapeutics. Oncotarget. 2016, 7, 23454–23467. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Wong, H.H.; Wang, J. Epstein-Barr virus positive diffuse large B-cell lymphoma of the elderly. Leuk. Lymphoma 2009, 50, 335–340. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Penas, P.F.; Nghiem, P. Clinical characteristics of merkel cell carcinoma at diagnosis in 195 patients: The aeiou features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef]
- Schwartz, J.L.; Wong, S.L.; McLean, S.A.; Hayman, J.A.; Lao, C.D.; Kozlow, J.H.; Malloy, K.M.; Bradford, C.R.; Frohm, M.L.; Fullen, D.R.; et al. NCCN guidelines implementation in the multidisciplinary Merkel cell carcinoma program at the University of Michigan. J. Natl. Compr. Cancer Netw. 2014, 12, 434–441. [Google Scholar] [CrossRef]
- Raju, S.; Vazirnia, A.; Totri, C.; Hata, T.R. Treatment of Merkel cell carcinoma of the head and neck: A systematic review. Dermatol. Surg. 2014, 40, 1273–1283. [Google Scholar] [CrossRef]
- Prewett, S.L.; Ajithkumar, T. Merkel cell carcinoma: Current management and controversies. Clin. Oncol. 2015, 27, 436–444. [Google Scholar] [CrossRef]
- Munoz, I.P.; Masferrer, J.P.; Vegas, J.O.; Montalvo, M.S.M.; Diaz, R.J.; Casas, A.M.P. Merkel cell carcinoma from 2008 to 2012: Reaching a new level of understanding. Cancer Treat. Rev. 2013, 39, 421–429. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Daud, A.; Friedlander, P.; Kluger, H.; Kohrt, H.; Kudchadkar, R.; Lipson, E.; Lundgren, L.; Margolin, K.; et al. Activity of PD-1 blockade with pembrolizumab as first systemic therapy in patients with advanced Merkel cell carcinoma [Abstract 22LBA: In Proceedings of the 18th ECCO-40th ESMA European Cancer Congress, Vienna, Austria, 25–29 September 2015]. Eur. J. Cancer 2015, 51, S720–S721. [Google Scholar] [CrossRef]
- Moshiri, A.S.; Nghiem, P. Milestones in the staging, classification, and biology of Merkel cell carcinoma. J. Natl. Compr. Canc. Netw. 2014, 12, 1255–1262. [Google Scholar] [CrossRef]
- Miller, N.J.; Bhatia, S.; Parvathaneni, U.; Iyer, J.G.; Nghiem, P. Emerging and mechanism-based therapies for recurrent or metastatic Merkel cell carcinoma. Curr. Treat. Options Oncol. 2013, 14, 249–263. [Google Scholar] [CrossRef]
- Aldabagh, B.; Joo, J.; Yu, S.S. Merkel cell carcinoma: Current status of targeted and future potential for immunotherapies. Semin. Cutan. Med. Surg. 2014, 33, 76–82. [Google Scholar] [CrossRef]
- Tothill, R.; Estall, V.; Rischin, D. Merkel cell carcinoma: Emerging biology, current approaches, and future directions. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharm. Ther. 2014, 39, 119–122. [Google Scholar]
- Stein, A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Oberemok, V.V.; Laikova, K.V.; Repetskaya, A.I.; Kenyo, I.M.; Gorlov, M.V.; Kasich, I.N.; Krasnodubets, A.M.; Gal’chinsky, N.V.; Fomochkina, I.I.; Zaitsev, A.S.; et al. A Half-Century History of Applications of Antisense Oligonucleotides in Medicine, Agriculture and Forestry: We Should Continue the Journey. Molecules 2018, 23, 1302. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Kirkwood, J.M. Oblimersen in the treatment of metastatic melanoma. Future Oncol. 2007, 3, 263–271. [Google Scholar] [CrossRef]
- De Smet, M.D.; Meenken, C.J.; van den Horn, G.J. Fomivirsen: A phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul. Immunol. Inflamm. 1999, 7, 189–198. [Google Scholar] [CrossRef]
- Moreno, P.M.; Pego, A.P. Therapeutic antisense oligonucleotides against cancer: Hurdling to the clinic. Front. Chem. 2014, 2, 87. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.E. Apoptosis in cancer therapy: Crossing the threshold. Cell 1994, 78, 539–542. [Google Scholar] [CrossRef]
- Olie, R.A.; Hafner, C.; Kuttel, R.; Sigrist, B.; Willers, J.; Dummer, R.; Hall, J.; Stahel, R.A.; Zangemeister-Wittke, U. BCL-2 and BCL-XL antisense oligonucleotides induce apoptosis in melanoma cells of different clinical stages. J. Investig. Dermatol. 2002, 118, 505–512. [Google Scholar] [CrossRef]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef]
- Bedikian, A.Y.; Millward, M.; Pehamberger, H.; Conry, R.; Gore, M.; Trefzer, U.; Pavlick, A.C.; DeConti, R.; Hersh, E.M.; Hersey, P.; et al. BCL-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The oblimersen melanoma study group. J. Clin. Oncol. 2006, 24, 4738–4745. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.; Cohen, G.M. BCL-2 inhibitors: Small molecules with a big impact on cancer therapy. Cell. Death Differ. 2009, 16, 360–367. [Google Scholar] [CrossRef]
- Jansen, B.; Schlagbauer-Wadl, H.; Brown, B.D.; Bryan, R.N.; van Elsas, A.; Müller, M.; Wolff, K.; Eichler, H.G.; Pehamberger, H. BCL-2 antisense therapy chemosensitizes human melanoma in SCID mice. Nat. Med. 1998, 4, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Klasa, R.J.; Gillum, A.M.; Klem, R.E.; Frankel, S.R. Oblimersen BCL-2 antisense: Facilitating apoptosis in anticancer treatment. Antisense Nucleic Acid Drug Dev. 2002, 12, 193–213. [Google Scholar] [CrossRef]
- Cheson, B.D. Oblimersen for the treatment of patients with chronic lymphocytic leukemia. Ther. Clin. Risk Manag. 2007, 3, 855–870. [Google Scholar]
- Mita, M.M.; Ochoa, L.; Rowinsky, E.K.; Kuhn, J.; Schwartz, G.; Hammond, L.A.; Patnaik, A.; Yeh, I.-T.; Izbicka, E.; Berg, K.; et al. A phase I, pharmacokinetic and biologic correlative study of oblimersen sodium (Genasense™, G3139) and irinotecan in patients with metastatic colorectal cancer. Ann. Oncol. 2006, 17, 313–321. [Google Scholar] [CrossRef]
- Houben, R.; Schrama, D.; Becker, J.C. Molecular pathogenesis of Merkel cell carcinoma. Exp. Dermatol. 2009, 18, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Grossman, D.; McNiff, J.M.; Li, F.; Altieri, D.C. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J. Investig. Dermatol. 1999, 113, 1076–1081. [Google Scholar] [CrossRef]
- Marusawa, H.; Matsuzawa, S.; Welsh, K.; Zou, H.; Armstrong, R.; Tamm, I.; Reed, J.C. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003, 22, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Pennati, M.; Folini, M.; Zaffaroni, N. Targeting survivin in cancer therapy: Fulfilled promises and open questions. Carcinogenesis 2007, 28, 1133–1139. [Google Scholar] [CrossRef]
- Talbot, D.C.; Ranson, M.; Davies, J.; Lahn, M.; Callies, S.; Andre, V.; Kadam, S.; Burgess, M.; Slapak, C.; Olsen, A.L.; et al. Tumor survivin is downregulated by the antisense oligonucleotide LY2181308: A proof-of-concept, first-in-human dose study. Clin. Cancer Res. 2010, 16, 6150–6158. [Google Scholar] [CrossRef]
- Ryan, B.M.; O’Donovan, N.; Duffy, M.J. Survivin: A new target for anti-cancer therapy. Cancer Treat. Rev. 2009, 35, 553–562. [Google Scholar] [CrossRef]
- Herrington, W.G.; Talbot, D.C.; Lahn, M.M.; Brandt, J.T.; Callies, S.; Nagle, R.; Winearls, C.G.; Roberts, I.S. Association of long-term administration of the survivin mRNA-targeted antisense oligonucleotide LY2181308 with reversible kidney injury in a patient with metastatic melanoma. Am. J. Kidney Dis. 2011, 57, 300–303. [Google Scholar] [CrossRef]
- Feinmesser, M.; Halpern, M.; Fenig, E.; Tsabari, C.; Hodak, E.; Sulkes, J.; Brenner, B.; Okon, E. Expression of the apoptosis-related oncogenes BCL-2, bax, and p53 in Merkel cell carcinoma. Can they predict treatment response and clinical outcome? Hum. Pathol. 1999, 30, 1367–1372. [Google Scholar] [CrossRef]
- Kennedy, M.M.; Blessing, K.; King, G.; Kerr, K.M. Expression of BCL-2 and p53 in Merkel cell carcinoma. An immunohistochemical study. Am. J. Dermatopathol. 1996, 18, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Schlagbauer-Wadl, H.; Klosner, G.; Heere-Ress, E.; Waltering, S.; Moll, I.; Wolff, K.; Pehamberger, H.; Jansen, B. BCL-2 antisense oligonucleotides (G3139) inhibit Merkel cell carcinoma growth in SCID mice. J. Investig. Dermatol. 2000, 114, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Varker, K.A.; Collamore, M.; Zwiebel, J.A.; Coit, D.; Kelsen, D.; Chung, K.Y. G3139 therapy (Genasense) in patients with advanced merkel cell carcinoma. Proc. Am. Assoc. Cancer Res. (AACR) 2007, 48, 147. [Google Scholar]
- Stein, C.A. Does antisense exist? Nature Med. 1995, 1, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Wraight, C.J.; White, P.J. Antisense oligonucleotides in cutaneous therapy. Pharmacol. Ther. 2001, 90, 89–104. [Google Scholar] [CrossRef]
- Dokka, S.; Cooper, S.R.; Kelly, S.; Hardee, G.E.; Karras, J.G. Dermal delivery of topically applied oligonucleotides via follicular transport in mouse skin. J. Investig. Dermatol. 2005, 124, 971–975. [Google Scholar] [CrossRef]
- Mehta, R.C.; Stecker, K.K.; Cooper, S.R.; Templin, M.V.; Tsai, Y.J.; Condon, T.P.; Bennett, C.F.; Hardee, G.E. Intercellular adhesion molecule-1 suppression in skin by topical delivery of antisense oligonucleotides. J. Investig. Dermatol. 2000, 115, 805–812. [Google Scholar] [CrossRef]
- Yacyshyn, B.R.; Bowen-Yacyshyn, M.B.; Jewell, L.; Tami, J.A.; Bennett, C.F.; Kisner, D.L.; Shanahan, W.R., Jr. A placebo-controlled trial of ICAM-1 antisense oligonucleotide in the treatment of Crohn’s disease. Gastroenterology 1998, 114, 113–142. [Google Scholar] [CrossRef]
- Beltinger, C.; Saragovi, H.U.; Smith, R.M.; LeSauteur, L.; Shah, N.; DeDionisio, L.; Christensen, L.; Raible, L.; Jarett, L.; Gewirtz, A.M. Binding uptake and intracellular trafficking of phosphorothioate modified oligodeoxynucleotides. J. Clin. Investig. 1995, 95, 1814–1823. [Google Scholar] [CrossRef]
- Leonetti, C.; Biroccio, A.; Benassi, B.; Stringaro, A.; Stoppacciaro, A.; Semple, S.C.; Zupi, G. Encapsulation of c-myc antisense oligodeoxynucleotides in lipid particles improves antitumoral efficacy in vivo in a human melanoma line. Cancer Gene Ther. 2001, 8, 459–468. [Google Scholar] [CrossRef]
- Fattal, E.; Bochot, A. State of the art and perspectives for the delivery of antisense oligonucleotides and siRNA by polymeric nanocarriers. Int. J. Pharm. 2008, 364, 237–248. [Google Scholar] [CrossRef]
- Brand, R.M.; Iversen, P.L. Transdermal delivery of antisense compounds. Adv. Drug Deliv. Rev. 2000, 44, 51–57. [Google Scholar] [CrossRef]
- Arora, V.; Hannah, T.L.; Iversen, P.L.; Brand, R.M. Transdermal use of phospharadiamidate morpholino oligomer AVI-4472 inhibits cytochrome P450 3A2 activity in male rats. Pharm. Res. 2002, 19, 1465–1470. [Google Scholar] [CrossRef]
- Hsu, T.; Mitragotri, S. Delivery of siRNA and other macromolecules into skin and cells using a peptide enhancer. Proc. Natl. Acad. Sci. USA 2011, 108, 15816–15821. [Google Scholar] [CrossRef]
- Regnier, V.; Morre, N.D.; Jadoul, A.; Preat, V. Mechanism of a phosphorothioate oligonucleotide delivery by skin electroporation. Int. J. Pharm. 1999, 184, 147–156. [Google Scholar] [CrossRef]
- Tezel, A.; Dokka, S.; Kelly, S.; Hardee, G.E.; Mitragotri, S. Topical delivery of anti-sense oligonucleotides using low frequency sonophoresis. Pharm. Res. 2004, 21, 2219–2225. [Google Scholar] [CrossRef]
- Lee, W.R.; Shen, S.C.; Liu, C.R.; Fang, C.L.; Hu, C.H.; Fang, J.Y. Erbium:YAG laser-mediated oligonucleotide and DNA delivery via the skin: An animal study. J. Control. Release 2006, 115, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Cormier, M.; Samiee, A.; Griffin, A.; Johnson, B.; Teng, C.L.; Hardee, G.E.; Daddona, P.E. Transdermal delivery of antisense oligonucleotides with microprojection patch (MacrofluxR) technology. Pharm. Res. 2001, 18, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Venuganti, V.V.; Saraswathy, M.; Dwivedi, C.; Kaushik, R.S.; Perumal, O.P. Topical gene silencing by iontophoretic delivery of an antisense oligonucleotide-dendrimer nanocomplex: The proof of concept in a skin cancer mouse model. Nanoscale 2015, 7, 3903–3914. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.S.; Chen, D.; Zheng, X.; Zhang, X.; Johnston, N.; Liu, Y.; Yuan, K.; Koropatnick, J.; Gillies, E.R.; Min, W.P. Non-covalently functionalized single-walled carbon nanotube for topical siRNA delivery into melanoma. Biomaterials 2014, 35, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, N.; Rosenthal, R.; Furuse, M.; Moll, I.; Fromm, M.; Brandner, J.M. Contribution of Tight Junction Proteins to Ion, Macromolecule, and Water Barrier in Keratinocytes. J. Investig. Dermatol. 2013, 133, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Zakrewsky, M.; Kumar, S.; Mitragotri, S. Nucleic acid delivery into skin for the treatment of skin disease: Proofs-of-concept, potential impact, and remaining challenges. J. Control. Release 2015, 219, 445–456. [Google Scholar] [CrossRef]
- Regnier, V.; Le Doan, T.; Preat, V. Parameters controlling topical delivery of oligonucleotides by electroporation. J. Drug. Target. 1998, 5, 275–289. [Google Scholar] [CrossRef]
- White, P.J.; Gray, A.C.; Fogarty, R.D.; Sinclair, R.D.; Thumiger, S.P.; Werther, G.A.; Wraight, C.J. C-5 propyne-modified oligonucleotides penetrate the epidermis in psoriatic and not normal human skin after topical application. J. Investig. Dermatol. 2002, 118, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lishko, V.; Hoffman, R.M. Liposome targeting of high molecular weight DNA to the hair follicles of histocultured skin: A model for gene therapy of the hair growth processes. In Vitro Cell. Dev. Biol. Anim. 1993, 29, 258–260. [Google Scholar] [CrossRef]
- Lieb, L.M.; Liimatta, A.P.; Bryan, R.N.; Brown, B.D.; Krueger, G.G. Description of the intrafollicular delivery of large molecular weight molecules to follicles of human scalp skin in vitro. J. Pharm. Sci. 1997, 86, 1022–1029. [Google Scholar] [CrossRef]
- Bastian, B.C.; Kashani-Sabet, M.; Hamm, H.; Godfrey, T.; Moore, D.H., 2nd; Bröcker, E.B.; LeBoit, P.E.; Pinkel, D. Gene amplifications characterize acral melanoma and permit the detection of occult tumor cells in the surrounding skin. Cancer Res. 2000, 60, 1968–1973. [Google Scholar]
- North, J.P.; Kageshita, T.; Pinkel, D.; LeBoit, P.E.; Bastian, B.C. Distribution and significance of occult intraepidermal tumor cells surrounding primary melanoma. J. Invest. Dermatol. 2008, 128, 2024–2030. [Google Scholar] [CrossRef]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K.; Rachkovsky, M.L.; Orlow, S.J.; Bolognia, J.L.; Sodi, S.A. Altered N-glycosylation in macrophage x melanoma fusion hybrids. Cell. Mol. Biol. (Noisy-le-grand) 1999, 45, 1011–1027. [Google Scholar]
- Zbytek, B.; Carlson, J.A.; Granese, J.; Ross, J.; Mihm, M.C.; Slominski, A. Current concepts of metastasis in melanoma. Expert Rev. Dermatol. 2008, 3, 569–585. [Google Scholar] [CrossRef]
- Oberemok, V.V.; Laikova, K.V.; Zaitsev, A.S.; Shumskykh, M.N.; Kasich, I.N.; Gal’chinsky, N.V.; Bekirova, V.V.; Makarov, V.V.; Agranovsky, A.A.; Gushchin, V.A.; et al. Molecular Alliance of Lymantria dispar Multiple Nucleopolyhedrovirus and a Short Unmodified Antisense Oligonucleotide of Its Anti-Apoptotic IAP-3 Gene: A Novel Approach for Gypsy Moth Control. Int. J. Mol. Sci. 2017, 18, 2446. [Google Scholar] [CrossRef] [PubMed]
- Oberemok, V.V.; Laikova, K.V.; Zaitsev, A.S.; Nyadar, P.M.; Gninenko, Y.I.; Gushchin, V.A.; Makarov, V.V.; Agranovsky, A.A. Topical treatment of LdMNPV-infected gypsy moth caterpillars with 18 nucleotides long antisense fragment from LdMNPV IAP-3 gene triggers higher level of apoptosis in the infected cells and mortality of the pest. J. Plant Prot. Res. 2017, 57, 18–24. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laikova, K.V.; Oberemok, V.V.; Krasnodubets, A.M.; Gal’chinsky, N.V.; Useinov, R.Z.; Novikov, I.A.; Temirova, Z.Z.; Gorlov, M.V.; Shved, N.A.; Kumeiko, V.V.; et al. Advances in the Understanding of Skin Cancer: Ultraviolet Radiation, Mutations, and Antisense Oligonucleotides as Anticancer Drugs. Molecules 2019, 24, 1516. https://doi.org/10.3390/molecules24081516
Laikova KV, Oberemok VV, Krasnodubets AM, Gal’chinsky NV, Useinov RZ, Novikov IA, Temirova ZZ, Gorlov MV, Shved NA, Kumeiko VV, et al. Advances in the Understanding of Skin Cancer: Ultraviolet Radiation, Mutations, and Antisense Oligonucleotides as Anticancer Drugs. Molecules. 2019; 24(8):1516. https://doi.org/10.3390/molecules24081516
Chicago/Turabian StyleLaikova, Kateryna V., Volodymyr V. Oberemok, Alisa M. Krasnodubets, Nikita V. Gal’chinsky, Refat Z. Useinov, Ilya A. Novikov, Zenure Z. Temirova, Mikhail V. Gorlov, Nikita A. Shved, Vadim V. Kumeiko, and et al. 2019. "Advances in the Understanding of Skin Cancer: Ultraviolet Radiation, Mutations, and Antisense Oligonucleotides as Anticancer Drugs" Molecules 24, no. 8: 1516. https://doi.org/10.3390/molecules24081516