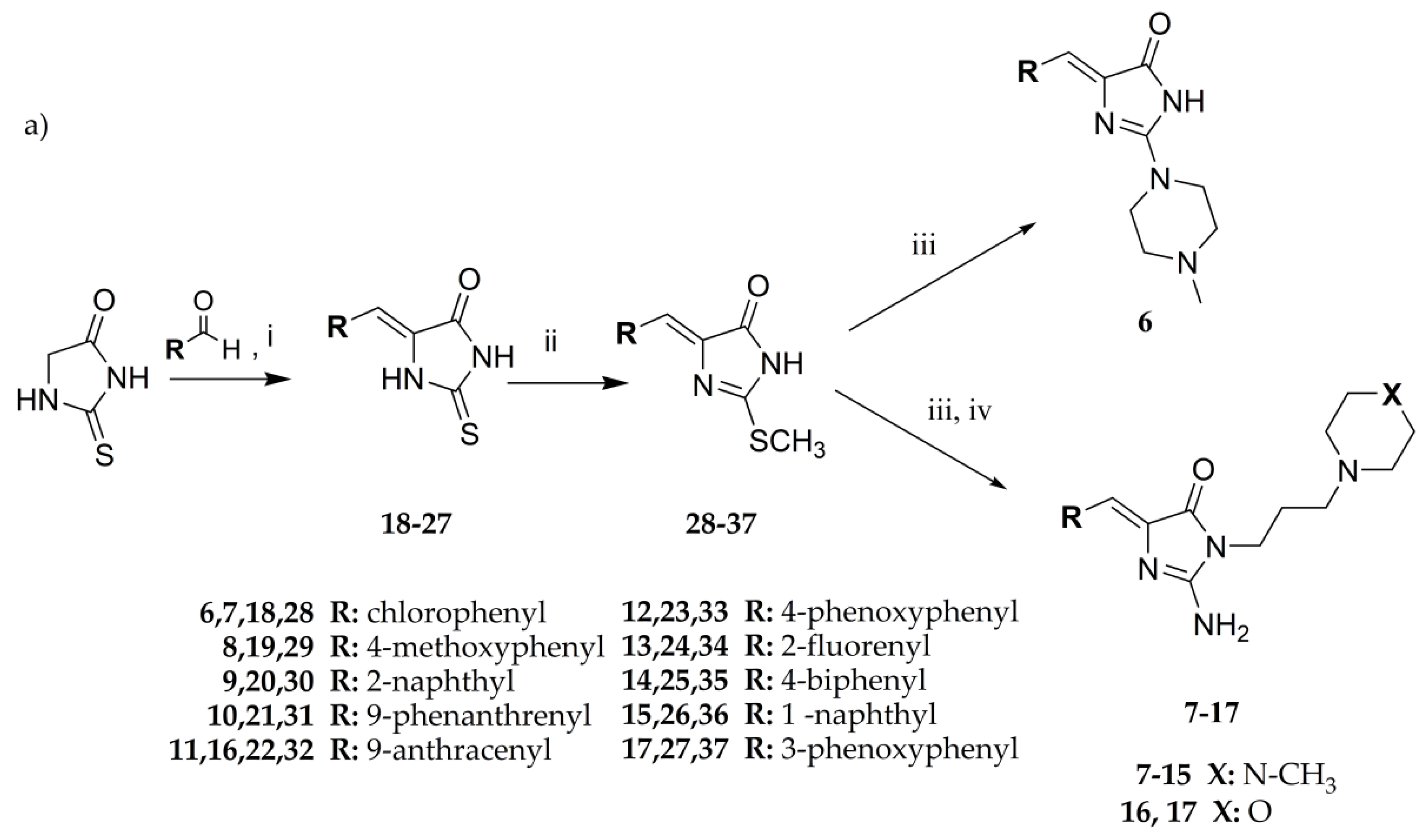
3.1.1. General Procedure to Obtain 5-Arylidenethiohydantoin (21, 23 and 24)
Thiohydantoin (2.90–5.80 g, 25–50 mmol), acetic acid (25–50 ml), sodium acetate (8.33–16.67 g, 100–200 mmol) with appropriate arylidene aldehyde (25–50 mmol) in flat-bottom flask were heated in boiling point for 4–6 h and then mixed for 20 h. Reaction was controlled by TLC-chloroform/ethyl acetate: 1/1. If necessary, purification was performed using crystallization from acetone or acetic acid.
(Z)-5-(phenanthren-9-ylmethylene)-2-thioxoimidazolidin-4-one (21) Phenanthrene-9-carbaldehyde (30.5 mmol, 6.29 g) and thiohydantoin (30.5 mmol, 3.54 g) were used. Yellow solid. Yield 99%; mp 283–285 °C. C18H12N2OS MW 304.37. LC/MS ±: purity 96.05% tR = 6.42, (ESI) m/z [M+H] 305.04. 1H-NMR δ [ppm]: 8.94–8.63 (m, 3H, N1-H, Ar-4,5-H), 8.30 (m, 1H, Ar-1-H), 8.03–7.87 (m, 1H, Ar-8-H), 7.80–7.61 (m, 4H, Ar-2,3,6,7-H), 7.12 (s, 1H, Ar-10-H), 6.76 (s, 1H, C = CH).
(Z)-5-(4-phenoxybenzylidene)-2-thioxoimidazolidin-4-one (23) 4-Phenoxybenzaldehyde (50 mmol, 9.91 g) and thiohydantoin (50 mmol, 5.81 g) were used. Yellow solid. Yield 81%; mp 233–235 °C. C16H12N2O2S MW 296.34. LC/MS ±: purity 99.11% tR = 6.52, (ESI) m/z [M+H]+ 297.07. 1H-NMR δ [ppm]: 12.00 (br.s, 1H, N3-H), 7.80–7.77 (d def., 2H, Ph-2,6-H), 7.41–7.39 (d def., 2H, Ph-3,5-H), 7.20–6.97 (m, 5H, Ph’-2,3,4,5,6-H), 6.42 (s, 1H, C = CH), 3.52 (br. s, 1H, taut. N1-H<->SH).
(Z)-5-((9H-fluoren-2-yl)methylene)-2-thioxoimidazolidin-4-one (24) 9H-Fluorene-2-carbaldehyde (25 mmol, 4.86 g) and thiohydantoin (25 mmol, 2.90 g) were used. Yellow solid. Yield 86%; mp 276–278 °C. C17H12N2OS MW 292.35. LC/MS ±: purity 94.88% tR = 6.45, (ESI) m/z [M+H]+ 293.01. 1H-NMR δ [ppm]: 12.38 (br.s, 1H, N3-H), 12.38 (br.s, 1H, N3-H), 12.22 (br.s, 1H, N1-H), 8.04 (s, 1H, Ar-1-H), 7.96–7.92 (m, 2H, Ar-4,5-H), 7.71 (d, J = 7.95 Hz, 1H, Ar-8-H), 7.60 (d, J = 6.92 Hz, 1H, Ar-3-H), 7.43–7.32 (m, 2H, Ar-6,7-H), 6.57 (s, 1H, CH = C), 3.96 (s, 2H, Ar-9-CH2).
3.1.2. General Procedure to Obtain 2-Methylthio-5-Arylidenethiohydantoins (31, 33, 34 and 37)
Sodium (0.48–0.87 g, 21.00–37.77 mmol) was put into ethanol (21.00–37.77 mL). Sodium ethoxide mixed with appropriate 5–arylidenethiohydantion (21.00–37.77 mmol) in flat–bottom flask for 3 min. Then, iodomethane (2.98–5.36 g, 21.00–37.77 mmol) was added and whole were mixed for 5–24 h. Reaction was controlled by TLC-chloroform/ethyl acetate: 1/1. Purification was performed using crystallization from acetone wherever necessary.
(Z)-2-(methylthio)-4-(phenanthren-9-ylmethylene)-1H-imidazol-5(4H)-one (31) (Z)-5-(Phenanthren-9-ylmethylene)-2-thioxoimidazolidin-4-one (21) (26.00 mmol, 7.91 g) with iodomethane (26.00 mmol, 3.69 g) was used. Yellow solid. Yield 99%; mp 271–274 °C. C19H14N2OS MW 318.39. LC/MS±: purity 97.70% tR = 6.77, (ESI) m/z [M+H]+ 319.06. 1H-NMR δ [ppm]: 9.24–9.10 (m, 1H, N3-H), 8.90–8.81 (m, 2H, Ar-4,5-H), 8.36–8.29 (m, 1H, Ar-8-H), 8.02–7.99 (m, 1H, Ar-1-H), 7.67–7.40 (m, 5H, Ar-2,3,6,7,10-H), 6.87 (m, 1H, C = CH), 2.71 (s, 3H, S-CH3).
(Z)-4-(4-phenoxybenzylidene)-2-(methylthio)-1H-imidazol-5(4H)-one (33) (Z)-5-(4-Phenoxybenzylidene)-2-thioxoimidazolidin-4-one (23) (37.00 mmol, 10.96 g) with iodomethane (37.00 mmol, 5.25 g) was used. Yellow solid. Yield 92%; mp 177–179 °C. C17H14N2O2S MW 310.37. LC/MS±: purity 96.15% tR = 7.39, (ESI) m/z [M+H]+ 311.09. 1H-NMR δ [ppm]: 11.80 (br.s, 1H, N3-H), 8.29–8.18 (d def., 2H, N3-H, Ph-2,6-H), 7.44–7.38 (t def., 2H, Ph-3,5-H), 7.21–7.13 (m, 1H, Ph’-4-H), 7.09–6.99 (m, 4H, Ph’-2,3,5,6-H), 6.71 (s, 1H, C = CH), 2.62 (s, 3H, S-CH3).
(Z)-5-((9H-fluoren-2-yl)methylene)-2-(methylthio)-3H-imidazol-4(5H)-one (34) (Z)-5-((9H-Fluoren-2-yl)methylene)-2-thioxoimidazolidin-4-one (24) (21.00 mmol, 6.14 g) with iodomethane (21.00 mmol, 2.98 g) was used. Yellow solid. Yield 90%; mp 240–242 °C. C18H14N2OS MW 306.38. LC/MS±: purity 91.26% tR = 7.58, (ESI) m/z [M+H]+ 307.10. 1H-NMR δ [ppm]: 8.39 (s, 1H, Ar-1-H), 8.27–8.18 (d def., 1H, Ar-8-H), 7.99–7.87 (m, 2H, Ar-4,5-H), 7.64–7.54 (d def., 1H, Ar-3-H), 7.45–7.20 (m, 3H, Ar-6,7-H), 6.80 ( s, 1H, CH = C), 3.96 (s, 2H, Ar-9-CH2), 2.70 (s, 3H, SCH3).
(Z)-5-(3-phenoxybenzylidene)-2-(methylthio)-3H-imidazol-4(5H)-one (37) (Z)-5-(3-Phenoxybenzylidene)-2-thioxoimidazolidin-4-one (27) (37.77 mmol, 11.19 g) with iodomethane (37.77 mmol, 5.36 g) was used. Yellow solid. Yield 95%; mp 214–216 °C. C17H14N2O2S MW 310.37. LC/MS±: purity 95.24% tR = 7.32, (ESI) m/z [M+H]+ 311.02. 1H-NMR δ [ppm]: 11.80 (s, 1H, N3-H), 8.06 (s, 1H, Ph-2-H), 7.64–7.61 (d def., 1H, Ph-6-H), 7.43–7.38 (m, 3H, Ph-5-H, Ph’-3,5-H), 7.19–7.05 (m, 4H, Ph-4-H, Ph’-2,4,6-H), 6.67 (s, 1H, C = CH), 2.35 (s, 3H, S-CH3).
3.1.3. General Procedure for Synthesis of Final Products (7–17)
(Z)-2-(methylthio)-5-arylidene-3H-imidazol-4(5H)-one (2–10 mmol) with appropriate amine (3–15 mmol) derivative in 50 mL flat-bottom flask were heated in oil bath with controlled temperature (120–130 °C) for 15 min. Then, ethanol (15-30 ml) was added and mixture was heated for 5–7 h and mixed for the next 20 h. Then, compounds were converted into hydrochloride forms by gaseous hydrochloride acid obtained from reaction between sodium chloride and sulphuric acid. Purification was performed using crystallization from ethanol wherever necessary.
(Z)-5-(4-chlorobenzylidene)-2-amino-3-(3-(4-methylpiperazin-1-yl)propyl)-3H-imidazol-4(5H)-one hydrochloride (7); (Z)-5-(4-Chlorobenzylidene)-2-(methylthio)-3H-imidazol-4(5H)-one (28) (10 mmol, 2.53 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (15 mmol, 2.36 g) were used. White solid. Yield 7.5%; mp 294 °C. C18H24ClN5Ox3HClxH2O MW 486.25. LC/MS ±: purity 100.00% tR = 3.34, (ESI) m/z [M+H]+ 362.26. 1H-NMR δ [ppm]: 11.82 (br. s, 1H, NH+), 9.22 (br. s, 2H, NH2-taut.: N1-H, C2 = NH), 7.84 (d, J = 8.46 Hz, 2H, Ar-2,6-H), 7.49 (d, J = 8.72 Hz, 2H, Ar-3,5-H), 6.77 (s, 1H, CH = C), 3.78 (t, J = 6.41 Hz, 2H, N3-CH2), 3.48 (br. s, 8H, Pip), 3.19 (br. s, 2H, Pip-CH2), 2.80 (br. s, 3H, CH3), 2.02 (br. s, 2H, N-CH2-CH2).
(Z)-5-(4-Methoxybenzylidene)-2-amino-3-(3-(4-methylpiperazin-1-yl)propyl)-3H-imidazol-4(5H)-one hydrochloride (8); (Z)-5-(4-Methoxybenzylidene)-2-(methylthio)-3H-imidazol-4(5H)-one (29) (10 mmol, 2.46 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (15 mmol, 2.36 g) were used. Yellow solid. Yield 15%; mp 262 °C. C19H27N5O2x3HCl·0.5H2O MW 475.93. LC/MS±: purity 100.00% tR = 2.58, (ESI) m/z [M+H]+ 358.34. 1H-NMR δ [ppm]: 11.8 (br. s, 1H, NH+), 9.5 (br. s, 2H, NH2-taut.: N1-H, C2 = NH), 7.76 (d, J = 8.72 Hz, 2H, Ar-2,6-H), 7.01 (d, J = 8.72 Hz, 2H, Ar-3,5-H), 6.84 (s, 1H, CH = C), 3.81 (s, 3H, O-CH3), 3.70–3.20 (m, 12H, Pip, Pip-CH2, N3-CH2), 2.79 (s, 3H, CH3), 2.00 (br. s, 2H, N-CH2-CH2).
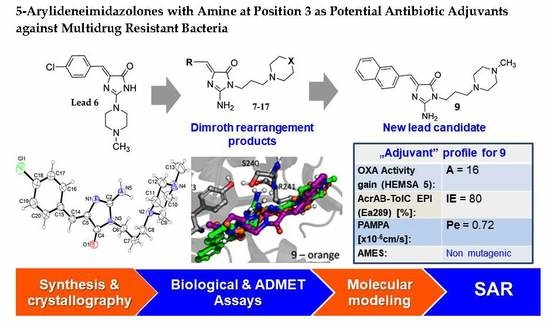
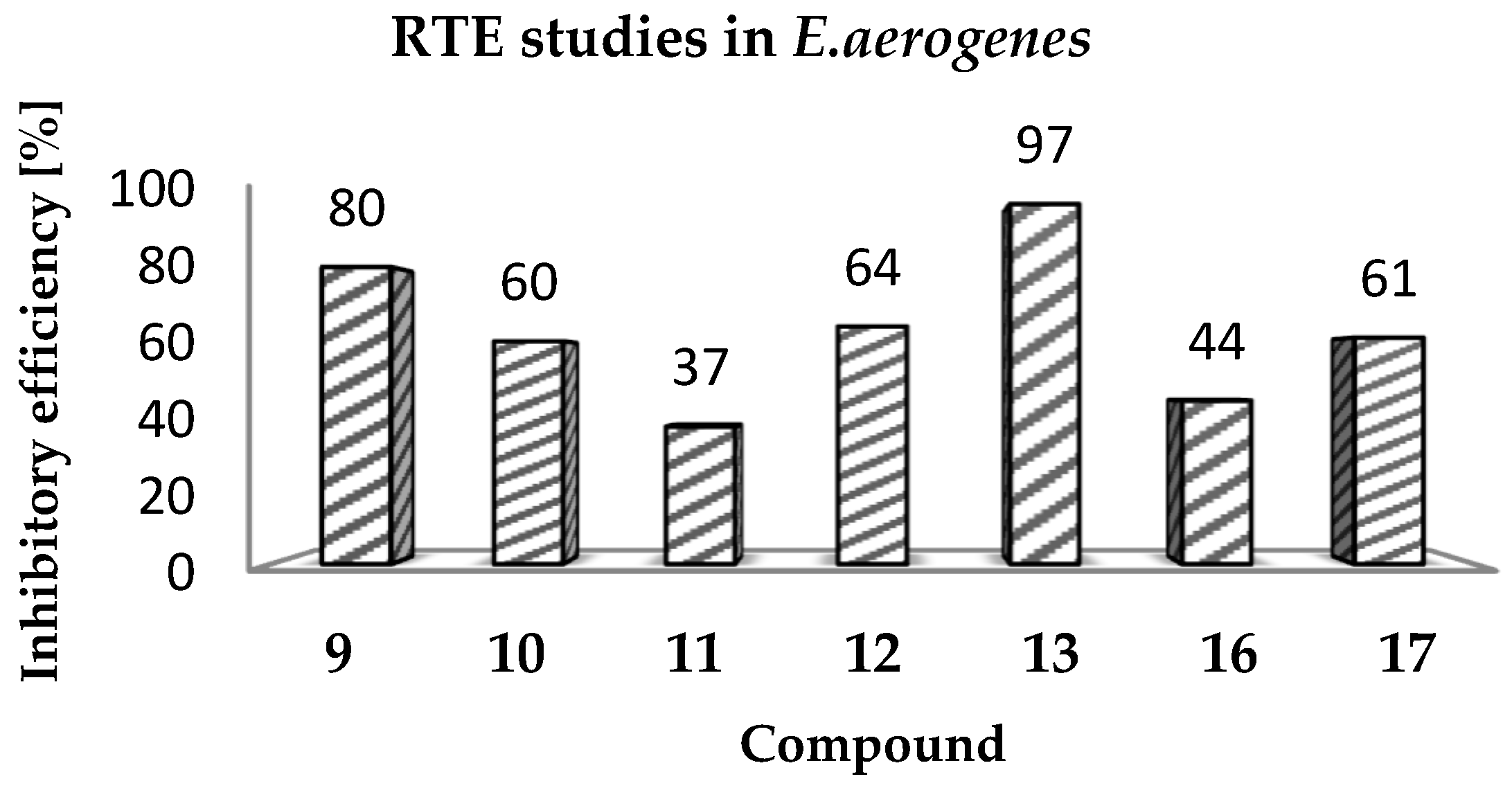
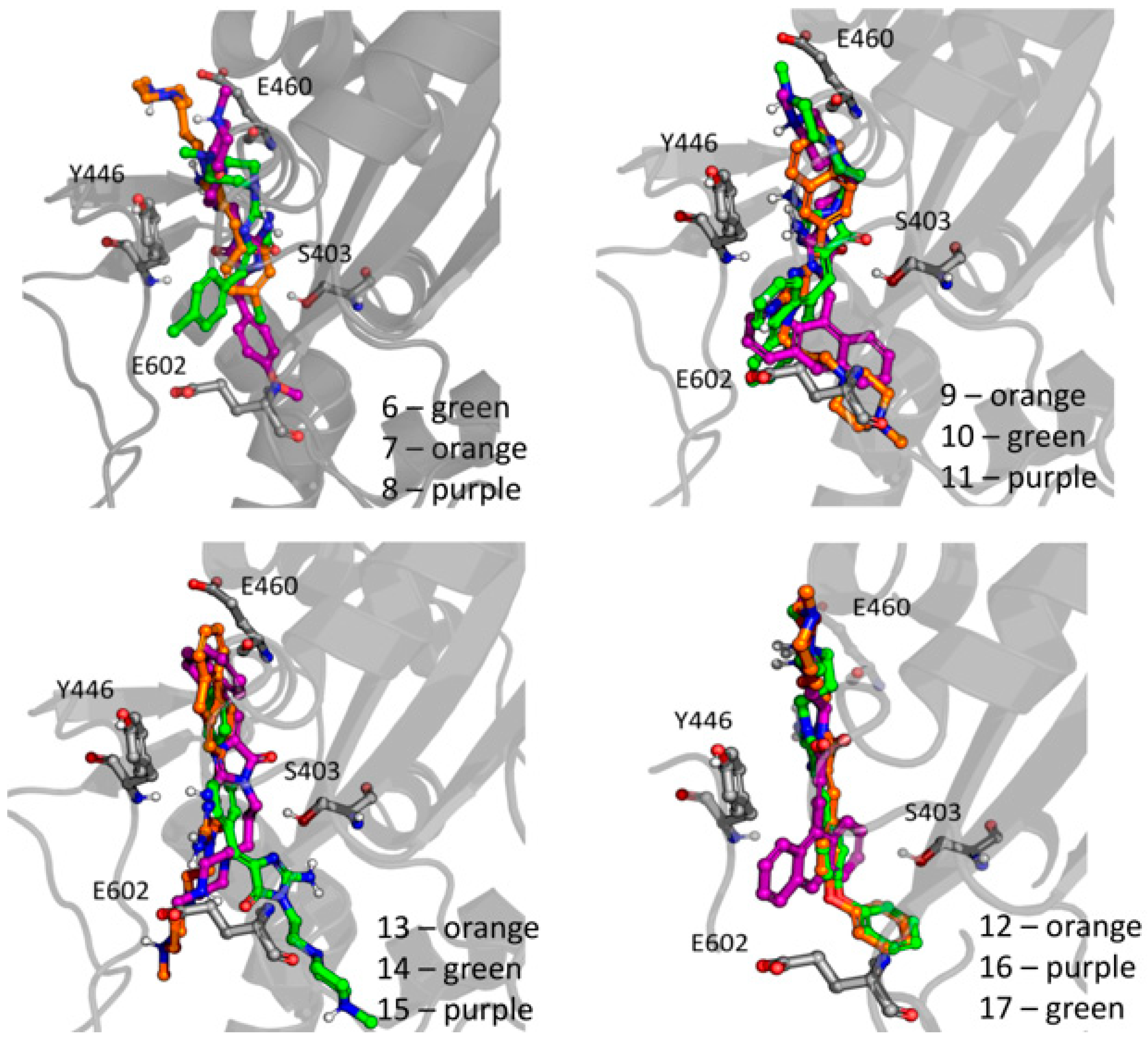
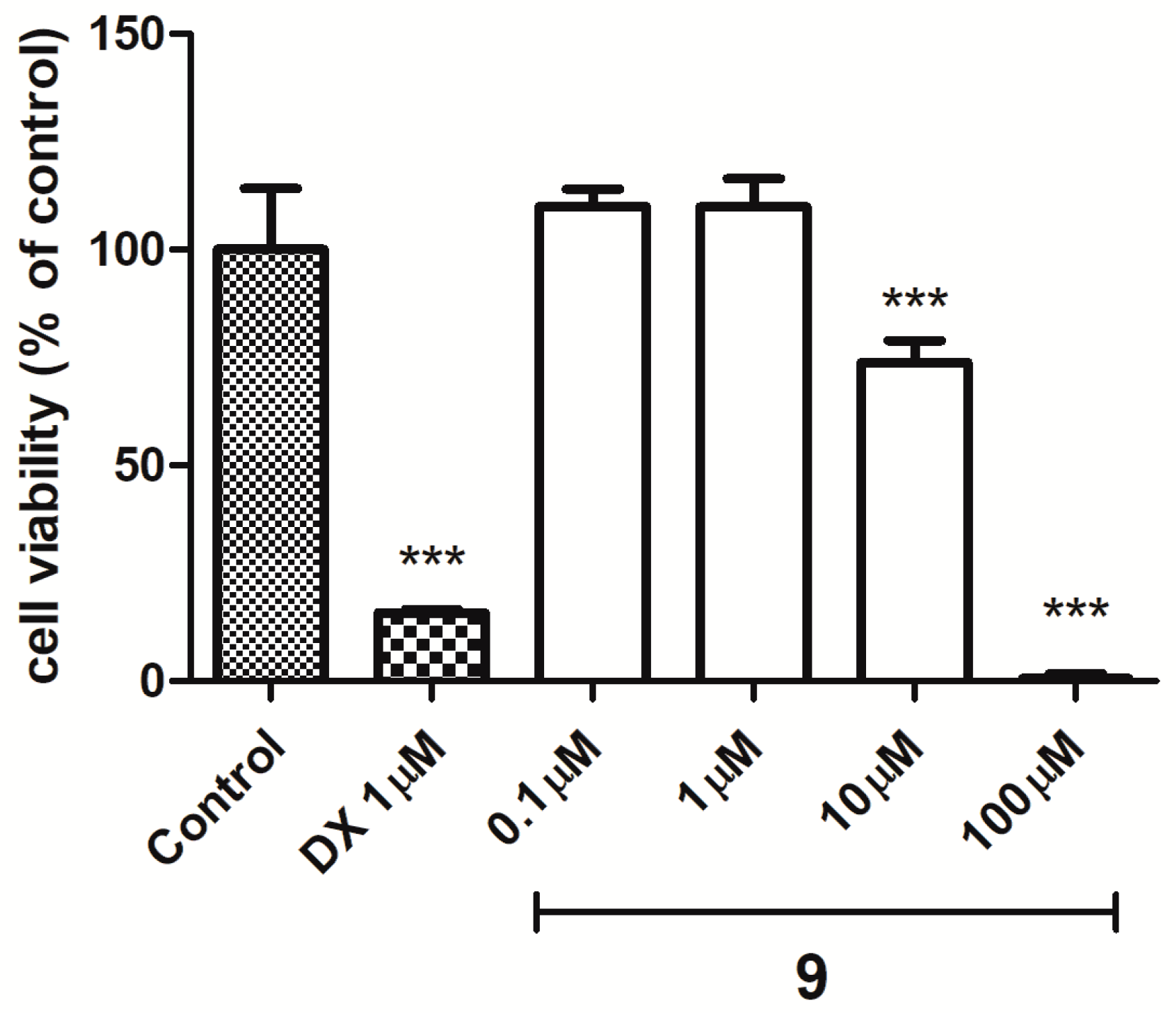
(Z)-2-Amino-3-(3-(4-methylpiperazin-1-yl)propyl)-5-(naphthalen-2-ylmethylene)-3H-imidazol-4(5H)-one hydrochloride (9) (Z)-2-(methylthio)-4-(naphthalen-2-ylmethylene)-1H-imidazol-5(4H)-one (30) (5 mmol, 1.34 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (10 mmol, 1.57 g) were used. Yellow solid. Yield 74%; mp 215-217 °C. C22H28ClN5O MW 413.94. LC/MS±: purity 100.00% tR = 3.34, (ESI) m/z [M+H]+ 378.28. 1H-NMR δ [ppm]: 8.39–8.34 (m, 2H, Ar-5,8-H), 7.85–7.78 (m, 5H, NH2, Ar-1,3,4-H), 7.47–7.45 (m, 2H, Ar-6,7-H), 6.50 (s, 1H, C = CH), 3.54 (t, J = 6.40Hz, N3-CH2), 2.45–2.20 (m, 10H, Pip, Pip-CH2), 2.09 (s, 3H, CH3), 1.68 (qui, J = 6.40Hz, 2H, N-CH2-CH2).
(Z)-2-Amino-3-(3-(4-methylpiperazin-1-yl)propyl)-5-(phenanthren-9-ylmethylene)-3H-imidazol-4(5H)-one hydrochloride (10) (Z)-2-(Methylthio)-5-(phenanthren-9-ylmethylene)-3H-imidazol-4(5H)-one (31) (2 mmol, 0.62 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (3 mmol, 0.47 g) were used. Yellow solid. Yield 17%; mp 266–268 °C. C26H30ClN5O MW 464.00. LC/MS±: purity 99.00% tR = 4.03, (ESI) m/z [M+H]+ 428.19. 1H-NMR δ [ppm]: 8.89–8.86 (m, 2H, Ar-1,10-H), 8.80–8.78 (m, 2H, Ar-4,7-H), 8.24–8.21 (m, 1H, Ar-6-H), 7.89 (s, 2H, NH2), 7.73–7.69 (m, 2H, Ar-2,5-H), 7.68–7.64 (m, 2H, Ar-3,8-H), 6.93 (s, 1H, C = CH), 3.60–3.20 (m, 2H, N3-CH2), 2.45–2.15 (m, 10H, Pip, Pip-CH2), 2.09 (s, 3H, CH3), 1.75 (m, 2H, N-CH2-CH2).
(Z)-2-Amino-5-(anthracen-10-ylmethylene)-3-(3-(4-methylpiperazin-1-yl)propyl)-3H-imidazol-4(5H)-one hydrochloride (11) (Z)-5-(Anthracen-10-ylmethylene)-2-(methylthio)-3H-imidazol-4(5H)-one (32) (4 mmol, 1.71 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (7.5 mmol, 1.18 g) were used. Yellow solid. Yield 61%; mp 246-248 °C. C26H30ClN5O MW 464.00. LC/MS±: purity 100.00% tR = 3.89, (ESI) m/z [M+H]+ 428.26. 1H-NMR δ [ppm]: 8.61 (br. s, 1H, Ar-5-H), 8.11-8.01 (m, 6H, Ar-1,4,6,9-H, NH2), 7.53–7.52 (m, 4H, Ar-2,3,7,8-H), 7.00 (s, 1H, C = CH), 3.40–3.15 (m, 2H, N3-CH2), 2.56–2.06 (m, 13H, Pip, Pip-CH2, CH3), 1.58–1.52 (m, 2H, N-CH2-CH2).
(Z)-5-(4-Phenoxybenzylidene)-2-amino-3-(3-(4-methylpiperazin-1-yl)propyl)-3H-imidazol-4(5H)-one (12) (Z)-5-(4-Phenoxybenzylidene)-2-(methylthio)-1H-imidazol-5(4H)-one (33) (4 mmol, 1.19 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (5 mmol, 0.78 g) were used. Yellow solid. Yield 68%; mp 220–230 °C. C24H30ClN5O2 MW 455.98. LC/MS±: purity 100.00% tR = 4.1, (ESI) m/z [M+H]+ 420.28. 1H-NMR δ [ppm]: 7.62–7.57 (m, 3H, NH2, Ph’-4-H), 7.46–7.36 (m, 4H, Ph-2,6-H, Ph’-3,5-H), 7.15–6.95 (m, 4H, Ph-3,5-H, Ph’-2,6-H), 6.72 (s, 1H, C = CH), 3.77 (br. s, 2H, N3-CH2), 3.70–3.14 (m, 10H, Pip, Pip-CH2), 2.79 (s, 3H, CH3), 1.99 (br. s, 2H, N-CH2-CH2). 13C-NMR (DMSO-d6, ppm): δ 157.23, 156.98, 130.56, 124.01, 118.93, 40.77, 40.49, 40.20, 39.92, 39.64, 39.36, 39.08.
(Z)-5-((9H-Fluoren-2-yl)methylene)-2-amino-3-(2-(4-methylpiperazin-1-yl)ethyl)-3H-imidazol-4(5H)-one hydrochloride (13) (Z)-5-((9H-Fluoren-2-yl)methylene)-2-(methylthio)-3H-imidazol-4(5H)-one (34) (3.5 mmol, 1.09 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (5 mmol, 0.78 g) were used. Orange solid. Yield 69.27%; mp 265–267 °C. C25H30ClN5O MW 451.99. LC/MS±: purity 99% tR = 3.92, (ESI) m/z [M+H]+. 1H-NMR δ [ppm]: 12.00 (br. s, 1H, NH+), 9.35 (br. s, 1H, NH2-taut: N1-H), 8.32–7.70 (m, 4H, NH2-taut: C2 = NH, Ar-1,4,5-H), 7.67–7.55 (m, 2H, Ar-3,8-H), 7.49–7.24 (m, 2H, Ar-6,7-H), 6.82 (br. s, 1H, CH = C), 4.08 (s, 2H, Ar-9-CH2), 3.90–3.01 (m, 12H, N3-CH2, Pip, Pip-CH2), 2.90 (s, 3H, CH3), 2.06 (m, 2H, N-CH2-CH2). 13C-NMR (DMSO-d6, ppm): δ 144.20, 143.96, 127.42, 126.88, 125.74, 125.51, 121.18, 121.00, 40.94, 40.89, 40.52, 40.24, 39.68, 39.39, 39.24, 38.95.
(Z)-2-Amino-5-(biphen-4-ylmethylene)-3-(2-(4-methylpiperazin-1-yl)ethyl)-3H-imidazol-4(5H)-one hydrochloride (14) (Z)-5-(Biphen-4-ylmethylene)-2-(methylthio)-3H-imidazol-4(5H)-one (35) (2.5 mmol, 0.72 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (4 mmol, 0.63 g) were used. Yellow solid. Yield 49%; mp 258–260 °C. C24H30ClN5O MW 439.98. LC/MS±: purity 96.39% tR = 3.78, (ESI) m/z [M+H]+. 1H-NMR δ [ppm]: 12.00 (br. s, 1H, NH+), 8.24–8.05 (t def., 1H, Ph’-4-H), 7.80 (br. s, 2H, NH2), 7.79–7.61 (m, 4H, Ph’-3,5-H, Ph-2,6-H), 7.52–7.30 (m, 4H, Ph’-2,6-H, Ph-3,5-H), 6.83 (br. s, 1H, CH = C), 4.26–2.97 (m, 12H, Pip, Pip-CH2, N3-CH2), 2.80 (s, 3H, CH3), 2.04 (br. s, 2H, N-CH2-CH2). 13C-NMR (DMSO-d6, ppm): δ 129.51, 127.11, 40.77, 40.49, 40.21, 39.92, 39.64, 39.36, 39.08.
(Z)-2-Amino-3-(3-(4-methylpiperazin-1-yl)propyl)-5-(naphthalen-1-ylmethylene)-3H-imidazol-4(5H)-one hydrochloride (15) (Z)-2-(Methylthio)-5-(naphthalen-1-ylmethylene)-3H-imidazol-4(5H)-one (36) (5 mmol, 1.34 g) and 3-(4-methylpiperazin-1-yl)propan-1-amine (7 mmol, 1.09 g) were used. Yellow solid. Yield 24%; mp 187–191 °C. C22H28ClN5O MW 413.94. LC/MS±: purity 99.07% tR = 3.21, (ESI) m/z [M+H]+ 378.21. 1H-NMR δ [ppm]: 8.83 (br. s, 1H, NH+), 8.19 (br. s, 2H, taut NH2), 7.96–7.88 (d def., 1H, Ar-8-H), 7.85–7.75 (d def., 2H, Ar-4,5-H), 7.61–7.44 (m, 4H, Ar-2,3,6,7-H), 6.98 (br. s, 1H, CH = C), 3.32 (br. s, 2H, N3-CH2), 2.47–2.17 (m, 10H, Pip, Pip-CH2), 2.12 (s, 3H, CH3), 1.70 (br. s, 2H, N-CH2-CH2). 13C-NMR (DMSO-d6, ppm): δ 133.79, 131.93, 131.62, 129.16, 128.53, 127.90, 126.89, 126.19, 126.05, 123.27, 55.09, 53.02, 46.12, 40.81, 40.52, 40.24, 39.96, 39.68, 39.20, 39.11.
(Z)-2-Amino-5-(anthracen-10-ylmethylene)-3-(3-morpholinopropyl)-3H-imidazol-4(5H)-one hydrochloride (16) (Z)-5-(Anthracen-10-ylmethylene)-2-(methylthio)-3H-imidazol-4(5H)-one (33) (4 mmol, 1.22 g) and 3-morpholinopropan-1-amine (5 mmol, 0.72 g) were used. Orange solid. Yield 70%; mp 244–246 °C. C25H27ClN4O2 MW 450.96. LC/MS±: purity 98.71% tR = 4.19, (ESI) m/z [M+H]+ 415.17. 1H-NMR δ [ppm]: 11.25 (br. s, 1H, NH+), 10.21 (br. s, 1H, NH2-taut: N1-H), 9.39 (br. s, 1H, NH2-taut: C2 = NH), 8.72 (s, 1H, Ar-9-H), 8.25–7.95 (m, 4H, Ar-1,4,5,8-H), 7.68–7.43 (m, 5H, Ar-2,3,6,7-H, CH = C), 4.16–2.69 (m, 10H, Mor, N3-CH2), 2.12 (br.s, 2H, Mor-CH2), 1.80 (s, 2H, N-CH2-CH2). 13C-NMR (DMSO-d6, ppm): δ 131.49, 129.42, 129.28, 126.77, 125.96, 66.60, 55.99, 53.71, 40.79, 40.50, 40.22, 39.94, 39.66, 39.37, 39.09.
(Z)-5-(3-phenoxybenzylidene)-2-amino-3-(3-morpholinopropyl)-3H-imidazol-4(5H)-one hydrochloride (17) (Z)-5-(3-Phenoxybenzylidene)-2-(methylthio)-3H-imidazol-4(5H)-one (37) (5 mmol, 1.55 g) and 3-morpholinopropan-1-amine (7 mmol, 1.01 g) were used. Yellow solid. Yield 41%; mp 227–230 °C. C23H27ClN4O3 MW 442.94. LC/MS±: purity 96.49% tR = 4.37, (ESI) m/z [M+H]+ 407.19. 1H-NMR δ [ppm]: 11.35 (br. s, 1H, NH+), 9.45 (br. s, 1H, NH2-taut: N1-H), 7.82-7.27 (m, 6H, NH2-taut: C2 = NH, Ph-5,6-H, Ph’-3,4,5-H), 7.26–6.85 (m, 4H, Ph-2,4-H, Ph’-2,6-H), 6.74 (br. s, 1H, CH = C), 4.03 (br. s, 4H, Mor-OCH2), 3.66–2.65 (m, 8H, N3-CH2, Mor-NCH2, Mor-CH2), 2.16–1.88 (t def., 2H, N-CH2-CH2). 13C-NMR (DMSO-d6, ppm): δ 157.67, 157.41, 130.55, 124.13, 118.97, 63.54, 51.44, 40.81, 40.53, 40.25, 39.97, 39.68, 39.40, 39.12.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











