
Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives


1
Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
2
Department of Chemistry and Biochemistry, Utah State University, Logan, UT 84322-0300, USA
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(2), 376; https://doi.org/10.3390/molecules24020376
Submission received: 5 January 2019
/
Revised: 18 January 2019
/
Accepted: 18 January 2019
/
Published: 21 January 2019
(This article belongs to the Special Issue σ and π Holes: A New Class of Non-Covalent Interactions)
Abstract
:When an N-base approaches the tetrel atom of TF4 (T = Si, Ge, Sn) the latter molecule deforms from a tetrahedral structure in the monomer to a trigonal bipyramid. The base can situate itself at either an axial or equatorial position, leading to two different equilibrium geometries. The interaction energies are considerably larger for the equatorial structures, up around 50 kcal/mol, which also have a shorter R(T··N) separation. On the other hand, the energy needed to deform the tetrahedral monomer into the equatorial structure is much higher than the equivalent deformation energy in the axial dimer. When these two opposite trends are combined, it is the axial geometry which is somewhat more stable than the equatorial, yielding binding energies in the 8–34 kcal/mol range. There is a clear trend of increasing interaction energy as the tetrel atom grows larger: Si < Ge < Sn, a pattern which is accentuated for the binding energies.
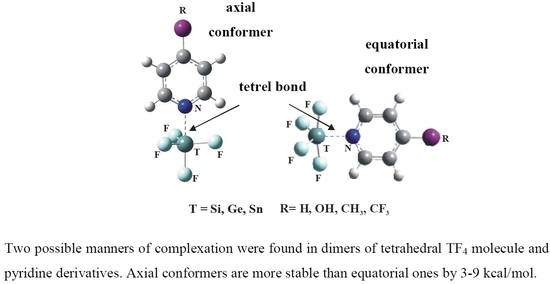
1. Introduction
The continuing development of modern chemistry is leading to improved understanding of microscale processes. An important part of this knowledge accrues to the rapidly accelerating understanding of noncovalent interactions. necessary to describe chemical and biological events occurring in living organisms as well as in vitro environment [1,2,3]. While the H-bond is certainly the most common and best understood noncovalent bond, it is becoming increasingly recognized that there are very similar interactions in which the bridging H is replaced by any of a wide range of other atoms. For example, the very electronegative halogen atoms may play the role of bridge in the eponymous bonds [4,5,6,7,8,9]. A central tenet of halogen and related bonds is the σ-hole [10,11,12,13,14,15] which refers to an electron-depleted region that lies directly opposite a covalent bond. With its positive electrostatic nature, this hole can attract a nucleophile [14,16,17], and the entire interaction is supplemented by polarization and dispersion forces. This idea is not limited to halogens, but has been extended to other electronegative atoms [18,19,20] and has recently expanded to transition metal interactions with Lewis bases, sometimes referred to as a “regium bond” [21]. It is not only the plane of the molecule which can house a positive region. When this phenomenon occurs above the plane of a molecule, it is commonly dubbed a π-hole [22,23,24].
The tetrel family of atoms (C, Si, Ge, etc.) is amongst those that can act as bridges [25,26,27,28] in analogies to H-bonds. The importance of tetrel bonds has been explored in a variety of forums including the SN2 reaction [29], stabilization of crystal structures [30,31], ability to serve as anion receptors [32], complexation of carbon dioxide with azoles [33] or cyclopenta- and cyclohexatetrelanes with selected anions [34]. In the biological realm [35] this sort of bond appears to be common in proteins [36,37] and involved in the enzymatic activity of S-adenosylmethionine-dependent methyltransferases [38]. Tetrel bonds were also considered in terms of their cooperativity [39], characterization in the solid-state [40], and interactions in general biological systems [41].
Halogen, chalcogen, and pnicogen atoms are typically covalently bonded to one, two, and three substituents, respectively. They thus offer little in the way of steric crowding to hinder the approach of a nucleophile. However, calculations have shown [42] how the larger number of substituents that accompany hypervalency of any of these sorts of central atoms can offer an impediment to noncovalent bonding and complicate the analysis of contributing factors. Even in its normal bonding situation, the tetrel atom is surrounded by four substituents. In such a case, the Lewis acid must significantly deform in order to provide a space for the nucleophile, accompanied by large deformation energies. This rearrangement has been shown to dramatically affect the molecule’s electrostatic potential, with ensuing repercussions for the interaction with a base.
These considerations lead to a central question. Can the internal rearrangements within a tetrel-containing molecule lead to alternative geometries for the complex with the base? While it is understood that a σ-hole is common along the extension of a R–T bond (where T and R represent a tetrel atom and a neighboring substituent atom) what would happen as the normal tetrahedral arrangement of the TR4 molecule is deformed so as to accommodate the base? A planar D4h sort of arrangement would preclude any σ-holes, leaving only the possibility of a π-hole. However, such a planar bonding situation is unlikely given its very high energy. If the TR4 were to remodel itself toward a trigonal bipyramid, as seems more likely, the vacant fifth site, awaiting the nucleophile, could lie at either an axial or equatorial position. The former represents the most common situation observed to date, wherein the base lies directly opposite one T–R bond, along a σ-hole, with the other three bonds occupying what might be termed equatorial sites. But one could certainly envision an alternative scenario, where the base occupies a vacant equatorial position. It is the goal of this work to consider these two options, to identify which is the more stable, and to understand the reasons for this preference. Another objective is the ability to predict in advance what particular sorts of systems will tend toward one geometry over the other. What conditions are necessary for both of these structures to coexist, and which pairs of molecules will only be present as one or the other?
In order to examine a range of different tetrel atoms, perfluorinated SiF4, GeF4, and SnF4 were taken as the Lewis acids. Pyridine and several of its p-substituted derivatives were used as Lewis base. Methyl and OH substituents were used to assess effects of electron-donating groups, and electron-withdrawing CF3 to analyze the opposite influence.
2. Results
2.1. Electrostatic Potentials of Monomers
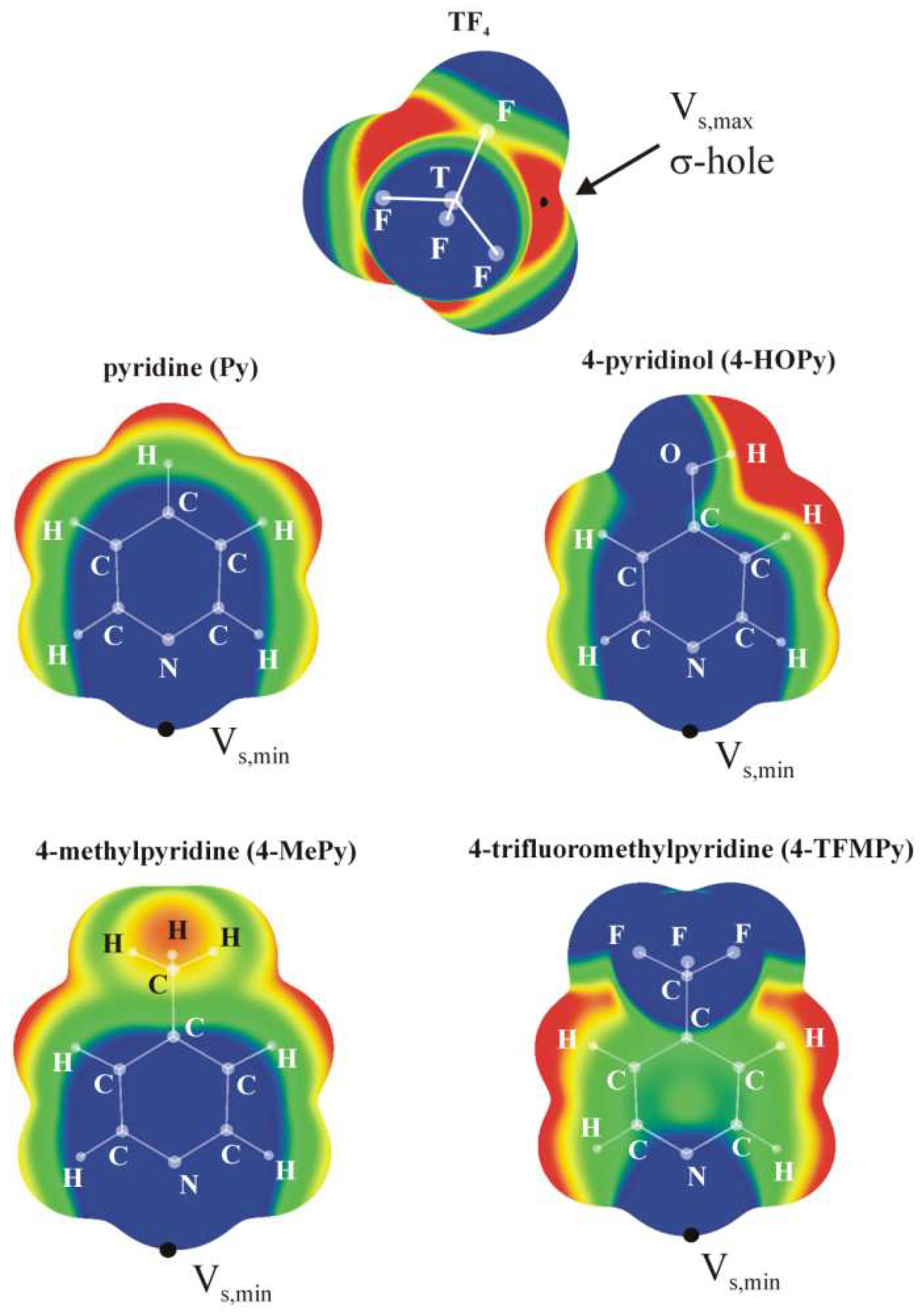
The molecular electrostatic potential (MEP) of the various TF4 monomers all have the same characteristics, as displayed in the top part of Figure 1. Within the context of a tetrahedral geometry there is a negative region surrounding each of the F atoms, and a positive area that lies directly opposite each T–F bond, known generally as a σ-hole. The magnitude of this hole can be characterized by the value of the maximum on a surface of isodensity, Vs,max, in this case ρ = 0.001 au. The values of Vs,max listed in Table 1 rise as the tetrel atom enlarges from a minimum of 41.3 kcal/mol for T = Si to a maximum of 70.1 kcal/mol for Sn. The MEPs of the four Lewis base derivatives of pyridine are also presented in Figure 1, and all contain a particularly negative region that is coincident with the N lone pair direction. As may be noted in the lower half of Table 1, the minimum on the 0.001 a.u. isodensity surface lies in the range between −30 and −38 kcal/mol; it is smallest for the R = CF3 substituent, which can be connected with the electron-withdrawing capacity of this species.
2.2. Structures of Complexes
The approach of the Lewis base to the originally tetrahedral TF4 can be thought of as the addition of a fifth ligand. The complex thus takes on some of the geometric characteristics of the penta-coordinated trigonal bipyramid. As such, the base can occupy either an axial or equatorial position on this framework. For most of the systems considered here, both such positions represent a minimum on the potential energy surface. These two geometries are displayed in Figure 2 and are denoted according to the axial or equatorial position of the base. The geometric aspects of the optimized complexes are reported in Table 2. As expected, the N atom lies very nearly directly opposite the Fax atom in the axial complexes, with θ (N··TFax) close to 180°. The angle between the N and the three equatorial F atoms is a bit less than the idealized 90° in a perfect trigonal bipyramid, due to the longer R (T··N) distance, as compared to R (T–Fax). The latter angle is of course quite different than the 109.5° in the uncomplexed tetrahedral TF4, suggesting a substantial monomer deformation upon complexation. With regard to the equatorial complexes, the N atom lies very close to 120° from the two equatorial F atoms. The two axial F atoms are both bent in a bit towards the N atom, leaving the θ (N··TFax) angle less than the idealized 90° by some 5–7°.
The axial complex is preferred over the equatorial structure in every case, and the margin of this energetic difference is contained in the last column of Table 2. This difference varies between 2.7 and 9.4 kcal/mol. It is greatest for Si and smallest for Sn, but shows little dependence upon the pyridine substituent R. It might finally be noted that in the single case of the 4-TFMPy∙∙∙SiF4 complex, with T = Si and R = CF3, there is no minimum corresponding to the equatorial geometry.
One may glean some insights into the energetics of the final complexes by consideration of the characteristics of the TF4 monomer, when organized in the trigonal bipyramid structure to which it tends within the complex. That is, each monomer was optimized but with the θ (F–T–F) angles characteristic of the trigonal bipyramid (90°, 120°, 180°) but with a vacancy in either an axial or equatorial position. There is a marked preference for an axial vacancy, as indicated by the second column of Table 3. This preference is equal to 10.7 kcal/mol for SnF4 and doubles as the tetrel atom is reduced in size to Si. Just on the basis of the energetics of the monomer, then, one would anticipate that the axial complexes with the pyridine derivatives should be more stable than the equatorial structures.
Each of the two monomer structures will have its own set of σ-holes, i.e., maxima in its MEP. The values of Vs,max in the vacant site listed in the last two columns of Table 3 are slightly larger for the equatorial vacancy. Another important issue compares the intensities of the σ-holes in the idealized trigonal bipyramid structures with those in the tetrahedral geometries of the unperturbed monomers. The values of Vs,max for the penta-coordinated structures in Table 3, both axial and equatorial vacancies, are far larger than those in the tetrahedral molecules in Table 1. This intensification of the σ-hole, and the ensuing magnification of the electrostatic attraction with an incoming nucleophile, provides an important impetus for the geometrical distortion of the TF4 that accompanies complexation.
An important facet of the geometries resides in the intermolecular distance R (T··N). These distances are contained in Table 2 and are in the range between 2.0 and 2.3 Å. In the axial structures, the distance is longest for T = Sn, understandable in light of its larger size, but it is also slightly longer for Si than for Ge, which can be understood in terms of energetics (see below). With respect to pyridine substituent R, the intermolecular distance elongates in the order CF3 > H > Me ~ OH, consistent with the pattern of more negative Vs,min in Table 1.
R (T··N) is a bit shorter for the equatorial structures as compared to axial; this decrement is sensitive to the nature of the tetrel atom. The closer contacts within the equatorial complexes is an important characteristic that has implications on the energetics discussed below. In short, an equatorial site within a trigonal bipyramid suffers from lesser steric repulsions than does an axial position. The Lewis base is thus freer to approach the tetrel atom from the equatorial direction, resulting in the smaller values of R in Table 2. Note that the difference in R between equatorial and axial dimers is equal to 0.17, 0.10, and 0.03 Å for T = Si, Ge, and Sn, respectively. This diminishing difference reflects the reduced steric repulsions for the larger tetrel atoms that move the F atoms further apart. The same effect is responsible for the lower energetic preference of the axial over the equatorial dimers for the larger T atoms in the last column of Table 2. The dependence on substituent in the equatorial dimers varies in the same manner as the axial structures. There is one minor difference in that R is always smaller for Si than for Ge, as opposed to nearly equal distances for the axial structures.
2.3. Energetics
The interaction energies of the various complexes reported in Table 4 show only a small dependence on the level of theory. Categories I and III both employed the cc-pVTZ basis set, (I) with MP2 and (III) with CCSD(T). The results are barely affected by the particular method of incorporating electron correlation. The switch to BLYP-D3 brand of DFT, coupled with a Def2TZVPP basis set (II) has a lowering effect on the interaction energy but only by a small amount. Most importantly, all levels of theory display the same trends from one system to the next.
The patterns in the energetic data are consistent with some of the trends in the geometries. The interactions within the axial complexes strengthen a great deal as the tetrel atom grows in size, whereas there is much less sensitivity noted in the equatorial structures, and it is the Ge that seems to be more strongly bound than either Si or Sn. The interaction, whether axial or equatorial, is strengthened slightly by either OH or Me substituent on the pyridine but weakened by CF3. This pattern is consistent with the idea that an electron-withdrawing agent will remove density from the N lone pair and thus weaken the interaction with a Lewis acid.
After taking proper account of the deformations that occur in each monomer as they engage one another to form the complex, one arrives at the overall energetics of the binding process, Eb. The binding energies in Table 5 are of course less negative than the interaction energies between the previously distorted monomers in Table 4 but nonetheless show many of the same patterns. In the first place the MP2(I) and DFT(II) quantities are similar and follow the same trends, although the latter are a few kcal/mol less exothermic than the former. The intensification of the binding along the Si < Ge < Sn order is present, now in the equatorial as well as the axial complexes. Just as in the case of the interaction energies, Eb is also intensified for the electron-donating OH and Me substituents, but weakened for CF3.
But there is one very important aspect of the energetics in Table 4 and Table 5. The interaction energies of the equatorial complexes in Table 4 are considerably larger than those of the axial geometries, in some cases nearly twice the magnitude. The energetic margin of equatorial over axial is most dramatic for the Si complexes, up as high as 25 kcal/mol, and is reduced as the tetrel atom grows in size. However, quite the opposite is true of the binding energies in Table 5 where it is the axial complexes which are preferred. The margins here are a little smaller, but still reach up near 10 kcal/mol. Despite this reversal between Eint and Eb, there remains the same Si > Ge > Sn ordering of the energy differences between axial and equatorial.
The opposite behavior of these two energetic measures of noncovalent bonding rests in the deformation energies. It was noted in Table 2 that the deformation of the tetrahedral TF4 molecule into either of the trigonal bipyramidal geometries requires quite a bit of energy. But it is more costly to leave the vacancy in the equatorial than in the axial position, in amounts varying from 10.7 kcal/mol for the larger Sn up to 22.6 kcal/mol for Si. It is the larger deformation energy of the equatorial structure that accounts for its reduced binding energy within the various complexes.
These deformation energies are recorded in Table 6, split out to show the deformation of both the Lewis acid and base individually. It is first apparent that the bulk of the deformation is associated with the Lewis acid TF4, since that of the base is only 1 kcal/mol or less. Secondly, the deformation required to accommodate the base in the axial position is quite a bit smaller than that for an equatorial association. The deformation energy is reduced as the tetrel atom becomes larger, which is especially true of the equatorial systems.
Comparison of Table 1 and Table 3 had indicated that in the idealized situations, the σ-hole of the trigonal bipyramid TF4 monomer is considerably more intense than in the optimized tetrahedral structure. The value of Vs,max in the geometries within the actual complexes is reported in the last column of Table 6. Since the geometries of the monomers are not quite fully formed trigonal bipyramids, the σ-holes are not quite as intense as those in Table 3, they nonetheless approach these values, and represent a huge increase over their magnitude within the tetrahedral geometry. Taking GeF4 as an example, Vs,max is equal to 50.9 kcal/mol in the tetrahedral monomer, vs. 116.4 and 120.1 kcal/mol for the idealized axial and equatorial geometries, respectively. Table 6 shows this quantity is roughly 100 kcal/mol in the axial complexes, slightly higher at 108 kcal/mol for the equatorial dimers. The increased electrostatic attraction resulting from this σ-hole magnification helps to compensate for the large geometrical deformation energy occurring within the Lewis acid molecule.
Decomposition of the total interaction energy offers a window into the factors that contribute to the stability of each complex. The various contributions are listed in Table 7 for both the axial and equatorial dimers, as well as the difference between these two structures. As an overall perspective, the electrostatic component accounts for roughly 60% of the total attractive force. The second most important factor is the orbital interaction which makes up between 34% and 41%. Dispersion makes only a minor contribution on the order of 5% or less. Focusing first on the electrostatic element, in the case of the axial dimers, it increases with heavier tetrel atom Si < Ge < Sn, while an opposite pattern is seen in the equatorial structures. The same preference for the heavier T atom occurs for the equatorial values of Eoi, while the axial structures fall into the Ge > Sn > Si ordering. As a result, the electrostatic component favors the axial over the equatorial structure for T = Si and Ge, but tends toward the equatorial for Sn. The orbital interactions lead to a heavy preference for axial for Si, a preference which is progressively abated for Ge and then for Sn.
Much of this trending information traces back to the intermolecular distances. Each component is of course heavily dependent upon the distance between the two subunits, rising dramatically as they come closer together. As noted in Table 2, R(T···N) varies only slowly with tetrel atom for the axial complexes. Hence, the electrostatic attraction can be closely related to the value of Vs,max of the TF4 molecule, which climbs in the order Si < Ge < Sn, as does Eelec. In the case of the equatorial dimers, R increases steadily along with the size of the T atom, thus acting to diminish Eelec, right along with Eoi, as T enlarges.
Previous energy decompositions of tetrel-bonded systems have observed a similar mix of terms. In one example [43], the electrostatic term was found most important for the majority of such dimers, followed by orbital interaction. It was only for the weakest complexes of CFH3 with π-systems (with interaction energies of only −1.3 kcal/mol) that the dispersion component played a significant role.
2.4. Analysis of Wave Function
One measure of the noncovalent bond strength originates in a AIM analysis of the topology of the electron density [44,45,46,47]. AIM molecular graphs for each complex are illustrated in Figure S1, where broken lines indicate intermolecular bond paths, with a small green dot marking the bond critical point. Each bond path between the two monomers is quantified via several aspects of the critical point. In particular, the density ρ, its Laplacian ∇2ρ, and the electron energy H [48], are all displayed in Table 8 for the various complexes fit into patterns that are only partially consistent with energetic and geometrical data. First focusing on the most important T···N bond path, ∇2ρ rises in the order Si < Ge < Sn for the axial complexes, the same order as observed for the interaction energies in Table 4. ρ and H display a somewhat different pattern, peaking for Ge. Consonant with the interaction energies, the AIM markers of noncovalent bond strength are larger for the equatorial complexes. Again there are different trends for ∇2ρ on one hand and ρ and H on the other. The latter are largest for Ge, while the peak of the former occurs for Si. In addition to the principal T···N bond path, there are indications of weaker bonding interactions between F atoms of TF4 and H of the pyridine derivatives, i.e., weak CH···F H-bonds. As measured by AIM, these bonds represent only fractions of the bond strength of T···N, but may account for a small amount of the total binding energy.
The behavior of the AIM descriptors is characteristic of tetrel bonded and related systems. For example, quite comparable values of the density at the bond critical point were derived when NH3 was paired with TF4 (T = C, Si, Ge, Sn), as well as other less fluorosubstituted Lewis acid molecules [49]. Complexes of TFH3 (T =Ge, Si, Sn, Pb) with various carbon π-electron systems displayed values of ρBCP up to 0.045 a.u. and all positive Laplacians [43]. Tetrel bonds between Tr5Cl10 and Tr6Cl12 (Tr = Si and Ge) and bases HCN, HF, OH− and Cl− [34] noted AIM markers similar to those observed here, with similarly positive Laplacians. In particularly strong tetrel bonds with anions, one can find values of ρBCP that surpass 0.1 a.u. [50].
The delocalization indices (DI) of T···N interactions [51,52] are collected in Table S1. These quantities specify the number of electrons in the interatomic bonding regions and can be interpreted as the bond order value [51,52]. The DI is much higher for complexes with Ge than for Si dimers which implies greater covalent nature of the former. This pattern is consistent with the other descriptors as well as interaction energies. (Values for Sn complexes were not possible to evaluate by virtue of the pseudopotential approach.)
An alternate means of analysis concerns charge transfer from the orbitals of one molecule to those of the other. Within the NBO framework, the energetic measure of such a transfer emerges as the perturbation energy E(2) [53,54,55,56,57,58,59]. The relevant transfers in these systems involve motion from the lone pair orbital of the pyridine N into the antibonding lone pair orbitals of the tetrel atom, designated LP* by NBO. (The latter can be thought of as alternate descriptions of σ* (F–T) orbitals which are centered largely on T as compared to F.) The total interorbital transfer energy is reported in Table 9. Whereas the interaction energies for the axial complexes obey a clear Si < Ge < Sn trend, E(2) is largest for Ge. The equatorial geometries lead to enlarged values of E(2) vs. axial, in common with the energetics. E(2) diminishes as the tetrel atom grows in size for these equatorial structures, inconsistent with the energetic trends.
The total charge transfers from the Lewis base molecule to the acid, encompassing all orbitals, is contained within the CT columns of Table 9. Just as for E(2), CT is much larger for equatorial than for axial complexes. With respect to the latter geometries, CT follows the Si < Ge < Sn trend of the energetics. The same can be said of the equatorial structures where CT and Eint both peak for Ge. Of the various NBO measures, it is the total intermolecular charge transfer that thus most closely parallels the energetics. The other general trend coming from presented analyses is that tetrel bonds studied have partly covalent nature. The amount of orbital interaction contribution presented by EDA (up to −70.50 kcal/mol), E(2) energies of intermolecular donation (reaching 169 kcal/mol), net charge transfer values exceeding 0.2 electrons and finally the negative values of H at the BCPs in AIM analysis are the apparent signs of the non-negligible influence of covalent character in interactions investigated here.
2.5. Conversion between Axial and Equatorial Complexes
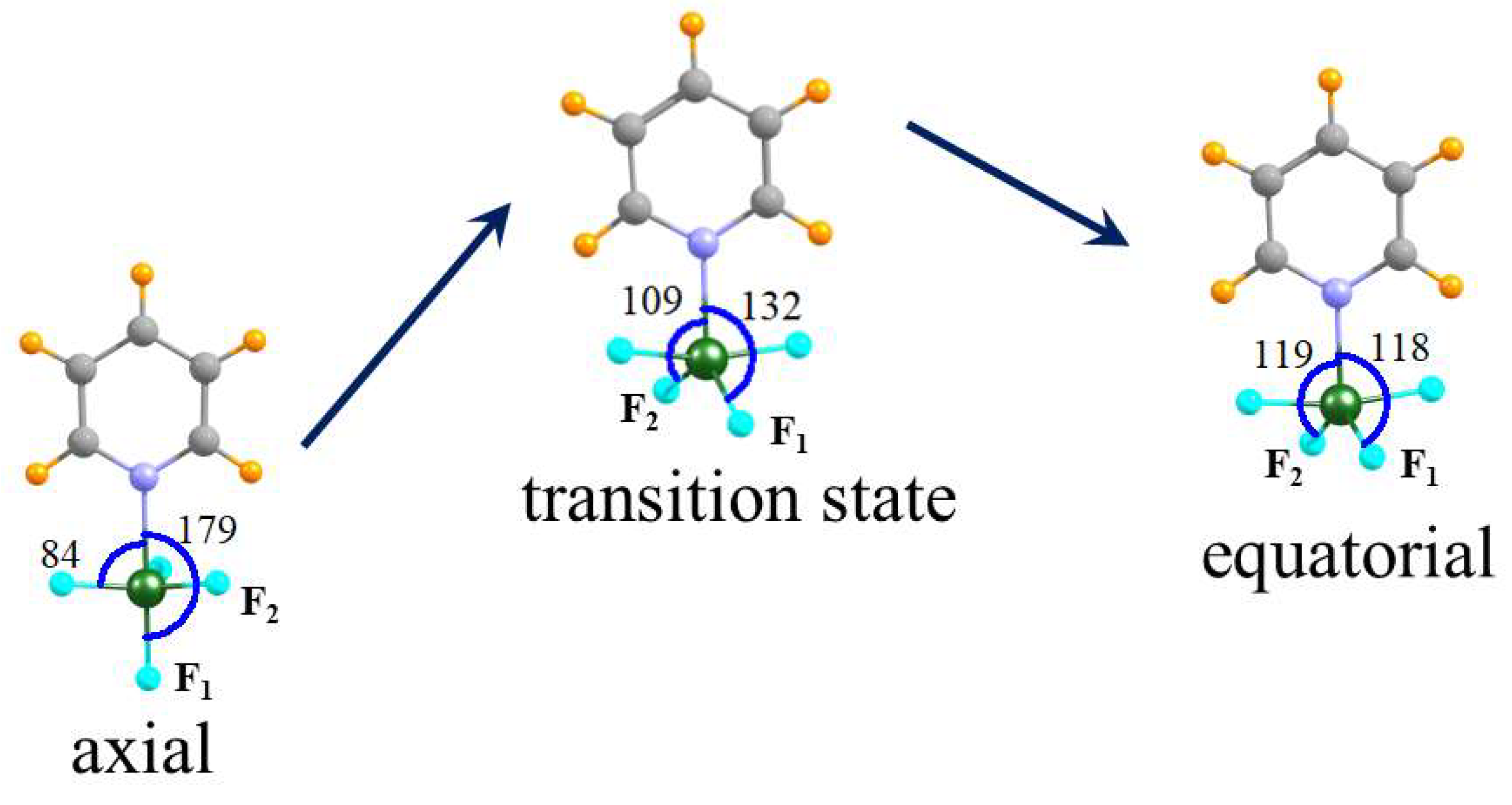
Given the presence of two minima on the potential energy surface, with comparable energies, the issue naturally arises concerning the ease of conversion from one to the other. For this purpose, focus was placed on the unsubstituted pyridine Lewis base. The transition state was located for this transition, as illustrated in Figure 3 for the Py···GeF4 system as an example. The shift of the GeF4 unit from axial to equatorial is an interesting one. Two of the three equatorial F atoms in the axial geometry retain their position fairly steadily, with the θ (NGeF) angle remaining nearly constant at 84°. The transition occurs as the axial F atom, F1, in Figure 3, moves in toward the N.
The θ (NGeF1) angle is 179° in the axial structure, diminishes to 132° in the transition state, until finally reaching 118° in the equatorial structure on the right side of Figure 3. In essence, it has shifted smoothly from an axial to an equatorial position relative to the pyridine. During this process, it has decreased its distance from the Ge progressively from 1.722 to 1.717 to 1.715 Å. The third equatorial F atom (F2) will ultimately become an equatorial ligand in the final structure. But in order to do, it must increase its angle with respect to N, with θ (NGeF2) beginning at 84°, increasing to 109° in the transition state, and finally to 119° in the equatorial geometry. The pertinent R (GeF2) bond length changes from 1.722 to 1.712 to 1.715 during this process. The barrier height G† for the transition from axial to equatorial is equal to 11.79, 8.38 and 1.46 kcal mol−1 for Py···SiF4, Py···GeF4 and Py···SnF4, respectively, as reported in Table S2. That is, the conversion becomes easier as the tetrel atom is enlarged.
2.6. Other Types of Geometry
While the geometries described above represent the principal minima identified on each potential energy surface, there are additional minima albeit of higher energy. These secondary minima are displayed in Table S3 along with their salient geometric characteristics. Most of these structures involve a weak interaction between the tetrel atom and the aromatic ring, at sites other than the N atom. The aromatic atom which is directly involved in this interaction is situated directly opposite one of the T–F bonds in what might be termed an axial site. The two molecules are spread apart, with intermolecular R(T···C) distances of 3 Å or more, with Sn engaging in the strongest such interactions of those identified. In no case is there a minimum found between the TF4 and a C atom ortho to N, due in part to the more positive natural charge on this C relative to the others.
3. Discussion
Within the context of classic ideas such as VSEPR theory, it is understood that the axial position of a trigonal bipyramid faces more steric repulsion than does its equatorial counterpart. This notion is based on the fact that each axial ligand has three close equatorial neighbors at an angle of 90°, while the equatorial site has only two such nearby neighbors. This distinction explains why the trigonal bipyramid geometry of TF4 with an equatorial vacancy is at an energetic disadvantage with respect to an axial vacancy. In fact, the calculated preference for the axial vacancy ranges from 10.7 to 22.6 kcal/mol (see Table 3), with the greatest difference associated with the smallest T atom Si. It is also for this reason that the deformation energies of the equatorial complexes with the pyridine derivatives are considerably larger than the same quantities for the axial structures.
The second issue where this axial vs. equatorial site distinction comes into play arises when the Lewis base approaches. For the reasons enunciated above, approach to an equatorial site will offer less steric repulsion than to an axial site with three close ligand neighbors. It is largely for this reason that the interaction energies for the equatorial complexes are more negative than for the axial geometries, and the N atom approaches more closely to the tetrel. Recall that the interaction energy involves pre-deformed monomers so it represents a pure interaction, unaffected by the energetic cost of preparing each monomer into the geometry it adopts within the dimer.
The binding energy, representing the exothermicity of the combination process, thus combines these two competing effects. It is largely the first factor, the monomer deformation energy, that controls the final energetics, making the axial complexes more stable than their equatorial counterparts. This preference varies between 3 and 10 kcal/mol, and is largest for the smallest Si atom. It is the mutual repulsions, both electrostatic and steric, that are largely responsible for the high deformation energies common to these TF4 molecules. For example, the replacement of some of these F atoms by H results in markedly lower deformation energies upon complexation with a Lewis base [25].
The dichotomy of two separate minima for tetrel bonded systems may be more common than previously thought. For example, replacement of the four F atoms of SiF4 by Br leads to a similar situation. DFT calculations show an equatorial placement of the pyridine base represents a true minimum on the surface, albeit higher in energy than its axial counterpart, by some 8 kcal/mol.
In order to delve more deeply into these ideas, the four F atoms of SiF4 were replaced by bulkier methyl groups. Of course, the optimal geometry of Si(CH3)4 is tetrahedral. But when forced to adopt a trigonal bipyramid structure, the one with an axial vacancy was favored by 26.5 kcal/mol over an equatorial vacancy, due to the steric repulsions between the methyl groups. In a second step a Lewis base NH3 was brought up to the vacancy of each structure. When the geometry of each dimer was fully relaxed, both optimized to the same sort of structure: Si(CH3)4 became tetrahedral, with the NH3 fitting into a hole between the methyl groups. In other words, the instability engendered by the steric repulsions makes the monomer with an equatorial vacancy so unstable that it quickly decays to a tetrahedral geometry, a derivative of axial with a linear R–T··N. The same can be said about tri and disubstitution: When only three or two of the F atoms of SiF4 were replaced by the bulkier methyl group, SiMe3F and SiMe2F2 only engage in an axial sort of tetrel bond with a N-base, with FSi··N close to linear.
The idea that a tetracoordinated TR4 molecule will deform into a trigonal bipyramid has become well recognized [60,61], as well as the idea that such a deformation leads to large distortion energies [62]. The concept that a tetrel bond can form in a direction that does not coincide with a T–R bond has some precedent [63]. For example, a prior work [64] found what was termed a π-tetrel bond when XC≡N was paired with a base. However, this bond did not substantially alter the geometry of the tetrel-containing molecule. Another bonding scenario where this out-of-plane situation can occur is the planar F2T=O molecule [26,65,66,67] or R2T=CH2 [25]. These sorts of geometries typically result in weaker binding, as for example, the replacement of TF4 by TF2=CF2 (T = Si, Ge, Sn) [25]. The reduction of substituents on the T atom from four to three has another effect: it markedly reduces the steric repulsions between them, leading to much reduced deformation energy [25].
The interaction energies computed here are in nice accord with prior calculations of related systems. Interactions between TF4 molecules and NH3 [38] are of comparable magnitude with those computed here for the pyridine derivatives; the values are reduced slightly for pyrazine. The complexation of TF4 molecules with a variety of bases was studied earlier [68] but in the context of only the axial complexes. Nevertheless, the deformation of the monomers was found to strongly intensify the σ-hole which provided a strong element in the large binding energies, consistent with the trends noted here. A similar effect was also observed for the pnicogen bonds involving ZF5 (Z = P, As, Sn) wherein the Lewis acid transitioned from a trigonal bipyramid to a square pyramid structure.
The comparative strengths of the various tetrel atoms Si, Ge, and Sn, as well as C, has also been a subject of a good deal of prior investigation [32,49,50,69,70,71] and this sort of noncovalent bond has been found in some cases to act as a superior Lewis acid when compared to other related bonds such as chalcogen, pnicogen, and halogen types [32,70,72,73,74] although this cannot be considered a universal rule [75,76]. Deformation energies in excess of 20 kcal/mol may be a common feature of tetrel bonding. It was noted earlier [70] in complexes of TF4 with NH3, which also confirmed the rise in this quantity as the tetrel atom becomes smaller. There was also an indication that the deformation energy would be a bit larger for THF3. It has been shown to play a major role in the tetrel bonding of other Lewis bases, for example in those containing methyl and larger alkyl substituents [70], again with this quantity reaching up toward 20 kcal/mol. In keeping with the findings here, the deformation effects are most dramatic for smaller tetrel atoms like Si where it can prevent the formation of a tetrel bond altogether.
One last functional that was tested here was the double-hybrid B2PLYP. Calculation of the interaction energies of the three complexes involving unsubstituted pyridine displayed in Table S4 suggests that B2PLYP outperforms BLYP-D3 and yields data comparable to MP2. However, since its computational cost is comparable to actual MP2 calculations, one might be better served with MP2 itself.
Finally with regard to C as central atom, it is well known that this atom is generally reluctant to engage in tetrel bonds of any strength. Indeed, even with four highly electron-withdrawing substituents, Vs,max for the tetrahedral CF4 is only 18.8 kcal/mol, less than half that of the other TF4 monomers. As such, CF4 is unable to form equatorial complexes with pyridine or any of its derivatives. The BLYP-D3 interaction energies for the axial structures are very small, only about −1.7 kcal/mol. AIM analysis suggests that these weak complexes are held together by secondary interactions, rather than a tetrel bond.
4. Methods
Full geometry optimization was performed for isolated monomers and complexes at the MP2 level in conjunction with the cc-pVTZ basis set [77,78]. For purposes of confirmation, the BLYP-D3/Def2TZVPP, B2PLYP (for pyridine complexes only) and CCSD(T)/cc-pVTZ levels were employed as well [79,80,81,82,83,84]. For Sn atoms, the cc-pVTZ-PP basis set which includes relativistic effects was chosen [85,86]. All minima were confirmed by vibrational analysis, transition states were located using a standard Berny algorithm while demanding the optimization to a transition state rather than a local minimum (TS keyword). Molecular coordinates for minima of primary TF4 complexes with pyridine and its derivatives are given in Table S5. The binding energy of each complex was calculated as the energy difference between the complex and the sum of the individually optimized monomers. The interaction energy takes as its reference the energies of the monomers measured in the geometries obtained within the complex. The difference between the former and latter is the deformation energy, defined as the energy required to distort each monomer from its optimized geometry to that within the dimer. The interaction and binding energies of the complexes were corrected for basis set superposition error (BSSE) via the Boys-Bernardi counterpoise procedure [87]. Geometry optimizations and energy calculations utilized the Gaussian 09 code [88]. Energy decomposition analysis (EDA) was performed at the BLYP-D3/ZORA/TZ2P level using DFT geometries with ADF software [89,90,91]. The molecular electrostatic potentials (MEPs) of the isolated monomers were calculated on the electron density isosurface of ρ = 0.001 a.u. at the MP2/cc-pVTZ level; the corresponding extrema were quantified using the WFA-SAS program [92]. MP2 electron density topology was analyzed in the context of the AIMAll program in order to visualize and characterize the bonding paths [93]. NBO analysis was employed to analyze the interactions in terms of atomic charges and orbital-orbital second-order energies using the DFT-generated wave function [94].
5. Conclusions
In summary, the approach of a Lewis base toward a tetrahedral TF4 molecule causes it to distort into a trigonal bipyramid shape. The base may come closer to T if it approaches along an equatorial direction, which also results in a higher interaction energy than approach toward an axial site. These interaction energies are quite high, surpassing 50 kcal/mol. One source of these large quantities is the very substantial intensification of the σ-hole that arises when the originally tetrahedral TF4 is distorted into a trigonal bipyramid shape. On the other hand, this same equatorial approach also induces a much larger deformation energy in the TF4 monomer. As a result of these two competing effects, it is the axial dimer which is the more stable of the two, by some 3–9 kcal/mol. It may be possible to isolate both sorts of structure in an experimental setting, although the energy barrier separating them is fairly low. The tetrel bond strength rises as the tetrel atom grows in size: Si < Ge < Sn; the effect of the substituent on the pyridine is considerably weaker.
Supplementary Materials
The following are available online. Figure S1: AIM molecular graphs of axial and equatorial TF4 (T = Si, Ge, Sn) complexes with pyridine derivatives at the MP2/cc-pVTZ level. Small green dots represent bond critical points. Table S1: The delocalization index (DI) of T···N interaction in TF4 complexes with pyridine and its derivatives. Data obtained at the MP2/cc-pVTZ level of theory. Table S2: Gibbs free energies of axial and equatorial conformers as well as TS for TF4 (T: Si, Ge, Sn) complexes with Py, calculated at the MP2/cc-pVTZ level of theory. Table S3: Secondary minima for TF4 complexes with pyridine and its derivatives. Data obtained at the MP2/cc-pVTZ level of theory. Eint corrected for BSSE (in kcal/mol). Parameters: distances in Å, angles in degrees. Table S4: Interaction energies (Eint, kcal mol−1) corrected for BSSE calculated at the MP2/cc-pVTZ (I), BLYP-D3/Def2TZVPP (II) and B2PLYP/def2TZVPP (III) levels of theory. Table S5: Molecular coordinates for minima of primary TF4 complexes with pyridine and its derivatives. Data obtained at the MP2/cc-pVTZ level of theory.
Author Contributions
Conceptualization, W.Z. and S.S.; Investigation, W.Z., M.M. and R.W.; Validation, W.Z., S.S., M.M. and R.W.; Data Curation, W.Z., S.S., M.M. and R.W.; Writing—Original Draft Preparation, W.Z., S.S. and M.M.; Writing—Review & Editing, W.Z. and S.S.; Visualization, W.Z., S.S., M.M. and R.W.; Project Administration, W.Z. and S.S.; Funding Acquisition, W.Z.
Funding
This work was financed in part by a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology.
Acknowledgments
A generous allocation of computer time from the Wroclaw Supercomputer and Networking Center is acknowledged.
Conflicts of Interest
There are no conflict to declare.
References
- Schneider, H.J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Yatsimirski, A. Principles and Methods in Supramolecular Chemistry; JohnWiley: Chichester, UK, 2000. [Google Scholar]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Wash, P.L.; Ma, S.; Obst, U.; Rebek, J. Nitrogen−Halogen Intermolecular Forces in Solution. J. Am. Chem. Soc. 1999, 121, 7973–7974. [Google Scholar] [CrossRef]
- Legon, A.C. Prereactive complexes of dihalogens XY with Lewis bases B in the gas phase: A systematic case for the Halogen analogue B small middle dot small middle dot small middle dotXY of the hydrogen bond B small middle dot small middle dot small middle dotHX. Angew. Chem. Int. Ed. Engl. 1999, 38, 2686–2714. [Google Scholar] [CrossRef]
- Caronna, T.; Liantonio, R.; Logothetis, T.A.; Metrangolo, P.; Pilati, T.; Resnati, G. Halogen bonding and π···π stacking control reactivity in the solid state. J. Am. Chem. Soc. 2004, 126, 4500–4501. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Chen, N.; Wu, H.; Knotts, N.; Kaupp, M. 3C NMR study of halogen bonding of haloarenes: Measurements of solvent effects and theoretical analysis. J. Am. Chem. Soc. 2004, 126, 4412–4419. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Bilewicz, E. Cooperativity halogen bonding effect–Ab initio calculations on H2CO⋯(ClF)n complexes. Chem. Phys. Lett. 2006, 427, 51–55. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the sigma-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. Chem. Phys. Chem. 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole strengths and interactions of F3MX molecules (M = C, Si, Ge and X = F., Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. σ-Hole interactions: Perspectives and misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. A new noncovalent force: Comparison of P···N interaction with hydrogen and halogen bonds. J. Chem. Phys. 2011, 134, 094315–094319. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen bonding interaction: A new supramolecular force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Stenlid, J.H.; Johansson, A.J.; Brinck, T. σ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. Phys. Chem. Chem. Phys. 2018, 20, 2676–2692. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. Σ-holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Directionality of π-holes in nitro compounds. Chem. Commun. 2015, 51, 1491–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Comparison between tetrel bonded complexes stabilized by σ and π hole interactions. Molecules 2018, 23, 1416. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, J.; Yang, X.; Liu, Z.; Li, W.; Li, Q. Comparison of σ-hole and π-hole tetrel bonds formed by pyrazine and 1,4-dicyanobenzene: The interplay between anion-π and tetrel bonds. Chem. Phys. Chem. 2017, 18, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Wenbo, D.; Xin, Y.; Jianbo, C.; Wenzuo, L.; Qingzhong, L. Comparison for σ-hole and π-hole tetrel-bonded complexes involving F2C=CFTF3 (T = C, Si, and Ge): Substitution, hybridization, and solvation effects. J. Fluorine Chem. 2018, 207, 38–44. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, J.; Bickelhaupt, F.M. Activation strain analysis of SN2 reactions at C, N, O, and F centers. J. Phys.Chem. A 2017, 121, 885–891. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Dronskowski, R. Tetrel bonds in infinite molecular chains by electronic structure theory and their role for crystal stabilization. J. Phys. Chem. A 2017, 121, 1381–1387. [Google Scholar] [CrossRef]
- Scheiner, S. Steric crowding in tetrel bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef]
- Scheiner, S. Tetrel Bonding as a vehicle for strong and selective anion binding. Molecules 2018, 23, 1147. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Elguero, J.; Alkorta, I. Complexes of CO2 with the azoles: Tetrel bonds, hydrogen bonds and other secondary interactions. Molecules 2018, 23, 906. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Tetrel bonding interactions in perchlorinated cyclopenta- and cyclohexatetrelanes: A combined DFT and CSD study. Molecules 2018, 23, 1770. [Google Scholar] [CrossRef] [PubMed]
- García-Llinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R–CF3···O tetrel bonding interactions in biological systems. J. Phys. Chem. A 2017, 121, 5371–5376. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Sahoo, D.K.; Bhaumik, S.; Jena, S.; Chandrakar, A.; Biswal, H.S. Noncovalent carbon-bonding interactions in proteins. Angew. Chem. Int. Ed. 2018, 57, 16496–16500. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Bauzá, A. S⋅⋅⋅Sn tetrel bonds in the activation of peroxisome proliferator-activated receptors (PPARs) by organotin molecules. Chem. Eur. J. 2018, 24, 16582–16587. [Google Scholar] [CrossRef]
- Trievel, R.C.; Scheiner, S. Crystallographic and computational characterization of methyl tetrel bonding in S-adenosylmethionine-dependent methyltransferases. Molecules 2018, 23, 2965. [Google Scholar] [CrossRef]
- Marin-Luna, M.; Alkorta, I.; Elguero, J. Cooperativity in tetrel bonds. J. Phys. Chem. A 2016, 120, 648–656. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. Characterization of the short O=C⋯O=C π-hole tetrel bond in the solid state. CrystEngComm 2018, 20, 3308–3312. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. RCH3···O Interactions in biological systems: Are they trifurcated H-bonds or noncovalent carbon bonds? Crystals 2016, 6, 26. [Google Scholar] [CrossRef]
- Scheiner, S.; Lu, J. Chalcogen, and pnicogen bonding involving hypervalent atoms. Chem. Eur. J. 2018, 24, 8167–8177. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bonds with π-electrons acting as lewis bases—Theoretical results and experimental evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms In Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A bond path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical-bond in terms of local properties of electron density and energy. Croat. Chem. Acta. 1984, 57, 1259. [Google Scholar]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridineTF3 and furan TF3 (T = C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef]
- Esrafili, M.; Mousavian, P. Strong tetrel bonds: Theoretical aspects and experimental evidence. Molecules 2018, 23, 2642. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Minaev, B.F.; Minaeva, V.A.; Podgornaya, A.T.; Ågren, H. Application of Bader’s atoms in molecules theory to the description of coordination bonds in the complex compounds of Ca2+ and Mg2+ with methylidene rhodanine and its anion. Russ. J. Gen. Chem. 2012, 82, 1254. [Google Scholar] [CrossRef]
- Karaush, N.; Baryshnikov, G.V.; Minaev, B.F. Alkali and alkaline-earth metal complexes with tetraoxa[8]circulene sheet: A computational study by DFT and QTAIM methods. RSC Adv. 2015, 5, 24299–24305. [Google Scholar] [CrossRef]
- De Paul, V.; Nziko, N.; Scheiner, S. Chalcogen bonding between tetravalent SF4 and amines. J. Phys. Chem. A 2014, 118, 10849–10856. [Google Scholar]
- Guo, X.; An, X.; Li, Q. Se···N chalcogen bond and Se···X halogen bond involving F2C=Se: Influence of hybridization, substitution, and cooperativity. J. Phys. Chem. A 2015, 119, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.; Mohammadirad, N. Substituent effects in cooperativity of chalcogen bonds. Mol. Phys. 2015, 113, 3282–3290. [Google Scholar] [CrossRef]
- Esrafili, M.; Nurazar, R. Chalcogen bonds formed through π-holes: SO3 complexes with nitrogen and phosphorus bases. Mol. Phys. 2016, 114, 276–282. [Google Scholar] [CrossRef]
- Azofra, L.; Alkorta, I.; Scheiner, S. Chalcogen bonds in complexes of SOXY (X, Y = F, Cl) with nitrogen bases. J. Phys. Chem. A 2015, 119, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Effects of charge and substituent on the S···N chalcogen bond. J. Phys. Chem. A 2014, 118, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Ramasami, P.; Ford, T. Chalcogen-bonded complexes of some carbon dioxide analogues. J. Mol. Struct. 2014, 1072, 28–31. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds-mechanisms protecting doublet and octet electron structures. Phys. Chem. Chem. Phys. 2017, 19, 29742–29759. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Asadollahi, S.; Mousavian, P. Anionic tetrel bonds: An Ab initio study. Chem. Phys. Lett. 2018, 691, 394–400. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Sokalski, W.A. Are various Σ-hole bonds steered by the same mechanisms? ChemPhysChem. 2017, 18, 1569–1577. [Google Scholar] [CrossRef]
- Azofra, L.M.; Scheiner, S. Chalcogen, and CH··O Hydrogen bonds in complexes pairing carbonyl-Containing molecules with 1, 2, and 3 molecules of CO2. J. Chem. Phys. 2015, 142, 034307–034317. [Google Scholar] [CrossRef] [PubMed]
- Nziko, V.d.P.N.; Scheiner, S. Comparison of p-hole tetrel bonding with s-hole halogen bonds in complexes of XCN (X = F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, Q.; Scheiner, S. The Π-tetrel bond and its influence on hydrogen bonding and proton transfer. ChemPhysChem. 2018, 19, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, Q. Comparison for Σ-hole and π-hole tetrel-bonded complexes involving cyanoacetaldehyde. Mol. Phys. 2018, 116, 222–230. [Google Scholar] [CrossRef]
- Shen, S.; Zeng, Y.; Li, X.; Meng, L.; Zhang, X. Insight into the π-Hole··· π-electrons tetrel bonds between F2ZO (Z = C, Si, Ge) and unsaturated hydrocarbons. Int. J. Quantum Chem. 2018, 118, e25521–25533. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Implications of monomer deformation for tetrel and pnicogen bonds. Phys. Chem. Chem. Phys. 2018, 20, 8832–8841. [Google Scholar] [CrossRef] [PubMed]
- Solel, E.; Kozuch, S. On the power of geometry over tetrel bonds. Molecules 2018, 23, 2742. [Google Scholar] [CrossRef]
- Scheiner, S. Systematic elucidation of factors that influence the strength of tetrel bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative assessment of tetrel bonding utilizing vibrational spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, Pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Scheiner, S. Highly selective halide receptors based on chalcogen, pnicogen, and tetrel bonds. Chem. Eur. J. 2016, 22, 18850–18858. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Assembly of effective halide receptors from components. Comparing hydrogen, halogen, and tetrel bonds. J. Phys. Chem. A 2017, 121, 3606–3615. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Legon, A. An Ab initio investigation of the geometries and binding strengths of tetrel-, pnictogen-, and chalcogen-bonded complexes of Co2, N2o, and Cs2 with simple Lewis bases: Some generalizations. Molecules 2018, 23, 2250. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Li, Q.; Scheiner, S. Comparative strengths of tetrel, pnicogen, chalcogen, and halogen bonds and contributing factors. Molecules 2018, 23, 1681. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–623. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, e034108. [Google Scholar] [CrossRef] [PubMed]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comp. Chem. 1996, 17, 1571–1587. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–968. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 19 January 2019).
- Bulat, F.; Toro-Labbe, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- AIMAll; Version 14.11.23; Todd A. Keith (TK Gristmill Software): Overland Park, KS, USA, 2014.
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
MEPs of isolated SiF4 and Lewis base ligands computed on the 0.001 a.u. isodensity surface at the MP2/cc-pVTZ level. Color ranges, in kcal/mol, are: red greater than 15, yellow between 8 and 15, green between 0 and 8, blue below 0 kcal/mol.
Figure 1.
MEPs of isolated SiF4 and Lewis base ligands computed on the 0.001 a.u. isodensity surface at the MP2/cc-pVTZ level. Color ranges, in kcal/mol, are: red greater than 15, yellow between 8 and 15, green between 0 and 8, blue below 0 kcal/mol.

Figure 2.
MP2-optimized structures of (a) axial and (b) equatorial complexes of TF4 (T = Si, Ge, Sn) with pyridine and its derivatives (R = H, OH, CH3, CF3).
Figure 2.
MP2-optimized structures of (a) axial and (b) equatorial complexes of TF4 (T = Si, Ge, Sn) with pyridine and its derivatives (R = H, OH, CH3, CF3).

Figure 3.
Gibbs free energy profile of transformation reaction between axial and equatorial conformers of the Py∙∙∙GeF4 complex. Data concerning energies and imaginary frequencies of TS are given in Table S2.
Figure 3.
Gibbs free energy profile of transformation reaction between axial and equatorial conformers of the Py∙∙∙GeF4 complex. Data concerning energies and imaginary frequencies of TS are given in Table S2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Molecular electrostatic potentials extrema (in kcal mol−1) on the 0.001 a.u. contour of the electron density (Vs,max and Vs,min) of isolated donors of σ-hole and π-hole as well as isolated Lewis bases used in this study, calculated at the MP2/cc-pvtz level of theory.
Table 1.
Molecular electrostatic potentials extrema (in kcal mol−1) on the 0.001 a.u. contour of the electron density (Vs,max and Vs,min) of isolated donors of σ-hole and π-hole as well as isolated Lewis bases used in this study, calculated at the MP2/cc-pvtz level of theory.
| Lewis Acid | Vs,max (σ-hole) |
|---|---|
| SiF4 | +41.3 |
| GeF4 | +50.9 |
| SnF4 | +70.1 |
| Lewis base | Vs,min a |
| Py | −36.3 |
| 4-HOPy | −37.3 |
| 4-MePy | −37.8 |
| 4-TFMPy | −29.9 |
a minimum coincides with N lone pair.
Table 2.
Structural parameters (R in Å, angles in degs) of complexes at the MP2/cc-pVTZ level.
| Axial Complexes | Equatorial Complexes | ||||||
|---|---|---|---|---|---|---|---|
| R (T···N) | θ (N··TFax) | θ (N··TFeq) | R (N∙∙∙T) | θ (N··TFeq) | θ (N··TFax) | ∆E a | |
| Py∙∙∙SiF4 | 2.143 | 179.6 | 82.9 | 1.973 | 119.1 | 83.7 | 9.42 |
| Py∙∙∙GeF4 | 2.120 | 179.5 | 84.3 | 2.024 | 118.4 | 84.5 | 5.91 |
| Py∙∙∙SnF4 | 2.244 | 178.2 | 83.1 | 2.218 | 118.6 | 83.3 | 3.27 |
| 4-HOPy∙∙∙SiF4 | 2.121 | 179.9 | 83.2 | 1.960 | 118.9 | 84.3 | 8.59 |
| 4-HOPy∙∙∙GeF4 | 2.104 | 179.9 | 84.4 | 2.012 | 118.4 | 85.2 | 5.15 |
| 4-HOPy∙∙∙SnF4 | 2.232 | 177.6 | 83.2 | 2.203 | 118.8 | 83.5 | 2.69 |
| 4-MePy∙∙∙SiF4 | 2.127 | 179.7 | 83.1 | 1.965 | 119.0 | 84.0 | 9.00 |
| 4-MePy∙∙∙GeF4 | 2.110 | 179.6 | 84.5 | 2.017 | 118.4 | 84.9 | 5.55 |
| 4-MePy∙∙∙SnF4 | 2.237 | 178.1 | 83.3 | 2.210 | 118.7 | 83.9 | 3.02 |
| 4-TFMPy∙∙∙SiF4 | 2.189 | 179.7 | 81.9 | - | - | - | - |
| 4-TFMPy∙∙∙GeF4 | 2.142 | 179.5 | 83.7 | 2.039 | 118.5 | 83.9 | 6.63 |
| 4-TFMPy∙∙∙SnF4 | 2.260 | 178.3 | 82.6 | 2.233 | 118.5 | 82.6 | 3.70 |
a Energetic preference of axial over equatorial complex (kcal/mol).
Table 3.
Difference in energy (kcal/mol) between axial and equatorial vacancy on an idealized trigonal bipyramid structure of TF4 molecule, along with maxima of molecular electrostatic potentials (kcal/mol) on the 0.001 a.u. isodensity surface.
Table 3.
Difference in energy (kcal/mol) between axial and equatorial vacancy on an idealized trigonal bipyramid structure of TF4 molecule, along with maxima of molecular electrostatic potentials (kcal/mol) on the 0.001 a.u. isodensity surface.
| Monomer | E(eq) − E(ax) | Vs,max, Axial | Vs,max, Equatorial |
|---|---|---|---|
| SiF4 | 22.56 | 120.6 | 126.5 |
| GeF4 | 17.39 | 116.4 | 120.1 |
| SnF4 | 10.68 | 124.3 | 131.4 |
Table 4.
Interaction energies (Eint, kcal mol−1) corrected for BSSE calculated at the MP2/cc-pVTZ (I), BLYP-D3/Def2TZVPP (II), and CCSD(T)/cc-pVTZ (III) levels of theory.
Table 4.
Interaction energies (Eint, kcal mol−1) corrected for BSSE calculated at the MP2/cc-pVTZ (I), BLYP-D3/Def2TZVPP (II), and CCSD(T)/cc-pVTZ (III) levels of theory.
| Axial Complexes | Equatorial Complexes | |||||
|---|---|---|---|---|---|---|
| (I) | (II) | (III) | (I) | (II) | (III) | |
| Py∙∙∙SiF4 | −26.75 | −22.50 | −27.07 | −50.09 | −47.38 | −50.98 |
| Py∙∙∙GeF4 | −34.73 | −34.37 | −34.93 | −52.02 | −48.71 | −52.62 |
| Py∙∙∙SnF4 | −39.68 | −37.87 | −39.85 | −50.66 | −44.97 | −51.13 |
| 4-HOPy∙∙∙SiF4 | −28.79 | −22.50 | −29.35 | −53.52 | −51.29 | −54.78 |
| 4-HOPy∙∙∙GeF4 | −36.62 | −34.37 | −37.10 | −55.30 | −51.93 | −56.26 |
| 4-HOPy∙∙∙SnF4 | −41.54 | −37.87 | −41.98 | −52.86 | −48.67 | −53.63 |
| 4-MePy∙∙∙SiF4 | −28.35 | −25.97 | −28.84 | −52.10 | −49.96 | −53.25 |
| 4-MePy∙∙∙GeF4 | −36.19 | −36.29 | −36.58 | −53.98 | −51.00 | −54.84 |
| 4-MePy∙∙∙SnF4 | −41.07 | −39.70 | −41.44 | −52.22 | −47.69 | −52.93 |
| 4-TFMPy∙∙∙SiF4 | −21.94 | −9.43 | −22.08 | - | - | - |
| 4-TFMPy∙∙∙GeF4 | −30.62 | −29.93 | −30.67 | −47.32 | −42.98 | −47.72 |
| 4-TFMPy∙∙∙SnF4 | −35.81 | −33.78 | −35.83 | −46.34 | −41.53 | −46.65 |
Table 5.
Binding energies (ΔEb, kcal mol−1) calculated at the MP2/cc-pVTZ (I) and BLYP-D3/Def2TZVPP (II) levels of theory.
Table 5.
Binding energies (ΔEb, kcal mol−1) calculated at the MP2/cc-pVTZ (I) and BLYP-D3/Def2TZVPP (II) levels of theory.
| Axial Complexes | Equatorial Complexes | |||
|---|---|---|---|---|
| (I) | (II) | (I) | (II) | |
| Py∙∙∙SiF4 | −10.39 | −7.85 | −0.98 | 1.67 |
| Py∙∙∙GeF4 | −20.51 | −18.06 | −14.60 | −12.97 |
| Py∙∙∙SnF4 | −32.41 | −27.64 | −29.14 | −24.59 |
| 4-HOPy∙∙∙SiF4 | −11.13 | −8.66 | −2.53 | −0.06 |
| 4-HOPy∙∙∙GeF4 | −21.52 | −19.29 | −16.36 | −14.86 |
| 4-HOPy∙∙∙SnF4 | −33.65 | −29.09 | −30.96 | −26.45 |
| 4-MePy∙∙∙SiF4 | −11.06 | −8.59 | −2.06 | 0.31 |
| 4-MePy∙∙∙GeF4 | −21.37 | −19.15 | −15.81 | −14.44 |
| 4-MePy∙∙∙SnF4 | −33.41 | −28.89 | −30.39 | −26.20 |
| 4-TFMPy∙∙∙SiF4 | −8.38 | −6.36 | - | - |
| 4-TFMPy∙∙∙GeF4 | −17.98 | −15.41 | −11.35 | −9.67 |
| 4-TFMPy∙∙∙SnF4 | −29.50 | −24.66 | −25.85 | −21.16 |
Table 6.
Deformation energies of complexes and maximum of MEP for TF4 in its geometry within the complex, calculated at the MP2/cc-pVTZ (I) level of theory. All quantities in kcal/mol.
Table 6.
Deformation energies of complexes and maximum of MEP for TF4 in its geometry within the complex, calculated at the MP2/cc-pVTZ (I) level of theory. All quantities in kcal/mol.
| Axial Complexes | ||||
|---|---|---|---|---|
| Edef of TF4 | Edef of LB | Sum | Vs,max | |
| Py∙∙∙SiF4 | 20.35 | 0.35 | 20.70 | 93.4 |
| Py∙∙∙GeF4 | 19.49 | 0.58 | 20.07 | 100.3 |
| Py∙∙∙SnF4 | 12.64 | 0.74 | 13.38 | 111.3 |
| 4-HOPy∙∙∙SiF4 | 21.72 | 0.41 | 22.13 | 95.0 |
| 4-HOPy∙∙∙GeF4 | 20.43 | 0.67 | 21.10 | 101.0 |
| 4-HOPy∙∙∙SnF4 | 13.28 | 0.85 | 14.13 | 112.1 |
| 4-MePy∙∙∙SiF4 | 21.35 | 0.37 | 21.72 | 94.6 |
| 4-MePy∙∙∙GeF4 | 20.15 | 0.60 | 20.75 | 101.0 |
| 4-MePy∙∙∙SnF4 | 13.09 | 0.76 | 13.85 | 111.9 |
| 4-TFMPy∙∙∙SiF4 | 17.35 | 0.33 | 17.68 | 89.6 |
| 4-TFMPy∙∙∙GeF4 | 17.70 | 0.58 | 18.28 | 98.5 |
| 4-TFMPy∙∙∙SnF4 | 11.49 | 0.75 | 12.24 | 110.0 |
| Equatorial Complexes | ||||
| Py∙∙∙SiF4 | 53.05 | 0.82 | 53.87 | 107.5 |
| Py∙∙∙GeF4 | 43.08 | 1.01 | 44.09 | 107.7 |
| Py∙∙∙SnF4 | 26.69 | 1.00 | 27.69 | 119.5 |
| 4-HOPy∙∙∙SiF4 | 54.93 | 0.94 | 55.87 | 109.1 |
| 4-HOPy∙∙∙GeF4 | 44.65 | 1.15 | 45.80 | 108.9 |
| 4-HOPy∙∙∙SnF4 | 27.06 | 1.17 | 28.23 | 119.8 |
| 4-MePy∙∙∙SiF4 | 54.02 | 0.84 | 54.86 | 108.3 |
| 4-MePy∙∙∙GeF4 | 43.90 | 1.03 | 44.93 | 108.4 |
| 4-MePy∙∙∙SnF4 | 27.05 | 1.04 | 28.08 | 119.8 |
| 4-TFMPy∙∙∙SiF4 | - | - | - | - |
| 4-TFMPy∙∙∙GeF4 | 41.43 | 1.04 | 42.47 | 106.4 |
| 4-TFMPy∙∙∙SnF4 | 25.52 | 1.04 | 26.56 | 118.2 |
Table 7.
EDA/BLYP-D3/ZORA/TZ2P decomposition of the interaction energy of complexes into Pauli repulsion (EPauli), electrostatic (Eelec), orbital interaction (Eoi) and dispersion (Edisp) components. All energies in kcal/mol.
Table 7.
EDA/BLYP-D3/ZORA/TZ2P decomposition of the interaction energy of complexes into Pauli repulsion (EPauli), electrostatic (Eelec), orbital interaction (Eoi) and dispersion (Edisp) components. All energies in kcal/mol.
| EPauli | Eelec | Eoi | Edisp | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ax | eq | eq − ax | ax | eq | eq − ax | ax | eq | eq − ax | ax | eq | eq − ax | |
| Py∙∙∙SiF4 | 89.72 | 117.10 | 27.38 | −70.23 | −93.70 | −23.47 | −40.00 | −67.04 | −27.04 | −4.53 | −4.38 | 0.15 |
| Py∙∙∙GeF4 | 108.13 | 110.56 | 2.43 | −84.50 | −90.48 | −5.98 | −49.68 | −64.82 | −15.14 | −4.63 | −4.19 | 0.44 |
| Py∙∙∙SnF4 | 99.74 | 87.36 | −12.38 | −85.48 | −80.05 | 5.43 | −45.43 | −48.91 | −3.48 | −4.52 | −3.98 | 0.54 |
| 4-HOPy∙∙∙SiF4 | 94.13 | 120.82 | 26.69 | −74.10 | −97.86 | −23.76 | −42.67 | −70.50 | −27.83 | −4.53 | −4.33 | 0.20 |
| 4-HOPy∙∙∙GeF4 | 112.02 | 116.15 | 4.13 | −88.08 | −95.58 | −7.50 | −51.93 | −68.40 | −16.47 | −4.63 | −4.20 | 0.43 |
| 4-HOPy∙∙∙SnF4 | 102.86 | 92.22 | −10.64 | −88.83 | −85.27 | 3.56 | −47.15 | −52.27 | −5.12 | −4.52 | −3.93 | 0.59 |
| 4-MePy∙∙∙SiF4 | 92.92 | 117.10 | 24.18 | −73.01 | −93.70 | −20.60 | −42.06 | −67.04 | −24.98 | −4.54 | −4.38 | 0.16 |
| 4-MePy∙∙∙GeF4 | 110.73 | 110.56 | −0.17 | −86.99 | −90.48 | −3.49 | −51.39 | −64.82 | −13.43 | −4.65 | −4.19 | 0.46 |
| 4-MePy∙∙∙SnF4 | 101.57 | 87.36 | −14.21 | −87.63 | −80.05 | 7.58 | −46.71 | −48.91 | −2.20 | −4.54 | −3.98 | 0.56 |
| 4-TFMPy∙∙∙SiF4 | 80.38 | - | - | −61.74 | - | - | −34.59 | - | - | −4.56 | - | - |
| 4-TFMPy∙∙∙GeF4 | 101.43 | 104.42 | 2.99 | −77.67 | −83.12 | −5.45 | −45.96 | −60.09 | −14.13 | −4.66 | −4.26 | 0.40 |
| 4-TFMPy∙∙∙SnF4 | 94.96 | 82.47 | −12.49 | −79.49 | −73.92 | 5.57 | −42.96 | −46.70 | −3.74 | −4.56 | −3.93 | 0.63 |
Table 8.
AIM bond critical point (BCP) properties: electron density ρ, Laplacian of electron density ∇2ρ (both in atomic units) and total electron energy (H, a.u.) obtained at the MP2/cc-pVTZ level.
Table 8.
AIM bond critical point (BCP) properties: electron density ρ, Laplacian of electron density ∇2ρ (both in atomic units) and total electron energy (H, a.u.) obtained at the MP2/cc-pVTZ level.
| Axial Complexes | Equatorial Complexes | |||||||
|---|---|---|---|---|---|---|---|---|
| Interaction | ρ | ∇2ρ | H | Interaction | ρ | ∇2ρ | H | |
| Py∙∙∙SiF4 | Si···N | 0.054 | 0.145 | −0.022 | Si···N | 0.077 | 0.276 | −0.032 |
| F···H | 0.015 | 0.073 | 0.003 | F···H | 0.019 | 0.094 | 0.003 | |
| - | - | - | F···H | 0.019 | 0.094 | |||
| Py∙∙∙GeF4 | Ge···N | 0.077 | 0.166 | −0.030 | Ge···N | 0.094 | 0.216 | −0.042 |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.020 | 0.093 | 0.004 | |
| F···H | 0.015 | 0.070 | 0.003 | F···H | 0.020 | 0.093 | 0.004 | |
| Py∙∙∙SnF4 | Sn···N | 0.073 | 0.211 | −0.020 | Sn···N | 0.077 | 0.230 | −0.022 |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.095 | 0.004 | |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.095 | 0.004 | |
| 4-HOPy∙∙∙SiF4 | Si···N | 0.057 | 0.159 | −0.023 | Si···N | 0.079 | 0.289 | −0.033 |
| F···H | 0.015 | 0.074 | 0.003 | F···H | 0.021 | 0.100 | 0.004 | |
| - | - | - | F···H | 0.020 | 0.099 | |||
| 4-HOPy∙∙∙GeF4 | Ge···N | 0.079 | 0.174 | −0.032 | Ge···N | 0.097 | 0.225 | −0.044 |
| F···H | 0.015 | 0.070 | 0.003 | F···H | 0.021 | 0.100 | 0.004 | |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.021 | 0.099 | 0.004 | |
| 4-HOPy∙∙∙SnF4 | Sn···N | 0.075 | 0.218 | −0.021 | Sn···N | 0.079 | 0.240 | −0.023 |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.095 | 0.004 | |
| F···H | 0.014 | 0.060 | 0.002 | F···H | 0.020 | 0.093 | 0.003 | |
| 4-MePy∙∙∙SiF4 | Si···N | 0.056 | 0.155 | −0.022 | Si···N | 0.079 | 0.284 | −0.032 |
| F···H | 0.015 | 0.073 | 0.003 | F···H | 0.019 | 0.095 | 0.003 | |
| - | - | - | F···H | 0.019 | 0.094 | |||
| 4-MePy∙∙∙GeF4 | Ge···N | 0.079 | 0.170 | −0.032 | Ge···N | 0.096 | 0.221 | −0.043 |
| F···H | 0.015 | 0.069 | 0.003 | F···H | 0.020 | 0.094 | 0.004 | |
| F···H | 0.015 | 0.069 | 0.003 | F···H | 0.020 | 0.094 | 0.004 | |
| 4-MePy∙∙∙SnF4 | Sn···N | 0.075 | 0.215 | −0.021 | Sn···N | 0.078 | 0.235 | −0.022 |
| F···H | 0.013 | 0.058 | 0.002 | F···H | 0.020 | 0.093 | 0.003 | |
| F···H | 0.013 | 0.058 | 0.002 | F···H | 0.020 | 0.093 | 0.003 | |
| 4-TFMPy∙∙∙SiF4 | Si···N | 0.049 | 0.118 | −0.020 | - | - | - | |
| 4-TFMPy∙∙∙GeF4 | Ge···N | 0.073 | 0.158 | −0.027 | Ge···N | 0.091 | 0.208 | −0.040 |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.020 | 0.094 | 0.003 | |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.020 | 0.094 | 0.003 | |
| 4-TFMPy∙∙∙SnF4 | Sn···N | 0.071 | 0.204 | −0.019 | Sn···N | 0.075 | 0.222 | −0.020 |
| F···H | 0.014 | 0.060 | 0.002 | F···H | 0.021 | 0.096 | 0.004 | |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.096 | 0.004 | |
Table 9.
NBO values of sum of E(2) (kcal/mol) for LP (N) → LP* (T), and total charge transfer (CT, me) from LB to TF4 obtained at the BLYP-D3/def2-TVZPP level.
Table 9.
NBO values of sum of E(2) (kcal/mol) for LP (N) → LP* (T), and total charge transfer (CT, me) from LB to TF4 obtained at the BLYP-D3/def2-TVZPP level.
| Axial Complexes | Equatorial Complexes | |||
|---|---|---|---|---|
| ∑ LP (N) → LP* (T) | CT | ∑ LP (N) → LP* (T) | CT | |
| Py∙∙∙SiF4 | 72.8 | 121 | 161.2 | 212 |
| Py∙∙∙GeF4 | 137.7 | 174 | 158.4 | 220 |
| Py∙∙∙SnF4 | 108.2 | 185 | 111.0 | 202 |
| 4-HOPy∙∙∙SiF4 | 98.5 | 139 | 169.0 | 221 |
| 4-HOPy∙∙∙GeF4 | 146.0 | 182 | 166.2 | 227 |
| 4-HOPy∙∙∙SnF4 | 113.5 | 191 | 116.6 | 210 |
| 4-MePy∙∙∙SiF4 | 90.7 | 138 | 166.9 | 218 |
| 4-MePy∙∙∙GeF4 | 143.8 | 180 | 163.6 | 225 |
| 4-MePy∙∙∙SnF4 | 110.4 | 190 | 114.9 | 209 |
| 4-TFMPy∙∙∙SiF4 | 17.9 | 36 | - | - |
| 4-TFMPy∙∙∙GeF4 | 123.4 | 159 | 149.2 | 206 |
| 4-TFMPy∙∙∙SnF4 | 100.6 | 172 | 103.5 | 192 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives. Molecules 2019, 24, 376. https://doi.org/10.3390/molecules24020376
AMA Style
Zierkiewicz W, Michalczyk M, Wysokiński R, Scheiner S. Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives. Molecules. 2019; 24(2):376. https://doi.org/10.3390/molecules24020376
Chicago/Turabian StyleZierkiewicz, Wiktor, Mariusz Michalczyk, Rafał Wysokiński, and Steve Scheiner. 2019. "Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives" Molecules 24, no. 2: 376. https://doi.org/10.3390/molecules24020376