Co-Delivery of Gemcitabine and Paclitaxel in cRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment

 ,
,
Abstract
:1. Introduction
2. Results
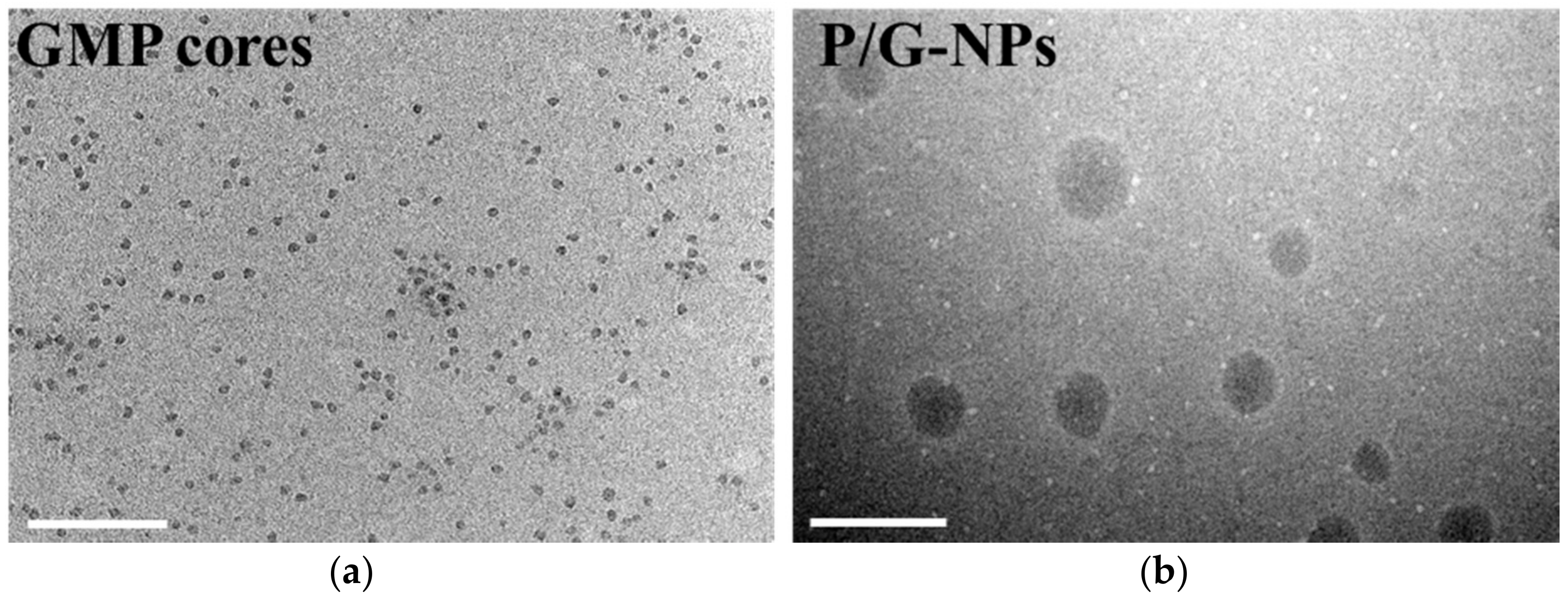
2.1. Characterization of Nanoparticles
2.2. Cellular Uptake
2.3. In Vitro Release
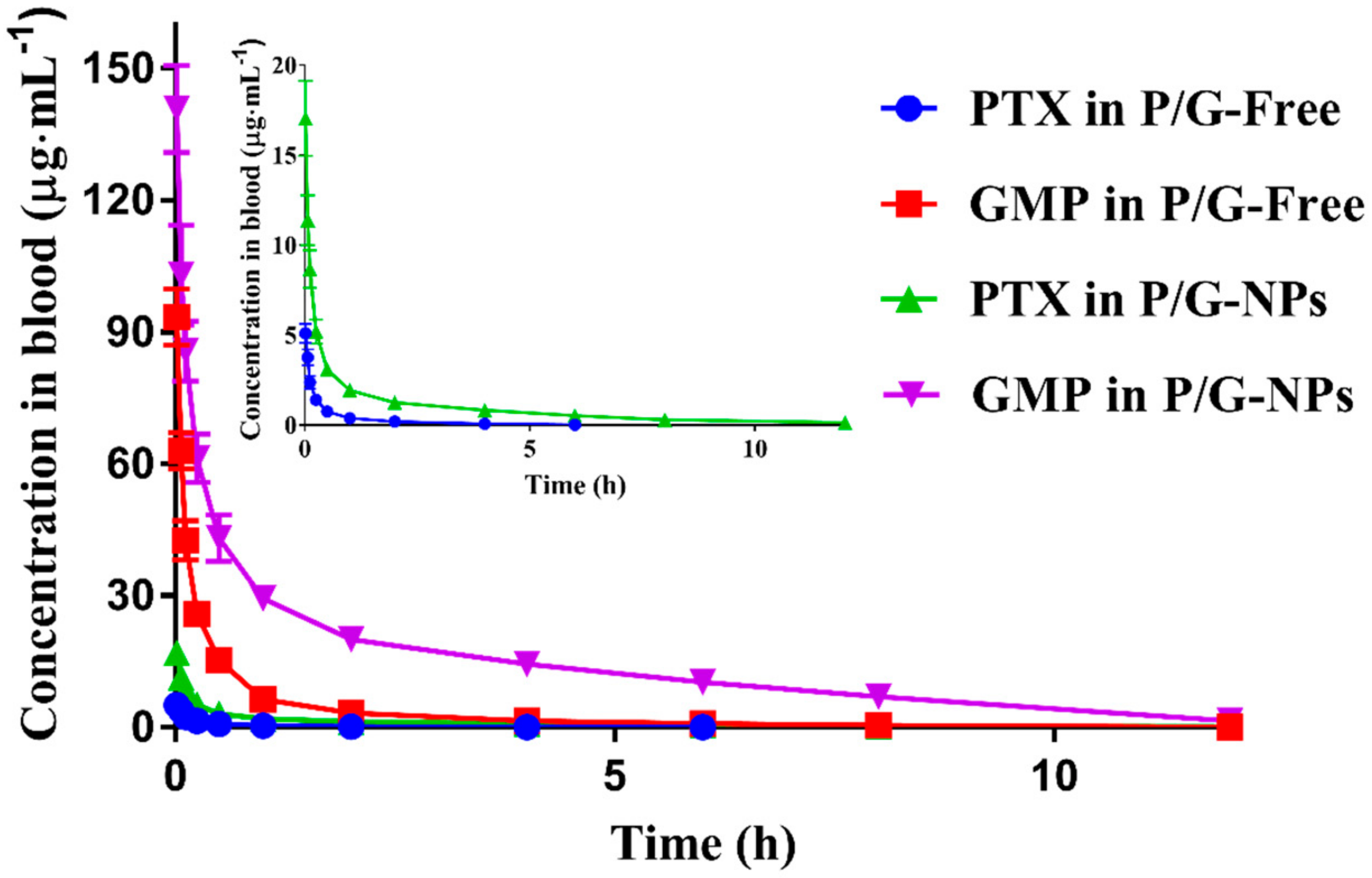
2.4. Pharmacokinetics of P/G Formulations
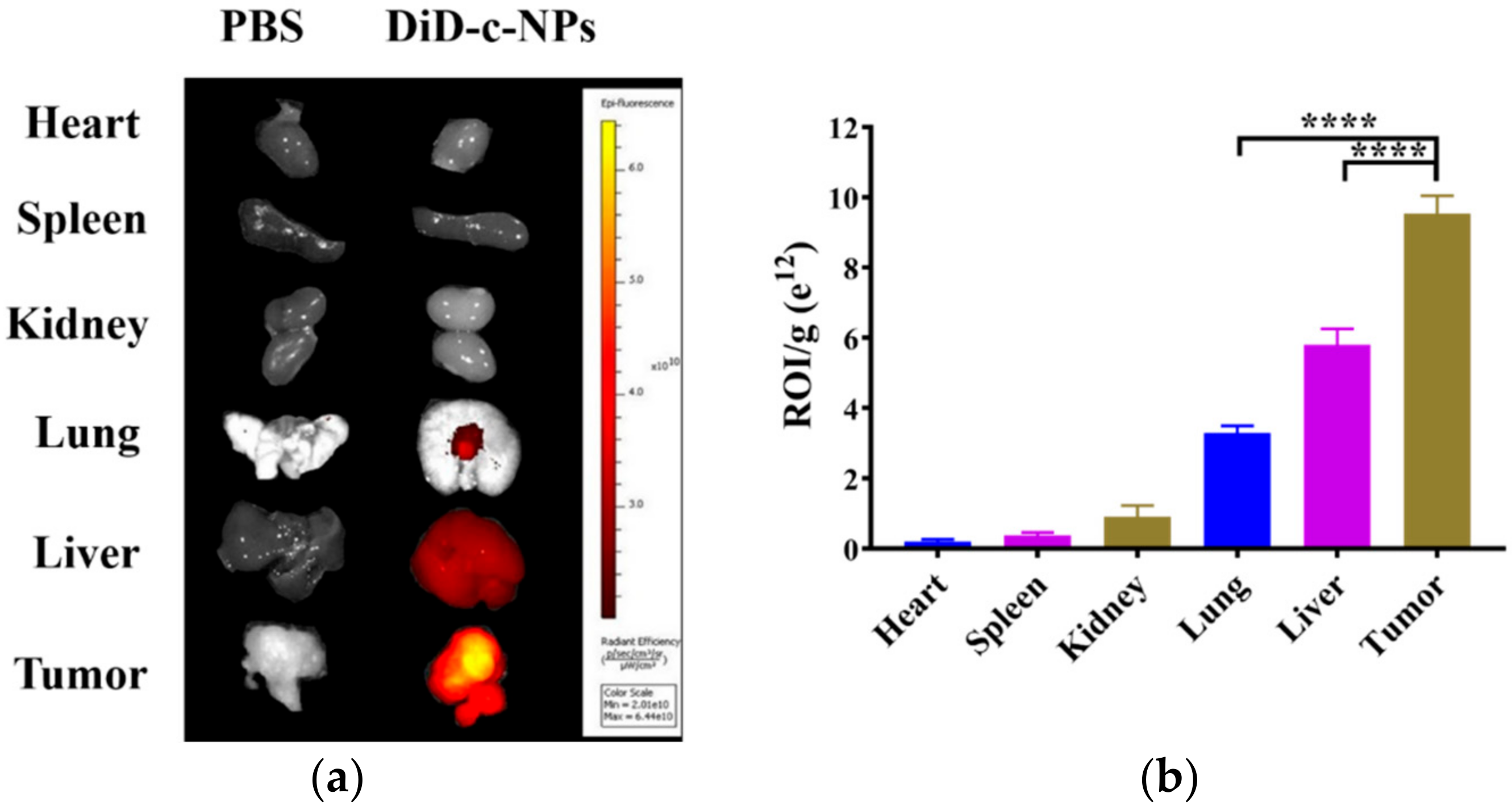
2.5. Tissue Distribution
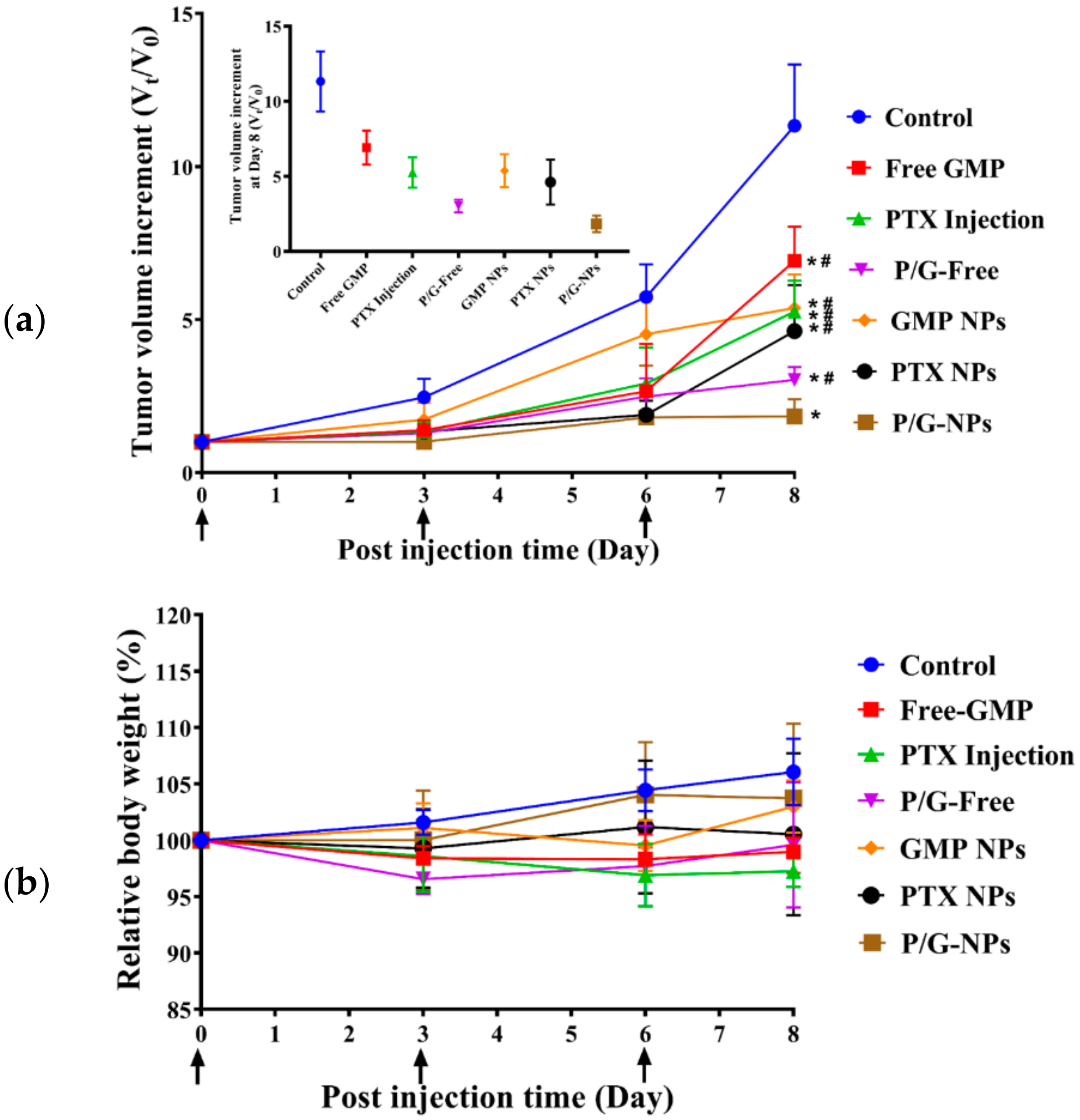
2.6. Antitumor Effect of P/G-NPs
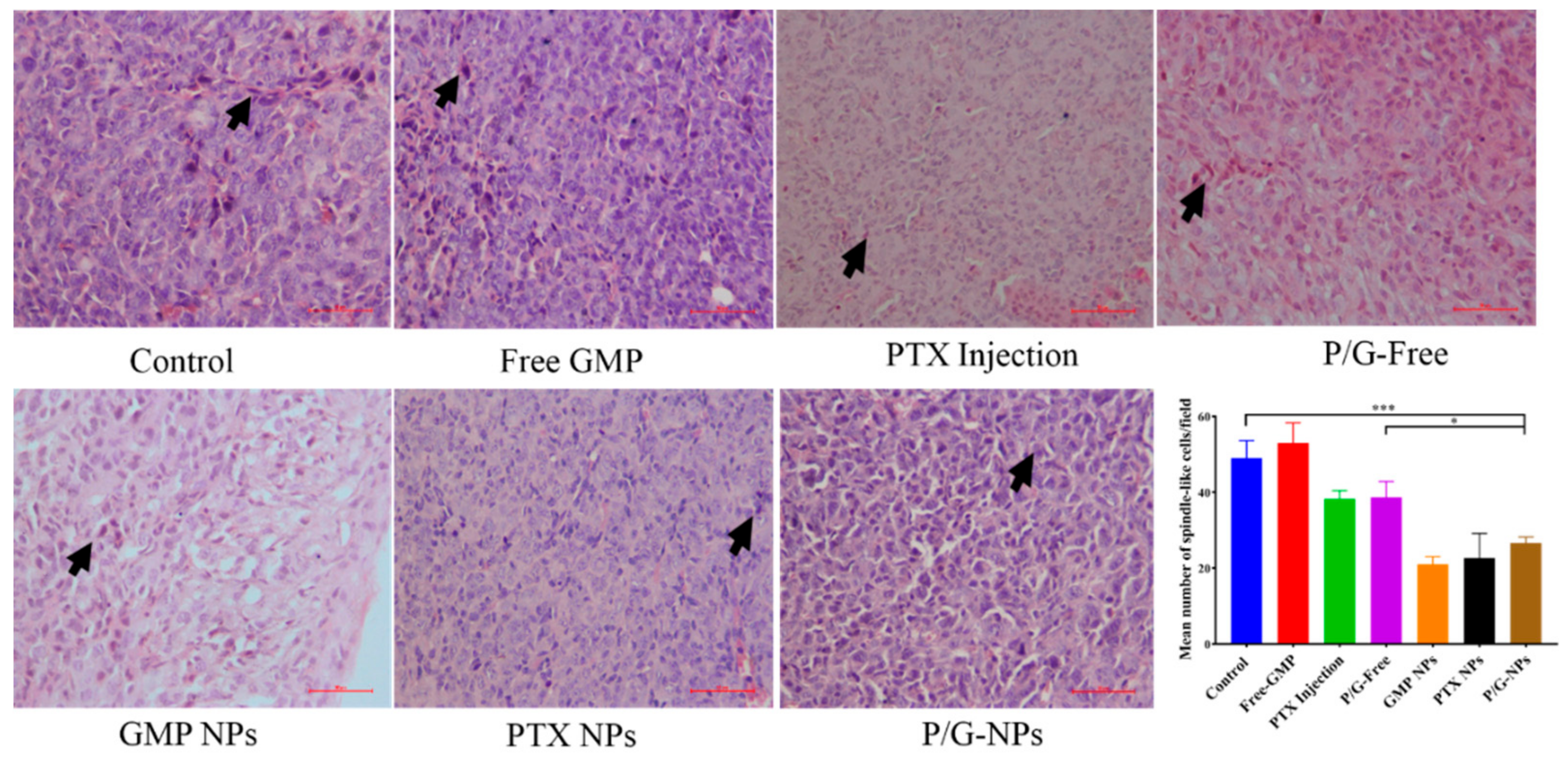
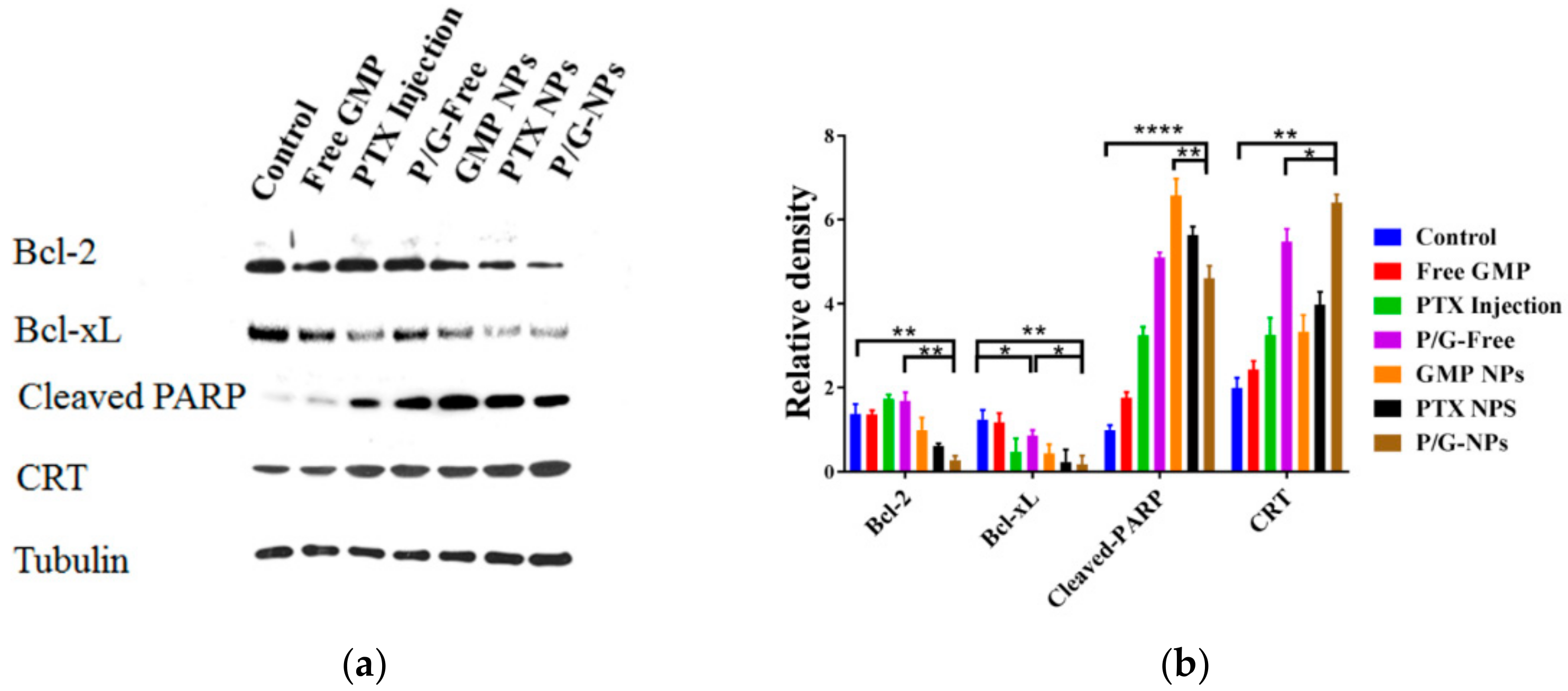
2.7. Induction of Significant Cell Apoptosis and Immunogenic Cell Death of P/G-NPs
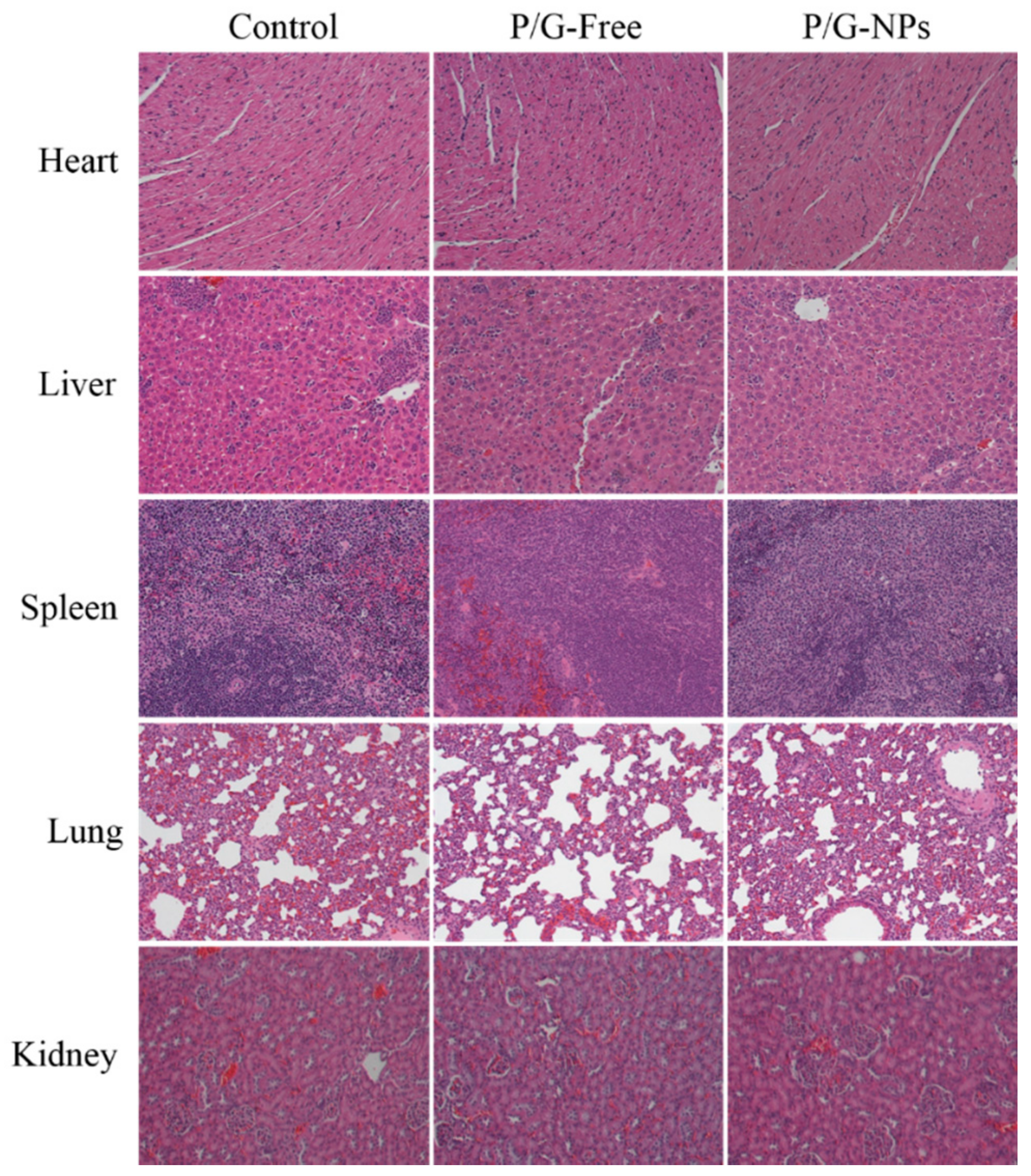
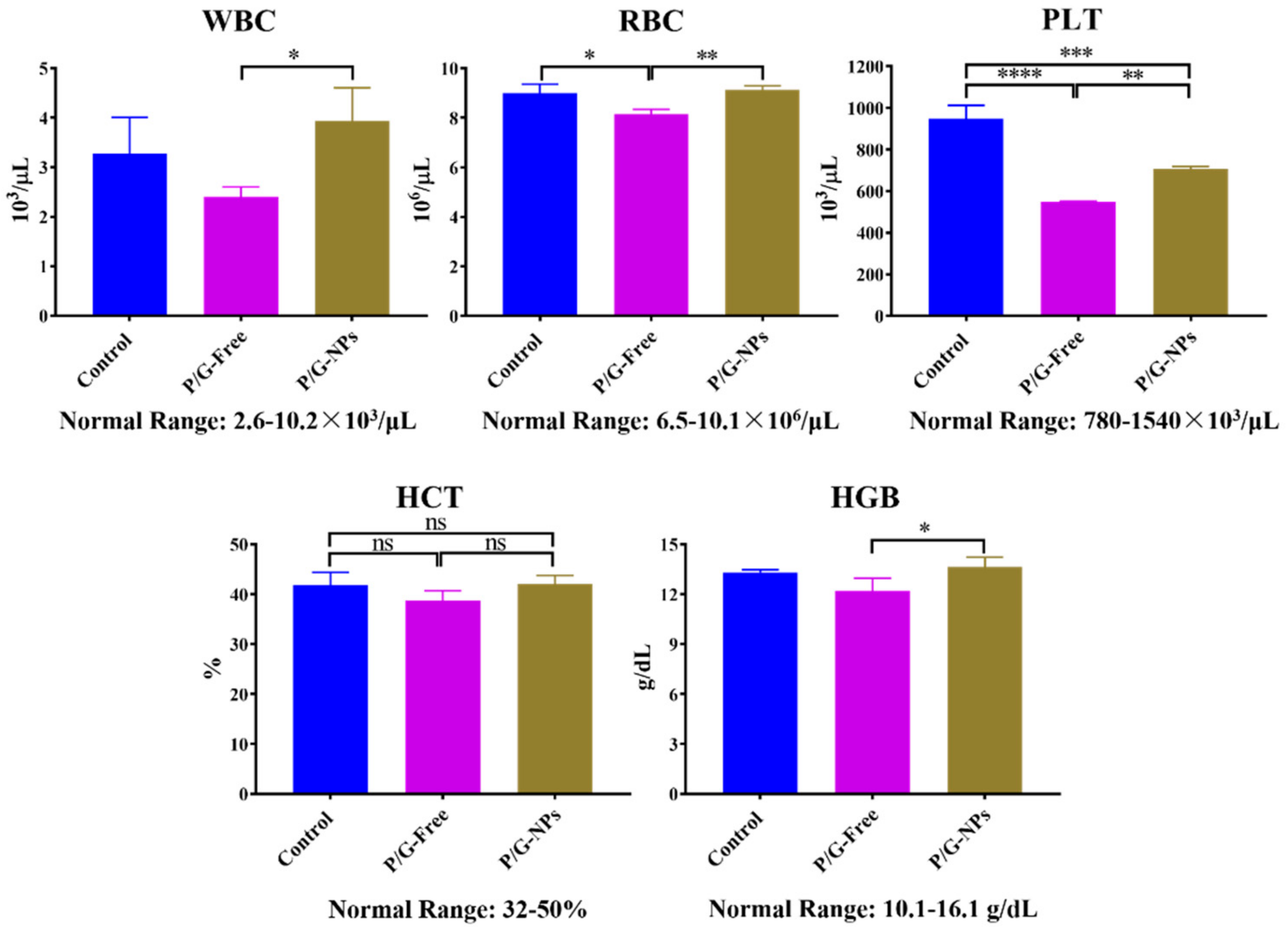
2.8. Toxicity Analysis In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Preparation of DOPA-Coated GMP Cores
4.4. Preparation of P/G-NPs Loads with Different Cargos
4.5. Characterization of P/G-NPs
4.6. Cellular Uptake
4.7. In Vitro Drug Release
4.8. Pharmacokinetics Studies
4.9. Antitumor Efficacy In Vivo
4.10. Tissue Distribution
4.11. Tissue Analysis
4.12. Toxicity Analyses In Vivo
4.13. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures 2018; American Cancer Society: Atlanta, GA, USA, 2018; pp. 1–76. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, K.; Ardavanis, A.; Kountourakis, P. Neoadjuvant therapy for locally advanced breast cancer: Focus on chemotherapy and biological targeted treatments’ armamentarium. J. Thorac. Dis. 2010, 2, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Hatam, N.; Askarian, M.; Javan-Noghabi, J.; Ahmadloo, N.; Mohammadianpanah, M. Cost-utility of “Doxorubicin and Cyclophosphamide” versus “Gemcitabine and Paclitaxel” for treatment of patients with breast cancer in Iran. Asian Pac. J. Cancer Prev. 2015, 16, 8265–8270. [Google Scholar] [CrossRef] [PubMed]
- Ghoncheh, M.; Pournamda, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Wang, J.; Lohman, G.J.S.; Stubbe, J. Mechanism of inactivation of human ribonucleotide reductase with p53R2 by gemcitabine 5′-diphosphate. Biochemistry 2009, 48, 11612–11621. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Tamam, H.; Yeo, Y. Mixed liposome approach for ratiometric and sequential delivery of paclitaxel and gemcitabine. AAPS PharmSciTech 2018, 9, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, W.; Huang, P.; Gandhi, V. Gemcitabine: Actions and Interactions. Nucleosides Nucleotides 1997, 1261–1270. [Google Scholar] [CrossRef]
- Abbruzzese, J.L.; Grunewald, R.; Weeks, E.A.; Gravel, D.; Adams, T.; Nowak, B.; Mineishi, S.; Tarassoff, P.; Satterlee, W.; Raber, M.N. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.; Mavroudis, D.; Nikolaidou, M.; Georgoulias, V.; Marselos, M. Coadministration of oxaliplatin does not influence the pharmacokinetics of gemcitabine. Anti-Cancer Drugs 2006, 17, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Sloat, B.R.; Sandoval, M.A.; Li, D.; Chung, W.G.; Lansakara, P.D.S.P.; Proteau, P.J.; Kiguchi, K.; DiGiovanni, J.; Cui, Z.R. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int. J. Pharm. 2011, 409, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albain, K.S.; Nag, S.M.; Calderillo-Ruiz, G.; Jordaan, J.P.; Llombart, A.C.; Pluzanska, A.; Rolski, J.; Melemed, A.S.; Reyes-Vidal, J.M.; Sekhon, J.S.; et al. Gemcitabine plus Paclitaxel versus Paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J. Clin. Oncol. 2008, 26, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Jung, K.H.; Im, S.A.; Sohn, J.H.; Ro, J.; Ahn, J.H.; Kim, S.B.; Nam, B.H.; Oh, D.Y.; Han, S.W.; et al. Phase III, multicenter, randomized trial of maintenance chemotherapy versus observation in patients with metastatic breast cancer after achieving disease control with six cycles of gemcitabine plus paclitaxel as first-line chemotherapy: KCSG-BR07-02. J. Clin. Oncol. 2013, 31, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Borsoi, C.; Leonard, F.; Lee, Y.; Zaid, M.; Elganainy, D.; Alexander, J.F.; Kai, M.; Liu, Y.T.; Kang, Y.; Liu, X.; et al. Gemcitabine enhances the transport of nanovector-albumin-bound paclitaxel in gemcitabine-resistant pancreatic ductal adenocarcinoma. Cancer Lett. 2017, 403, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.M.; Li, W.; Zhang, Y.Y.; Zhang, B. Anti-tumor efficiency of paclitaxel and DNA when co-delivered by pH responsive ligand modified nanocarriers for breast cancer treatment. Biomed. Pharmacother. 2016, 83, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Noh, I.; Kim, H.O.; Choi, J.; Choi, Y.; Lee, D.K.; Huh, Y.M.; Haam, S. Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials 2015, 53, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Brusa, P.; Rocco, F.; Arpicco, S.; Ceruti, M.; Cattel, L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J. Control. Release 2004, 100, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Mumper, R.J. Paclitaxel Nano-Delivery Systems: A Comprehensive Review. J. Nanomed. Nanotechnol. 2013, 4, 1000164. [Google Scholar] [CrossRef] [PubMed]
- Fogli, S.; Danesi, R.; Gennari, A.; Donati, S.; Conte, P.F.; Del Tacca, M. Gemcitabine, epirubicin and paclitaxel: Pharmacokinetic and pharmacodynamic interactions in advanced breast cancer. Ann. Oncol. 2002, 13, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Persidis, A. Cancer multidrug resistance. Nat. Biotechnol. 1999, 17, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Vrignaud, S.; Benoit, J.P.; Saulnier, P. Strategies for the nanoencapsulation of hydrophilic molecules in polymer-based nanoparticles. Biomaterials 2011, 32, 8593–8604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, Y.; Gao, Y.Y.; Gai, X.M.; Wang, D.; Wang, Y.Y.; Yang, X.G.; Zhang, D.; Pan, W.S.; Yang, X.G. Co-delivery of hydrophilic gemcitabine and hydrophobic paclitaxel into novel polymeric micelles for cancer treatment. RSC Adv. 2017, 7, 24030–24039. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Guo, S.T.; Zhang, J.; Kim, W.Y.; Huang, L. Nanoparticles with precise ratiometric co-loading and co-delivery of gemcitabine monophosphate and cisplatin for treatment of bladder cancer. Adv. Funct. Mater. 2014, 24, 6601–6611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schwerbrock, N.M.; Rogers, A.B.; Kim, W.Y.; Huang, L. Codelivery of VEGF siRNA and Gemcitabine monophosphate in a single nanoparticle formulation for effective treatment of NSCLC. Mol. Ther. 2013, 21, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Miao, L.; Guo, S.T.; Zhang, Y.; Zhang, L.; Satterlee, A.; Kim, W.Y.; Huang, L. Synergistic anti-tumor effects of combined gemcitabine and cisplatin nanoparticles in a stroma-rich bladder carcinoma model. J. Control. Release 2014, 182, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Zhang, J.M.; Fu, C.M.; Xie, X.M.; Peng, F.; You, J.S.; Tang, H.L.; Wang, Z.Y.; Li, P.; Chen, J.P. iRGD-modified lipid–polymer hybrid nanoparticles loaded with isoliquiritigenin to enhance anti-breast cancer effect and tumor-targeting ability. Int. J. Nanomed. 2017, 12, 4147–4162. [Google Scholar] [CrossRef] [PubMed]
- Hirsjärvi, S.; Belloche, C.; Hindré, F.; Garcion, E.; Benoit, J.P. Tumour targeting of lipid nanocapsules grafted with cRGD peptides. Eur. J. Pharm. Biopharm. 2014, 87, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the role of RGD-recognizing integrins in cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, B.P.; Cheresh, D.A. The Role of αv integrins during angiogenesis: Insights into potential mechanisms of action and clinical development. J. Clin. Investig. 1999, 103, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, Y.Y.; Shen, Y.Q.; Wang, A.M.; Wang, S.L.; Xie, T. The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Slee, A.M.; Mousa, S.A. The αv integrin antagonists as novel anticancer agents: An update. Expert Opin. Investig. Drugs 2002, 11, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Gajbhiye, K.R.; Gajbhiye, V.; Siddiqui, I.A.; Gajbhiye, J.M. cRGD functionalised nanocarriers for targeted delivery of bioactives. J. Drug Target. 2018, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Murugan, C.; Rayappan, K.; Thangam, R.; Bhanumathi, R.; Shanthi, K.; Vivek, R.; Thirumurugan, R.; Bhattacharyya, A.; Sivasubramanian, S.; Gunasekaran, P.; et al. Combinatorial nanocarrier based drug delivery approach for amalgamation of anti-tumor agents in breast cancer cells: An improved nanomedicine strategies. Sci. Rep. 2016, 6, 34503. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Z.; Song, Y.L.; Di, Y.; He, H.; Fu, D.L.; Jin, C. Enhanced tumor targeting of cRGD peptide-conjugated albumin nanoparticles in the BxPC-3 cell line. Sci. Rep. 2016, 6, 31539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [CrossRef] [PubMed]
- Gaber, M.H.; Hong, K.; Huang, S.K.; Papahadjopoulos, D. Thermosensitive Sterically Stabilized Liposomes: Formulation and In Vitro Studies on Mechanism of Doxorubicin Release by Bovine Serum and Human Plasma. Pharm. Res. 1995, 12, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Goel, H. Oral leiomyoma extending in retromolar region. J. Indian Soc. Pedod. Prev. Dent. 2011, 29, S61–S65. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. 1994, 5, S3–S6. [Google Scholar] [PubMed]
- Zhang, R.; Yang, J.Y.; Sima, M.; Zhou, Y.; Kopeček, J. Sequential combination therapy of ovarian cancer with degradable N-(2-hydroxypropyl) methacrylamide copolymer paclitaxel and gemcitabine conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 12181–12186. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.C.; Tseng, Y.C.; Mozumdar, S.; Huang, L. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J. Control. Release 2010, 142, 416–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.L.; Chen, H.Z.; Gao, X.L. Lipid-coated calcium phosphate nanoparticle and beyond: A versatile platform for drug delivery. J. Drug Target. 2018, 26, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Wang, M.Y.; Liu, H.Y.; Liu, X.S.; Situ, A.; Wu, B.; Ji, Z.X.; Chang, C.H.; Nel, A.E. Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano 2015, 9, 3540–3557. [Google Scholar] [CrossRef] [PubMed]
- Wilschut, J.; Hoekstra, D. Membrane fusion: From liposomes to biological membranes. Trends Biochem. Sci. 1984, 9, 479–483. [Google Scholar] [CrossRef]
- Foldvari, M. Observations of membrane fusion in a liposome dispersion: The missing fusion intermediate? F1000Res 2015, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- William, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.W.; Ellis, I.O.; Robertson, J.F.R.; Willsher, P.; McClelland, R.A.; Hewitt, K.N.; Blamey, R.W.; Nicholson, R.I. Immunocytochemical localization of BCL-2 protein in human breast cancers and its relationship to a series of prognostic markers and response to endocrine therapy. Int. J. Cancer 1994, 59, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mandal, M.; Lipton, A.; Harvey, H.; Thompson, C.B. Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifeninduced apoptosis in human MCF-7 breast cancer cells. Clin. Cancer Res. 1996, 2, 1215–1219. [Google Scholar] [PubMed]
- Fiebig, A.A.; Zhu, W.J.; Hollerbach, C.; Leber, B.; Andrews, D.W. Bcl-xL is qualitatively different from and ten times more effective than Bcl-2 when expressed in a breast cancer cell line. BMC Cancer. 2006, 6, 213. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Mu, H.X.; Dai, K.; Yi, L. Calreticulin: A potential anti-cancer therapeutic target. Pharmazie 2017, 72, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2006, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Prathyuman, S.; Sellappa, S.; Joseph, S.; Keyan, K.S. Enhanced calreticulin expression triggers apoptosis in the MCF-7 cell line. Asian Pac. J. Cancer Prev. 2010, 11, 1133–1136. [Google Scholar] [PubMed]
- Chen, P.; Luo, S.; Wen, Y.J.; Li, Y.H.; Li, J.; Wang, Y.S.; Du, L.C.; Zhang, P.; Tang, J.; Yang, D.B.; et al. Low-dose paclitaxel improves the therapeutic efficacy of recombinant adenovirus encoding CCL21 chemokine against murine cancer. Cancer Sci. 2014, 5, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Greish, K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J. Drug Target. 2004, 15, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Y.C.; Li, X.; Liang, X.L.; Luo, X.J. The influence of different long-circulating materials on the pharmacokinetics of liposomal vincristine sulfate. Int. J. Nanomed. 2016, 11, 4187–4197. [Google Scholar] [CrossRef]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle size (nm) | Zeta Potential (mV) | EE 1 of GMP 2 (%) | EE of PTX 3(%) | DL 4 of GMP (wt % 5) | DL of PTX (wt %) | |
|---|---|---|---|---|---|---|
| Day 1 | 85.1 ± 8.1 | 18.3 ± 0.63 | 93.6 ± 1.2 | 98.7 ± 0.5 | 6.3 ± 0.1 | 0.8 ± 0.004 |
| Day 15 | 86.8 ± 6.9 | 17.4 ± 0.88 | 95.8 ± 3.8 | 97.5 ± 0.6 | 6.4 ± 0.3 | 0.8 ± 0.005 |
| P/G-Free | P/G-NPs | |||
|---|---|---|---|---|
| PTX | GMP | PTX | GMP | |
| AUC0→ t (μg·mL−1·min−1) | 114.53 ± 13.53 | 2265.97 ± 293.34 | 684.97 ± 99.93 * | 10,324.06 ± 1801.59 § |
| MRT0→ t (min) | 59.25 ± 4.13 | 92.45 ± 11.25 | 197.28 ± 17.08 * | 210.68 ± 16.32 § |
| CL1 (mL·min−1) | 0.017 ± 0.003 | 0.0070 ± 0.0023 | 0.0028 ± 0.0011 * | 0.0015 ± 0. 0004 § |
| Vz2 (L/kg) | 1.49 ± 0.31 | 1.22 ± 0.35 | 0.74 ± 0.23 * | 0.34 ± 0.11 § |
| Cmax (μg·mL−1) | 5.10 ± 1.76 | 93.38 ± 18.87 | 17.04 ± 4.22 * | 140.80 ± 25.22 § |
| AST 1 (U/L) | ALT 2 (U/L) | LDH 3 (U/L) | Creatinine (mg/dL) | BUN 4 (mmol/L) | |
|---|---|---|---|---|---|
| Control | 103.3 ± 49.2 | 79.0 ± 6.4 | 95.3 ± 30.5 | 0.2 ± 0.2 | 8.4 ± 1.4 |
| P/G-Free | 178.3 ± 51.6 | 110.2 ± 21.7 | 100.1 ± 28.9 | 0.5 ± 0.1 | 10.3 ± 2.9 |
| P/G-NPs | 206.6 ± 38.4 | 98.6 ± 9.4 | 120.7 ± 41.2 | 0.4 ± 0.3 | 15.9 ± 2.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Zhang, P.; Zou, Q.; Li, X.; Fu, J.; Luo, Y.; Liang, X.; Jin, Y. Co-Delivery of Gemcitabine and Paclitaxel in cRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment. Molecules 2018, 23, 2906. https://doi.org/10.3390/molecules23112906
Zhang J, Zhang P, Zou Q, Li X, Fu J, Luo Y, Liang X, Jin Y. Co-Delivery of Gemcitabine and Paclitaxel in cRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment. Molecules. 2018; 23(11):2906. https://doi.org/10.3390/molecules23112906
Chicago/Turabian StyleZhang, Jing, Peng Zhang, Qian Zou, Xiang Li, Jianjiang Fu, Ying Luo, Xinli Liang, and Yi Jin. 2018. "Co-Delivery of Gemcitabine and Paclitaxel in cRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment" Molecules 23, no. 11: 2906. https://doi.org/10.3390/molecules23112906