
Unfractionated and Low Molecular Weight Heparin Reduce Platelet Induced Epithelial-Mesenchymal Transition in Pancreatic and Prostate Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
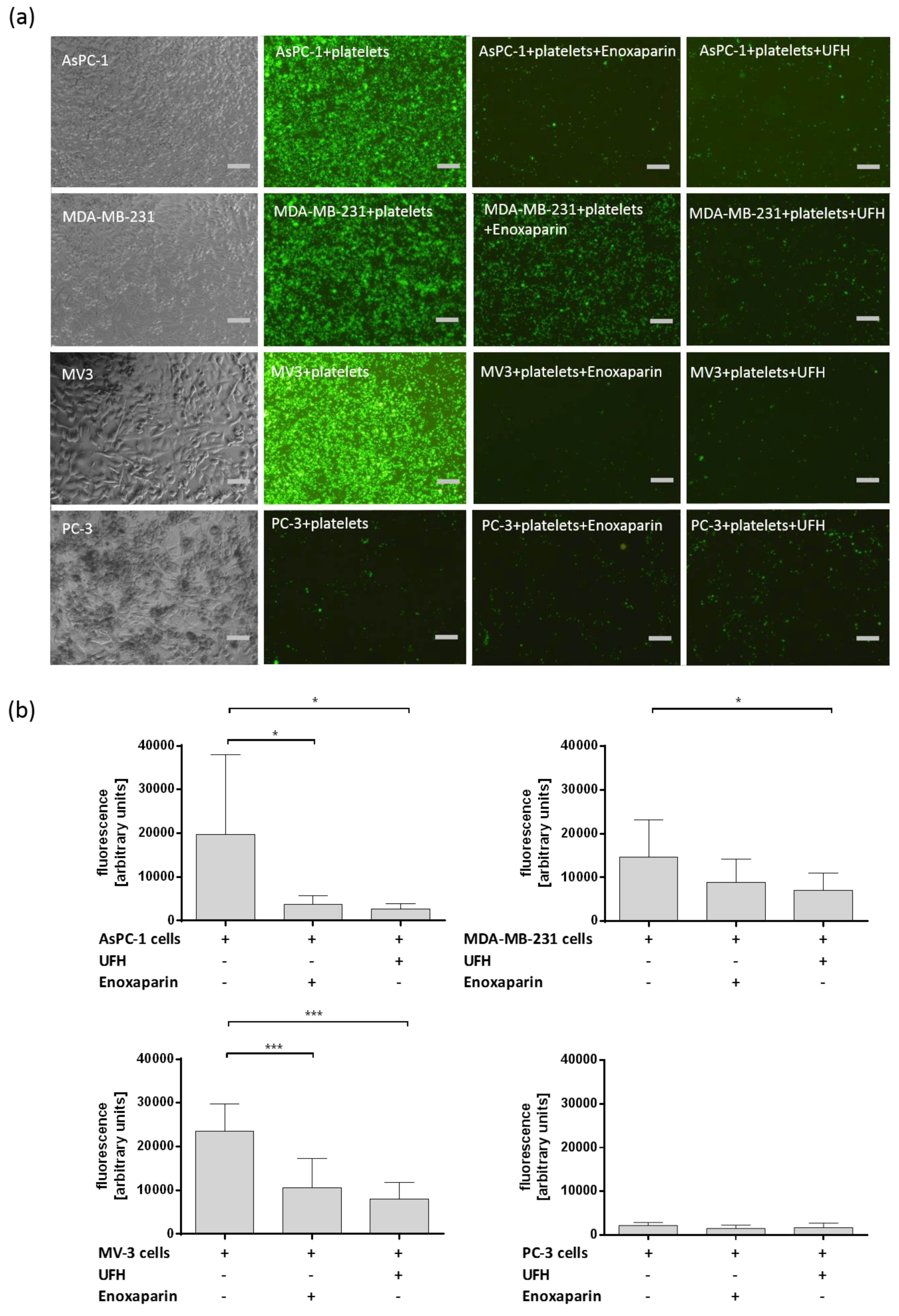
2.1. Interaction between Different Tumor Cells and Platelets and the Impact of UFH and LMWH
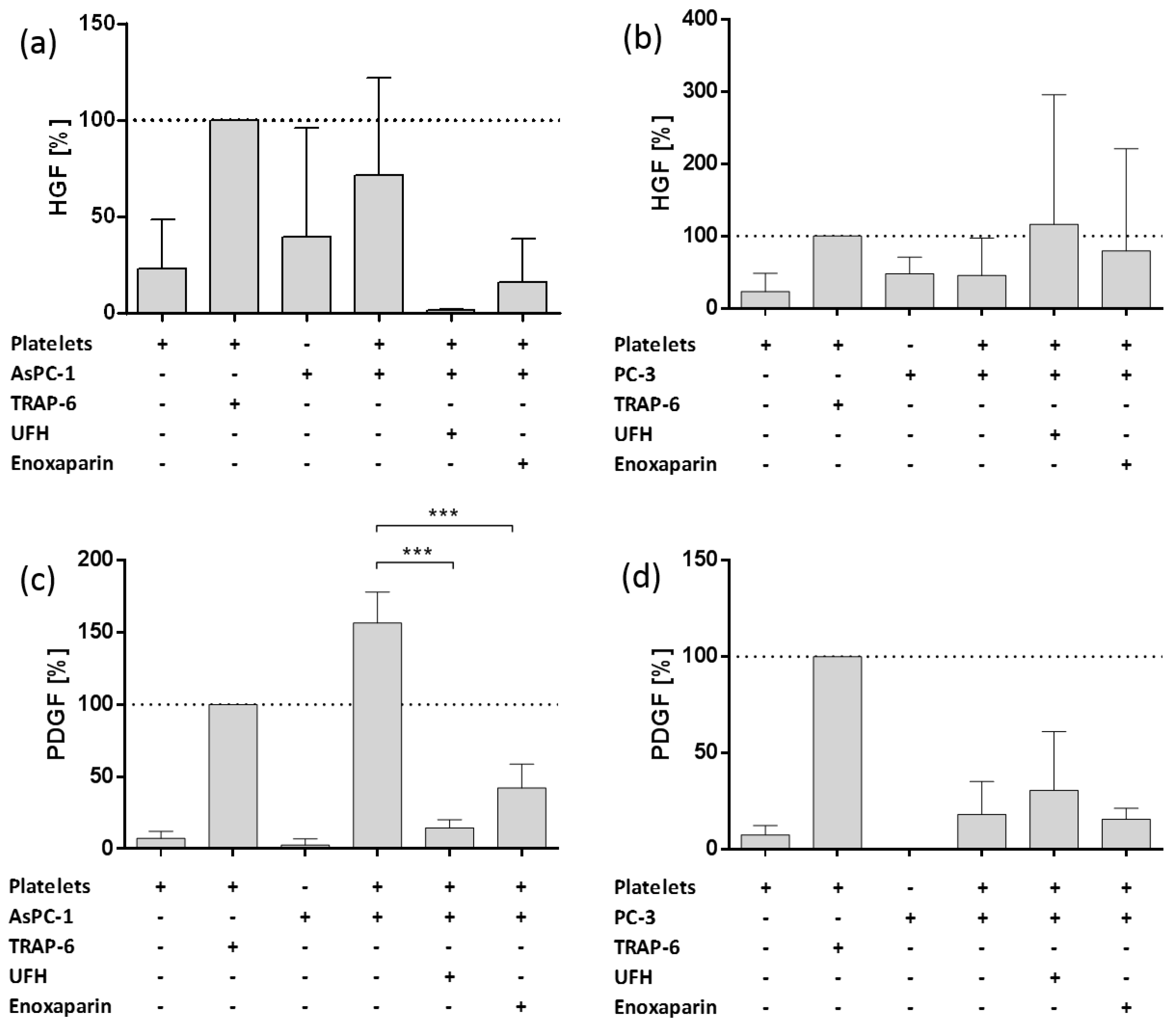
2.2. Impact of AsPC-1 and PC-3 Cell Induced Platelet Activation on Hepatocyte Growth Factor (HGF) and Platelet-Derived Growth Factor (PDGF) Granule Secretion
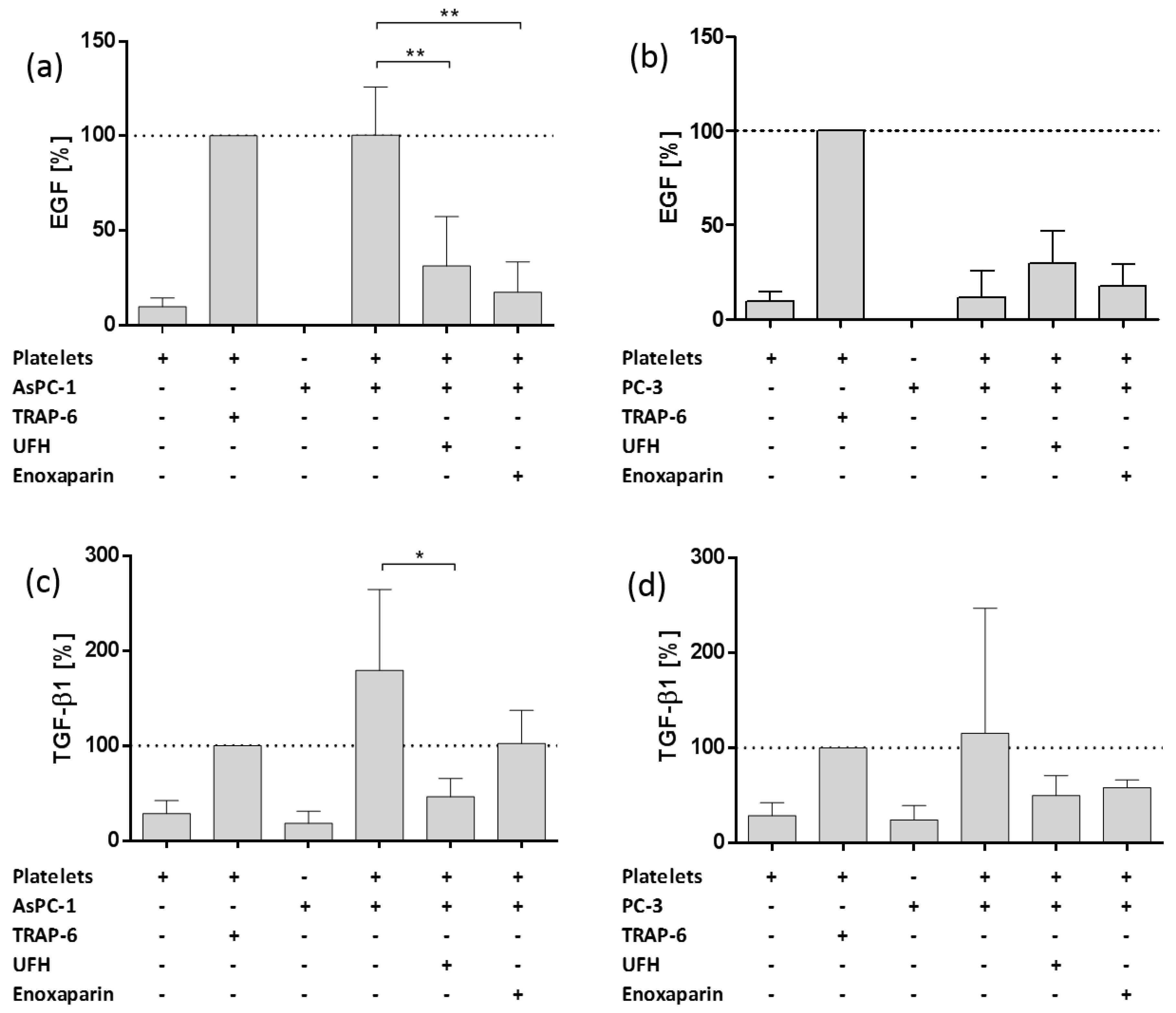
2.3. Impact of AsPC-1 and PC-3 Cell Induced Platelet Activation on Epidermal Growth Factor and Transforming Growth Factor Beta 1 Granule Release
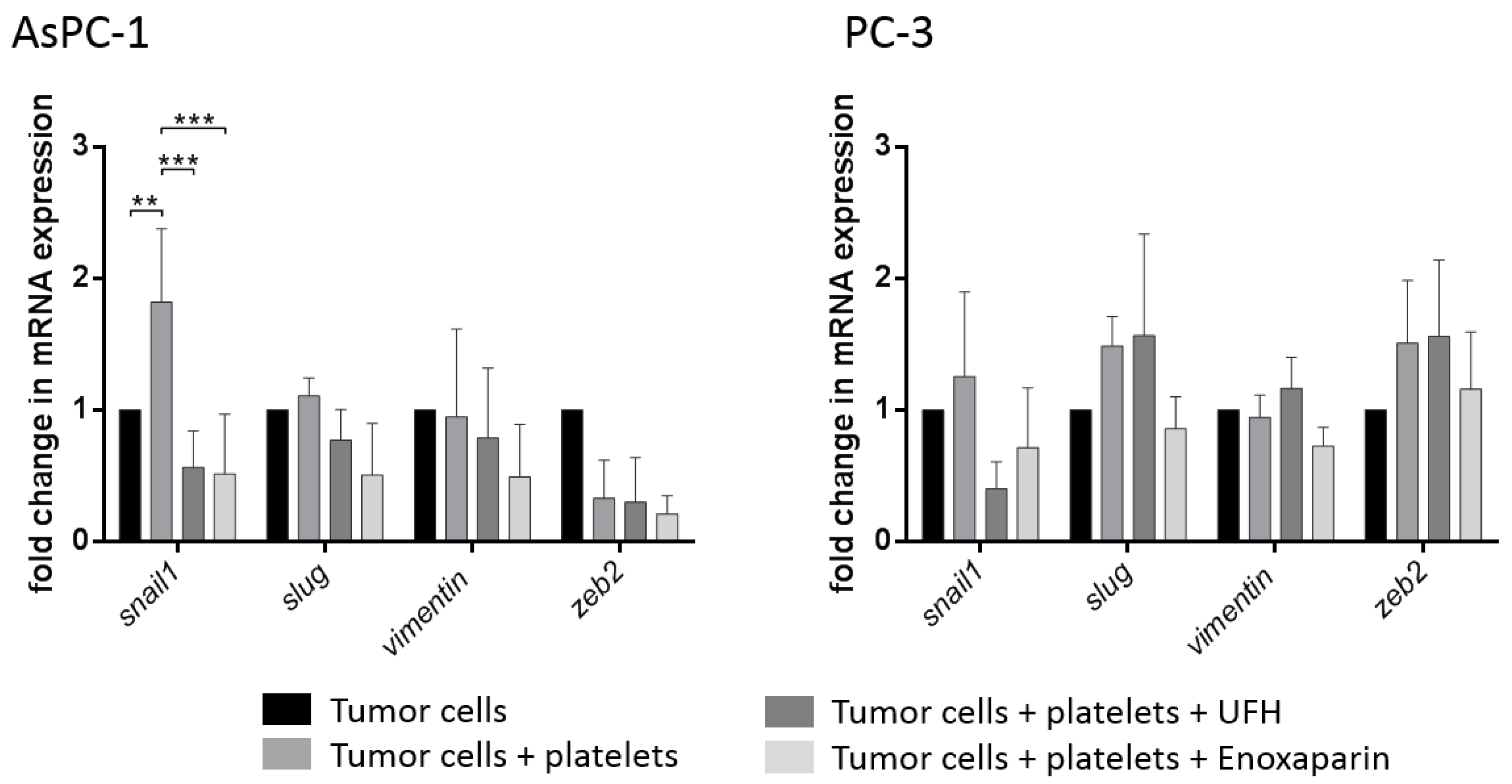
2.4. Interaction of Platelets with Tumor Cells Induces a Mesenchymal Phenotype
2.5. Platelet Treatement Induces an Invasive and Promigratory Phenotype in Tumor Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Platelet Isolation and Activation
4.3. EMT Induction
4.4. Adhesion Assay
4.5. ELISA
4.6. qRT-PCR
4.7. Western Blot
4.8. Wound Healing/Scratch Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Männel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar] [PubMed]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.-G.; Kopp, H.-G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, L.; Schön, M.P. Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Li, N. Platelets in cancer metastasis: To help the “villain” to do evil. Int. J. Cancer 2016, 138, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Coupland, L.A.; Chong, B.H.; Parish, C.R. Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res. 2012, 72, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Skolnik, G.; Ericson, L.E.; Bagge, U. The effect of thrombocytopenia and antiserotonin treatment on the lodgement of circulating tumor cells. A vital fluorescence microscopic, electron microscopic and isotope study in the rat. J. Cancer Res. Clin. Oncol. 1983, 105, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Skolnik, G.; Bagge, U.; Dahlström, A.; Ahlman, H. The importance of 5-HT for tumor cell lodgement in the liver. Int. J. Cancer 1984, 33, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B.Z.; Kahn, M.L. Platelets and tumor cells: A new form of border control. Cancer Cell 2013, 24, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Honn, K.V.; Grossi, I.M.; Diglio, C.A.; Wojtukiewicz, M.; Taylor, J.D. Enhanced tumor cell adhesion to the subendothelial matrix resulting from 12(S)-HETE-induced endothelial cell retraction. FASEB J. 1989, 3, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Janowska-Wieczorek, A.; Marquez-Curtis, L.A.; Wysoczynski, M.; Ratajczak, M.Z. Enhancing effect of platelet-derived microvesicles on the invasive potential of breast cancer cells. Transfusion 2006, 46, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Battinelli, E.M.; Markens, B.A.; Italiano, J.E. Release of angiogenesis regulatory proteins from platelet alpha granules: Modulation of physiologic and pathologic angiogenesis. Blood 2011, 118, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Sabrkhany, S.; Griffioen, A.W.; Oude Egbrink, M.G.A. The role of blood platelets in tumor angiogenesis. Biochim. Biophys. Acta 2011, 1815, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möhle, R.; Green, D.; Moore, M.A.; Nachman, R.L.; Rafii, S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. USA 1997, 94, 663–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ezra, J.; Sheibani, K.; Hwang, D.L.; Lev-Ran, A. Megakaryocyte synthesis is the source of epidermal growth factor in human platelets. Am. J. Pathol. 1990, 137, 755–759. [Google Scholar] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, A.; Okitaka, M.; Takagi, S.; Takami, M.; Sato, S.; Nishio, M.; Okumura, S.; Fujita, N. A critical role of platelet TGF-β release in podoplanin-mediated tumour invasion and metastasis. Sci. Rep. 2017, 7, 42186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrugno, A.; Tormoen, G.W.; Kuhn, P.; McCarty, O.J.T. The prothrombotic activity of cancer cells in the circulation. Blood Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gil-Bernabé, A.M.; Lucotti, S.; Muschel, R.J. Coagulation and metastasis: What does the experimental literature tell us? Br. J. Haematol. 2013, 162, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.W.; Doggen, C.J.M.; Osanto, S.; Rosendaal, F.R. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005, 293, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Spek, C.A.; Versteeg, H.H.; Borensztajn, K.S. Anticoagulant therapy of cancer patients: Will patient selection increase overall survival? Thromb. Haemost. 2015, 114. [Google Scholar] [CrossRef]
- García-Escobar, I.; Beato-Zambrano, C.; Muñoz Langa, J.; Brozos Vázquez, E.; Obispo Portero, B.; Gutiérrez-Abad, D.; Muñoz Martín, A.J. Pleiotropic effects of heparins: Does anticoagulant treatment increase survival in cancer patients? Clin. Transl. Oncol. 2018, 20, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, B.; Chastang, C.; Brechot, J.M.; Capron, F.; Dautzenberg, B.; Delaisements, C.; Mornet, M.; Brun, J.; Hurdebourcq, J.P.; Lemarie, E. Subcutaneous heparin treatment increases survival in small cell lung cancer. “Petites Cellules” Group. Cancer 1994, 74, 38–45. [Google Scholar] [CrossRef]
- van Doormaal, F.F.; Di Nisio, M.; Otten, H.-M.; Richel, D.J.; Prins, M.; Buller, H.R. Randomized trial of the effect of the low molecular weight heparin nadroparin on survival in patients with cancer. J. Clin. Oncol. 2011, 29, 2071–2076. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Julian, J.A.; Laperriere, N.J.; Geerts, W.; Agnelli, G.; Rogers, L.R.; Malkin, M.G.; Sawaya, R.; Baker, R.; Falanga, A.; et al. PRODIGE: A randomized placebo-controlled trial of dalteparin low-molecular-weight heparin thromboprophylaxis in patients with newly diagnosed malignant glioma. J. Thromb. Haemost. 2010, 8, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Ornstein, D.L.; Zacharski, L.R. The use of heparin for treating human malignancies. Haemostasis 1999, 29 (Suppl. 1), 48–60. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L. Heparin as an inhibitor of cancer progression. Prog. Mol. Biol. Transl. Sci. 2010, 93, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wong, R.; Feramisco, J.; Nadeau, D.R.; Varki, N.M.; Varki, A. Heparin and cancer revisited: Mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 3352–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsig, L. Selectins facilitate carcinoma metastasis and heparin can prevent them. Physiology 2004, 19, 16–21. [Google Scholar] [CrossRef]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 2193–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, Y.; Gao, Y.; Shen, J.; Zheng, S.; Wei, M.; Zeng, X. Modified heparins inhibit integrin alpha(IIb)beta(3) mediated adhesion of melanoma cells to platelets in vitro and in vivo. Int. J. Cancer 2009, 125, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J.; Boehme, B.; Podda, M.; Henschler, R.; Jager, E.; Tandi, C.; Boehncke, W.-H.; Zollner, T.M.; Kaufmann, R.; Gille, J. Endothelial P-selectin as a target of heparin action in experimental melanoma lung metastasis. Cancer Res. 2004, 64, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Battinelli, E.M.; Markens, B.A.; Kulenthirarajan, R.A.; Machlus, K.R.; Flaumenhaft, R.; Italiano, J.E. Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 2014, 123, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells-what challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonde, A.-K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Pozo Martin, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Tabor, V.; Wikell, A.; Jalkanen, S.; Fuxe, J. TGF-β1-Induced Epithelial-Mesenchymal Transition Promotes Monocyte/Macrophage Properties in Breast Cancer Cells. Front. Oncol. 2015, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.-H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Al-Azayzih, A.; Gao, F.; Somanath, P.R. P21 activated kinase-1 mediates transforming growth factor β1-induced prostate cancer cell epithelial to mesenchymal transition. Biochim. Biophys. Acta 2015, 1853, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Minami, K.; Hayashi, N.; Yokoyama, Y.; Mori, S.; Yamamoto, H.; Koizumi, M. The CD44 standard isoform contributes to radioresistance of pancreatic cancer cells. J. Radiat. Res. (Tokyo) 2017, 58, 816–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, T.; Tajima, H.; Makino, I.; Nakagawara, H.; Kitagawa, H.; Fushida, S.; Harmon, J.W.; Ohta, T. Metastasis-promoting role of extravasated platelet activation in tumor. J. Surg. Res. 2015, 193, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Akl, E.A.; van Doormaal, F.F.; Barba, M.; Kamath, G.; Kim, S.Y.; Kuipers, S.; Middeldorp, S.; Yosuico, V.; Dickinson, H.O.; Schünemann, H.J. Parenteral anticoagulation may prolong the survival of patients with limited small cell lung cancer: A Cochrane systematic review. J. Exp. Clin. Cancer Res. 2008, 27, 4. [Google Scholar] [CrossRef] [PubMed]
- Altinbas, M.; Coskun, H.S.; Er, O.; Ozkan, M.; Eser, B.; Unal, A.; Cetin, M.; Soyuer, S. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J. Thromb. Haemost. 2004, 2, 1266–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds UFH and Enoxaparin are available from the authors. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponert, J.M.; Gockel, L.M.; Henze, S.; Schlesinger, M. Unfractionated and Low Molecular Weight Heparin Reduce Platelet Induced Epithelial-Mesenchymal Transition in Pancreatic and Prostate Cancer Cells. Molecules 2018, 23, 2690. https://doi.org/10.3390/molecules23102690
Ponert JM, Gockel LM, Henze S, Schlesinger M. Unfractionated and Low Molecular Weight Heparin Reduce Platelet Induced Epithelial-Mesenchymal Transition in Pancreatic and Prostate Cancer Cells. Molecules. 2018; 23(10):2690. https://doi.org/10.3390/molecules23102690
Chicago/Turabian StylePonert, Jan Moritz, Lukas Maria Gockel, Svenja Henze, and Martin Schlesinger. 2018. "Unfractionated and Low Molecular Weight Heparin Reduce Platelet Induced Epithelial-Mesenchymal Transition in Pancreatic and Prostate Cancer Cells" Molecules 23, no. 10: 2690. https://doi.org/10.3390/molecules23102690