1. Introduction
Cancer is still the second leading cause of death in developing and developed countries [
1,
2]. Despite the increasing rate of survival in the last 40 years, the severe side effects of radiation and chemotherapy cancer treatment have been acknowledged to be of major importance, and could cause a number of problems, including systemic toxicity, mild cognitive impairments, and mouth ulcerations [
3]. Moreover, long-term childhood-cancer survivors’ follow-up studies have observed the incidence of many side effects, such as endocrine disruption, cardiac function, auditory dysfunction, and the possibility of developing other types of cancer due to long-term chemotherapy treatment [
4], let alone the recurrence of the disease.
However, new types of treatment have been established. These new approaches include gene therapy [
3,
5], nanoparticulate vectors [
6,
7,
8], modified bacteria [
9,
10], and the utility of the immune system [
11,
12]. Recently, several approaches have been conducted using no-viral anticancer therapy. Cheng et al. (2018) used a self-assembled supramolecular host–guest delivery system to deliver chemotherapeutics to drug-resistant cancer cells and tumors [
13]. In another study by Liu and coworkers (2018), a supramolecular hydrogel was used for drug-resistant cancer therapy [
14]. Moreover, viruses that have the ability to replicate specifically inside cancer cells and subsequently causing death to these infected cells have shown great promise; these are called oncolytic viruses [
15,
16,
17,
18]. In recent years, there has been an increasing interest in oncolytic adenoviral vector anticancer therapy due to these viruses’ ability to efficiently infect a variety of both dividing and nondividing cells, and because of their high-affinity binding site for attaching to the coxsackie virus and adenovirus (Ad) receptor (CAR) of susceptible cells. Ad can also easily produce high titers in vitro. By simply replacing part of the genome of the virus with a therapeutic gene, viruses (retrovirus and adenovirus) can also be used as gene-delivery carriers. They have been widely used in reported gene-delivery studies and in the majority of ongoing clinical trials due to their efficiency in delivering genetic materials [
19]. However, the induction of an immune response is the major barrier towards Ad vector applications. This limitation results in the reduction of viral transduction and thus decreased therapeutic efficiency [
20,
21].
Several attempts have been made to overcome the induction of immune responses, such as the use of pharmacological inhibitors, genetic engineering, and carrier cells. All these strategies significantly decreased overall inflammatory response, yet did not inhibit the production of neutralizing antibodies [
22]. More recently, the use of biopolymers as drug-delivery systems for anticancer therapy have received wide attention because of their nonimmunogenicity, nontoxicity, and biodegradability. One such biopolymer is poly-gamma-glutamic acid (γ-PGA). Polymer γ-PGA is a biocompatible, biodegradable, nontoxic, and nonimmunogenic biopolymer [
23,
24,
25,
26];
γ-PGA is a homopolyamide composed of glutamic acid monomers connected by amide linkages between α-amino and
γ-carboxyl groups, with D and L isomeric units distributed in repeated blocks [
27,
28]. The γ-PGA biopolymer has been widely used as a drug-delivery platform because of its free carboxyl groups on the side chains in the α-position, which offers attachment points for the conjugation of therapeutic agents. This enhances aqueous solubility and controls the release of the drug [
23,
29].
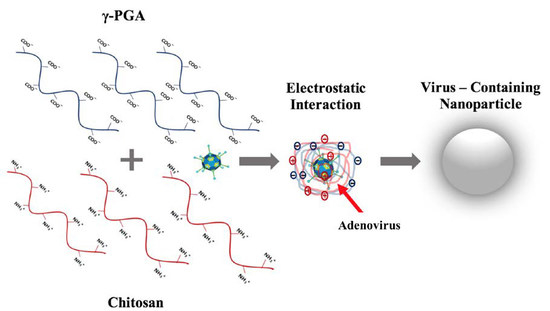
One long-term goal is to develop ‘stealth’ vectors of low immunogenicity carrying destructive cargoes capable of destroying the replication abilities of, for example, the Epstein–Barr virus (EBV), the major cause of Hodgkin′s lymphoma [
30]. The main aim of this study was to see if polymer encapsulation of such vectors helps to lower their immunogenicity.
3. Discussion
The limitations associated with conventional anticancer drug formulations have led to numerous attempts in developing more effective therapies. In general, recombinant Ad vectors have many properties that make them useful for alternative cancer therapies [
34]. However, the induction of the immune response to recombinant Ad has appeared as a significant concern in the success of anticancer therapies. Along with the encapsulation of biopolymer approaches, as used in this study, many other approaches are in place to reduce the activation of host-immune responses. These approaches include serotype switching, silencing mediators of hepatocyte injury, immunosuppression, use of high-dose vectors, and genetic manipulation of capsid-encoding sequences [
35,
36]. These approaches have many successful aspects, yet there are still limitations to their applicability. For instance, the immunosuppression approach has side effects that are unwanted in cancer-treatment clinical settings. Adenovirus immunostimulation is mediated mainly through viral capsid proteins, and is not dependent on transduction or viral gene expression [
37]. To control Ad immunostimulatory properties, the encapsulation of Ad in γ-PGA NPs is an attractive approach.
The most widespread synthetic polymers for micro/nanoencapsulation are poly (lactic-co-glycolic acid) (PLGA), poly(glycolic acid) (PGA), and poly(lactic acid) (PLA). All these polymers have been approved by the U.S. Food and Drug Administration (FDA) for particular medical applications, as they are degradable [
38]. Nevertheless, these synthetic polymers may reduce the surrounding pH during polymer degradation, which subsequently affects cellular function by generating a highly acidic microenvironment, thus limiting their in vivo application [
39]. Increased attention has been turned to the use of biopolymers to overcome this problem. Naturally occurring biopolymers produced by living micro-organisms during cell growth may be the key, as they can be produced in the laboratory and they show obvious synthesizing flexibility. One of the most recently discovered biopolymers for nanoparticle applications is γ-PGA.
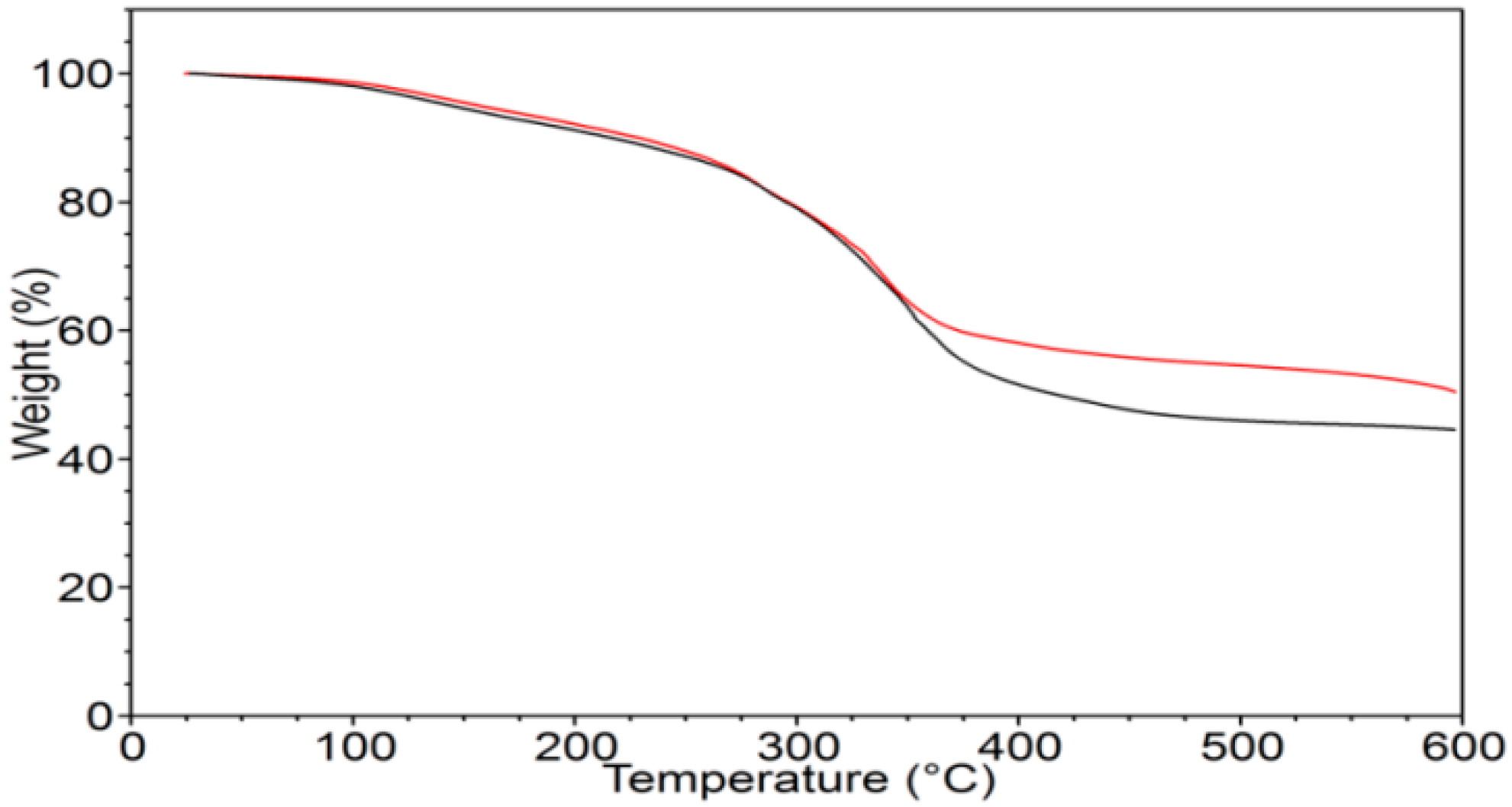
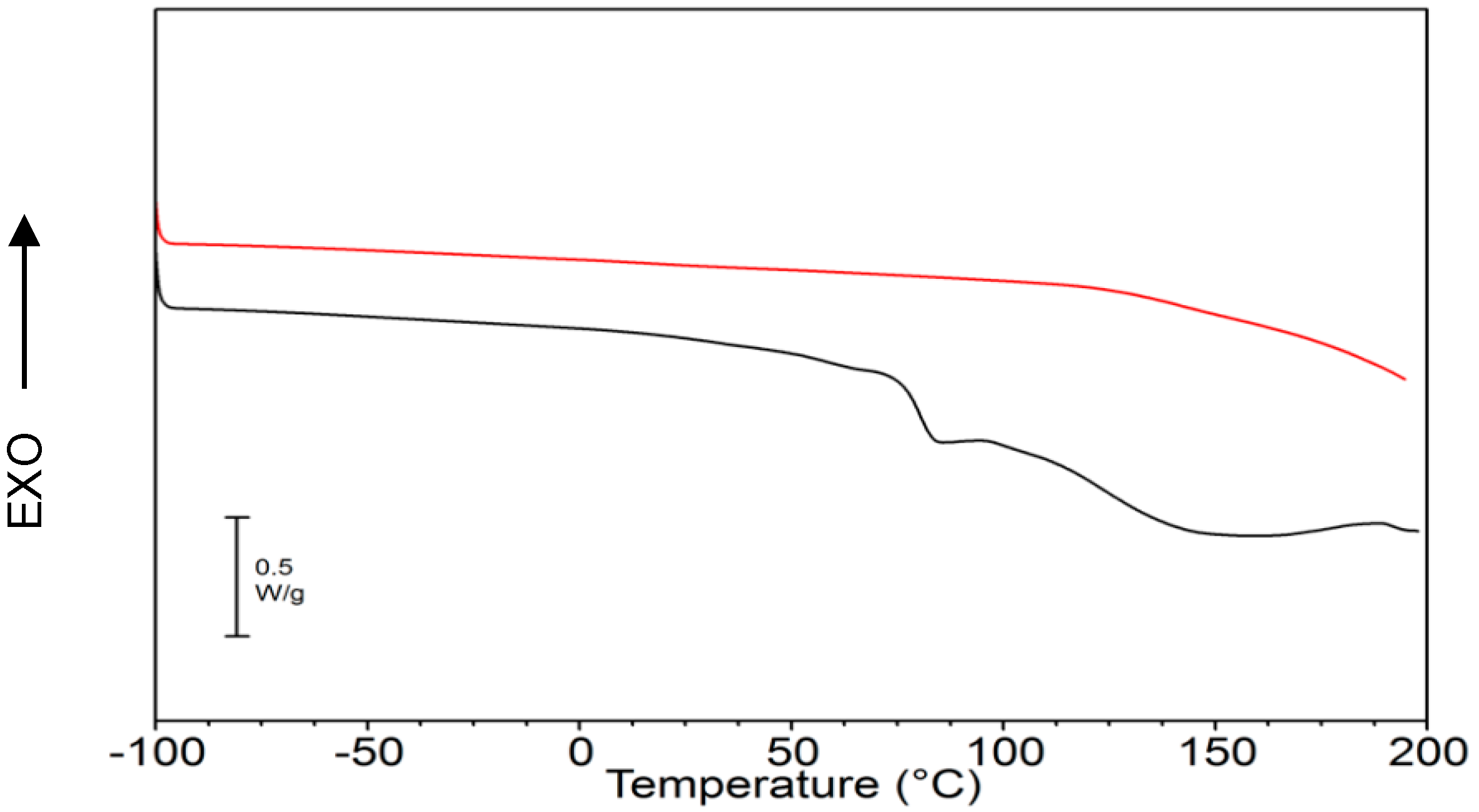
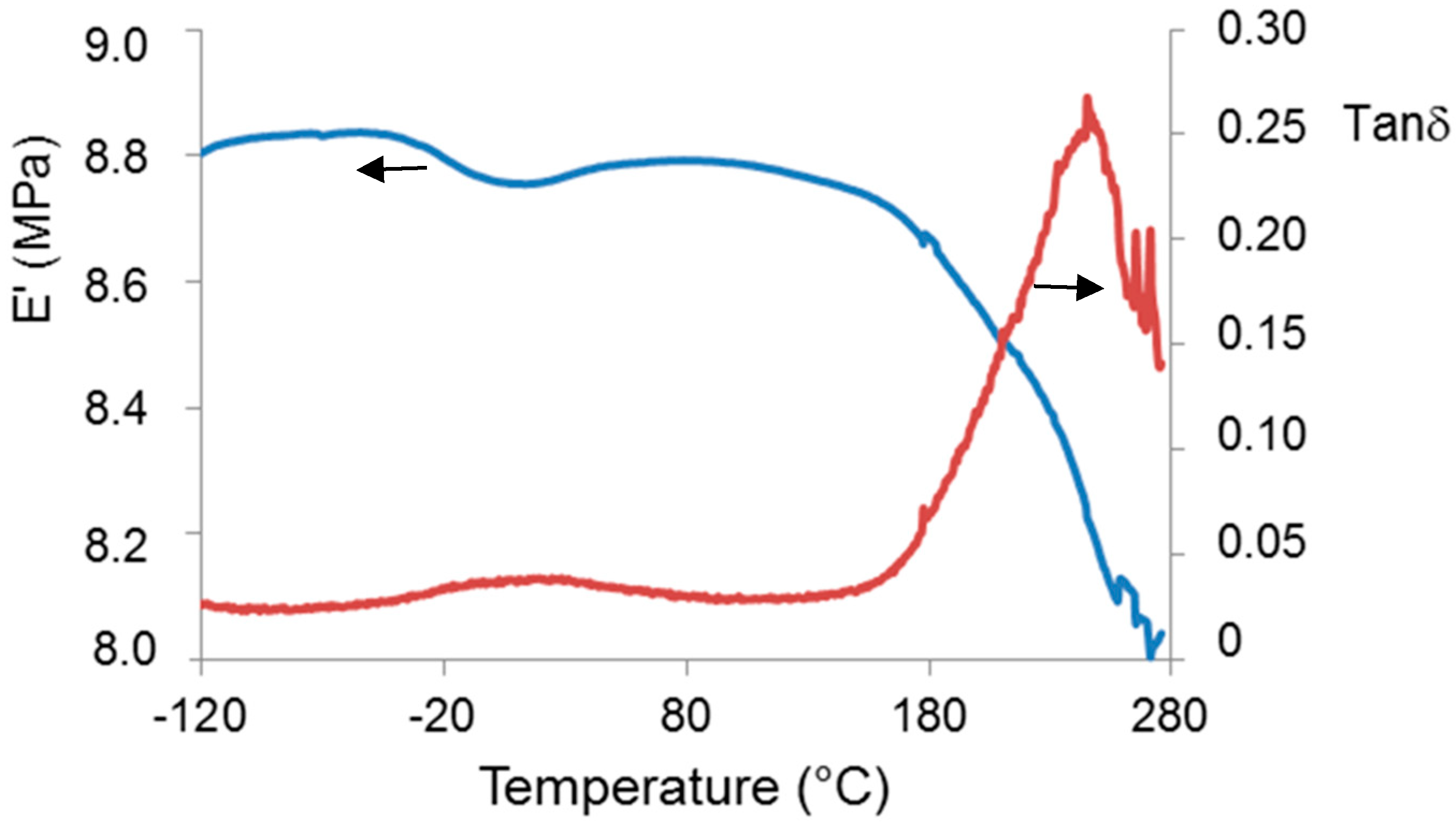
Biopolymer γ-PGA has been used in many therapeutic applications in the last few decades, as it is nonimmunogenic, biodegradable, and nontoxic. γ-PGA is an amorphous polymer with a T
g around 80 °C, as demonstrated by DSC and dynamic mechanical (DMTA) analyses (
Figure 3 and
Figure 4). It has a hydrophilic character (
Figure 6), as expected on the basis of its chemical structure (
Figure 1 and
Figure 2), which justifies the presence of absorbed water in the sample. This is evidenced by the initial loss in the TGA curve (
Figure 2) and by the elastic-modulus increase in DMTA measures (
Figure 6), associated with an increase of sample rigidity due to absorbed-water evaporation. Additionally, the polymer has many beneficial properties, including a shielding surface that does not require chemical alteration or genetic manipulation, enhancement of vector stability, and eliminating nonspecific interaction. To reserve vector bioactivity, adenovirus encapsulation inside γ-PGA NPs can be performed in minor reaction conditions. Besides γ-PGA, CH and its derivatives are widely used for drug delivery; this has been attributed to their biodegradability and biocompatibility, as well as CH being able to be degraded into nontoxic products in vivo [
40]. As the fabrication of γ-PGA NPs began in the last decade, there are very few data regarding their effect on mammalian cells. However, a study by Hsieh et al. (2005) demonstrated that the addition of γ-PGA and CH into the medium enhances hydrophilic properties and serum protein intake, and increases the attachment and growth of rat osteosarcoma cells [
40]. Physical stability of the virus offered by γ-PGA against harsh environmental conditions was also demonstrated by our team in a previous study [
41]. Some therapeutic agents have been encapsulated into polymeric NPs to protect them from neutralization in patients receiving repeated doses of this kind of treatment [
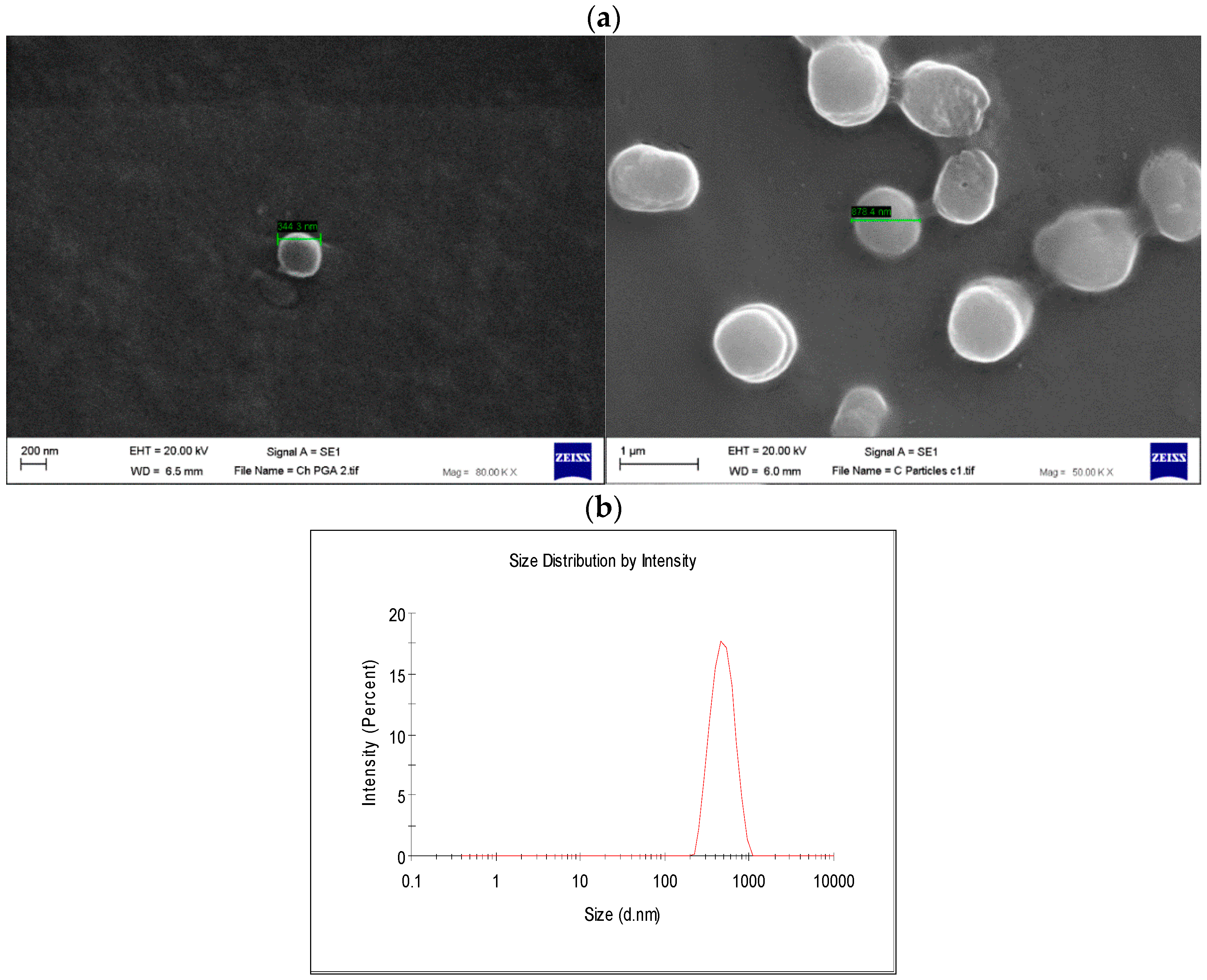
22]. This application was the driving force for the development of an encapsulated Ad vector. After determining that Ad can be effectively encapsulated into γ-PGA-CH NPs, these NPs were characterized (
Figure 7) and tested for their ability to avoid in vitro antibody neutralization. The Ad-loaded γ-PGA-CH NPs were fabricated using the ionic gelation technique. Encapsulation efficiency was 93% (
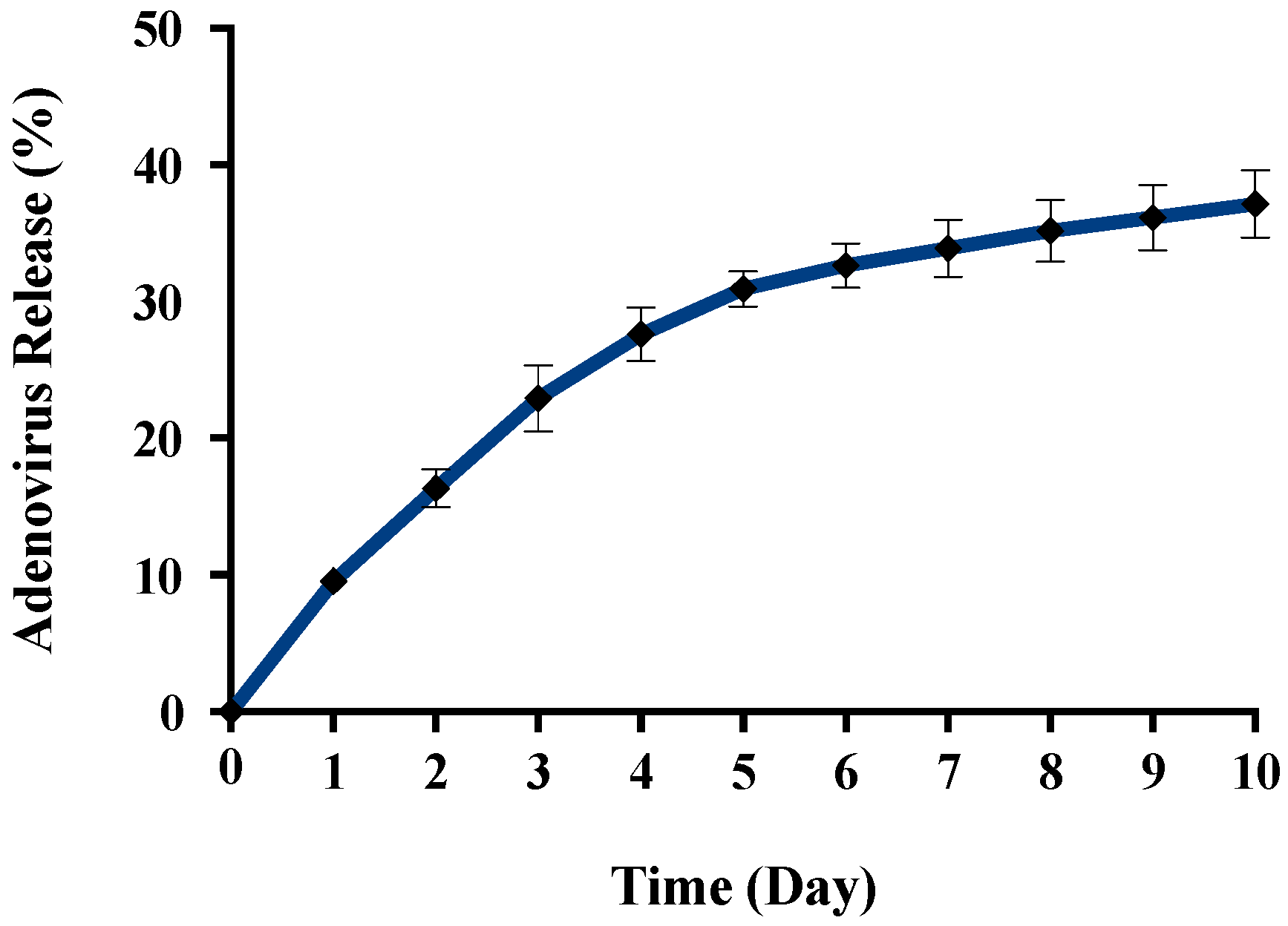
Figure 8). The in vitro release behavior of Ad from γ-PGA-CH NPs was evaluated in physiological buffers at pH = 7 (
Figure 9). Polymer degradation in an aqueous solution generates pores on the NP surface; thus, the release rate of smaller molecules is higher than that of bigger molecules. However, the entire released Ad retained full infectivity when introduced to HEK293 cells. The number of released Ad in this study was measured by an infectivity assay. In contrast, other mentioned studies evaluated the amount of released Ad by measuring the radioactivity of the radiolabeled Ad [
42,
43]. However, the observed Ad-release profiles in this study cannot be directly compared with others reported earlier, as different polymers, sizes, formulation recipes, and fabrication conditions were used for the preparation of Ad encapsulated in polymeric particles.
As an infectivity assay was used to measure the amount of released virus (
Figure 8 and
Figure 9), it is worth noting that all released Ad in our study retained full bioactivity, while the maximum bioactivity reported by Wang and coworkers (2007) was 11.4% [
44]. A solvent evaporation process has been the most used method to date for encapsulating Ad. This procedure involves a series of critical damaging phases for Ad stability. Viral particles can be denatured and aggregated upon exposure to organic solvents. High shear forces may also cause irreversible viral inactivation through physical aggregation. Puapermpoonsiri et al. (2009) stated that organic solvent used in a solvent-evaporation method such as dichloromethane has a strong denaturing effect on the protein of the virus during the dichloromethane–water interface [
45]. The results generated here are encouraging, as the formulation of encapsulated Ad vectors is feasible with retention of bioactivity due to the absence of organic solvent and less-damaging reaction conditions used in the ionic gelation method.
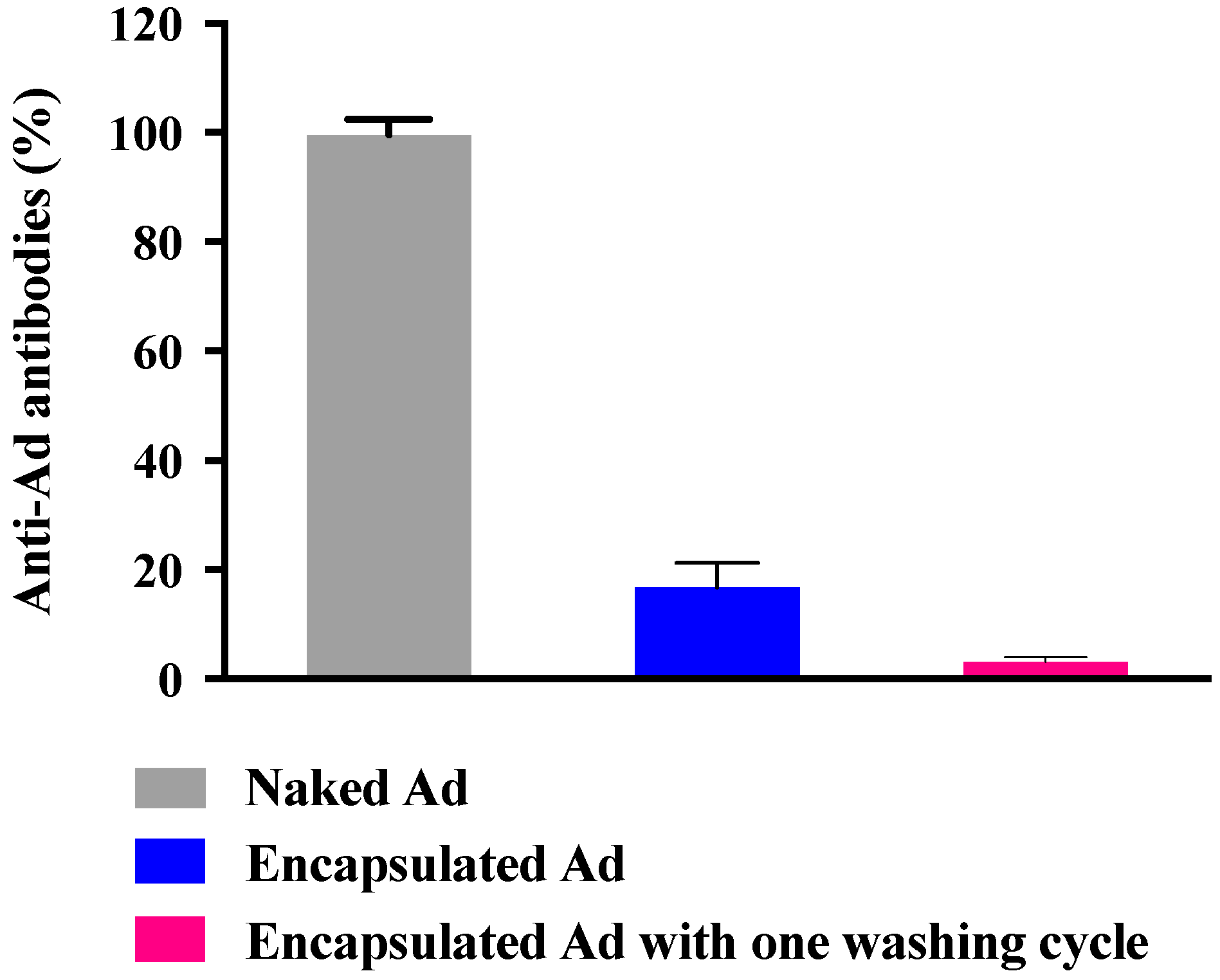
The present study also implies that a neutralizing antibody response against viral capsid proteins can be somewhat reduced by encapsulating Ad into γ-PGA NPs, as only 3.1% were detected by antiadenovirus antibodies (
Figure 10). This finding is similar to that of the conjugation of Polyethylene Glycol (PEG) molecules to adenovirus capsids. Croyle et al. (2001) found that the modification of viral capsids with PEG can reduce the immune response generated against viral proteins [
22]. However, a study by Shimizu and coworkers (2012) revealed that intravenous administration of PEG-coated Ad elicits anti-PEG antibodies, which reached the maximum level after five days of PEGylated Ad injection [
46]. Moreover, unlike Ad encapsulation, modification of viral capsids with PEG can cause loss of infectivity due to the denaturation of viral surface proteins that mediate viral binding to the host cells [
44]. However, these Ad-containing NPs have not been tested for generation of a cellular immune response. Thus, the exact nature of the response to these NPs must be extensively characterized in vitro and in vivo.
Further examination was conducted to evaluate the prospective kinetics of NPs towards the HEK293 cell line and cellular uptake. In addition, one clear advance allowing location and treatment of tumors more efficiently is through more precise targeting of tumor tissue with fluorescently labeled NPs. Radiation doses could be reduced through clearer imaging of the tumor. Drug wastage could similarly be reduced, along with concomitant side effects if the therapeutic agent was specifically directed at tumor tissue, with consequent minimization of collateral damage to adjacent healthy tissue. Conjugation of FITC to chitosan would give stability to the fluorescent dye that is usually applied for cellular uptake studies in vitro and in vivo. Inaccurate results may be obtained if FITC molecules are encapsulated inside the particle, as it is dissociated and subsequently released from the particle during the experiment. For that it is a very important consideration to certify that FITC-NPs measurement for NP kinetics and cellular uptake studies belong to the particle itself and not to the dissociated FITC molecules [
47]. However, fluorescently labeled γ-PGA-CH NPs were successfully created in this study and introduced to mammalian cells. The images in
Figure 11 from an inverted fluorescent microscope revealed that the labeled NPs were clearly attached/internalised on/into the cells. A study conducted by Kuo and coworkers (2011) suggested that γ-PGA could increase permeability by boosting the cellular uptake, possibly through its own specific receptor-mediated pathway [
48].
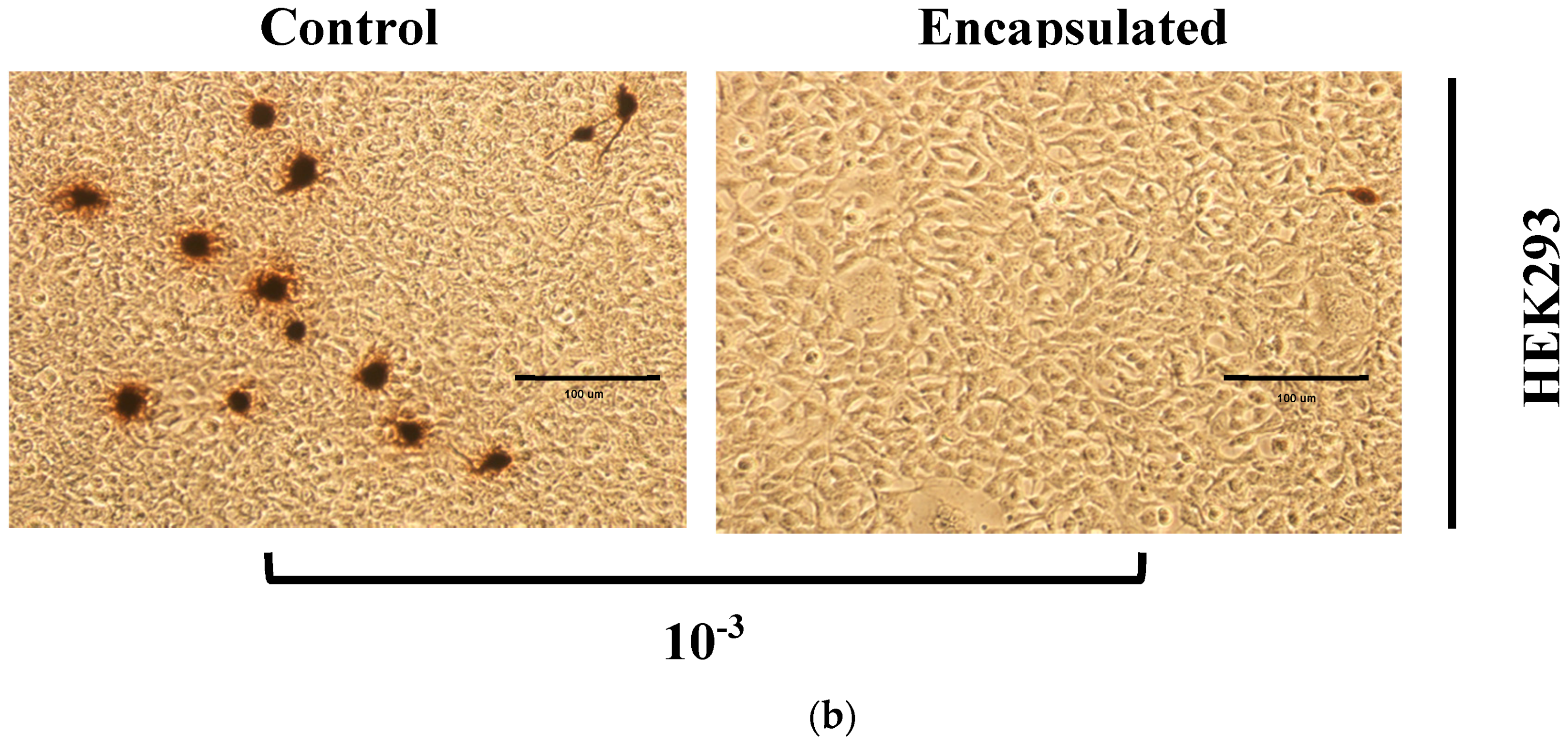
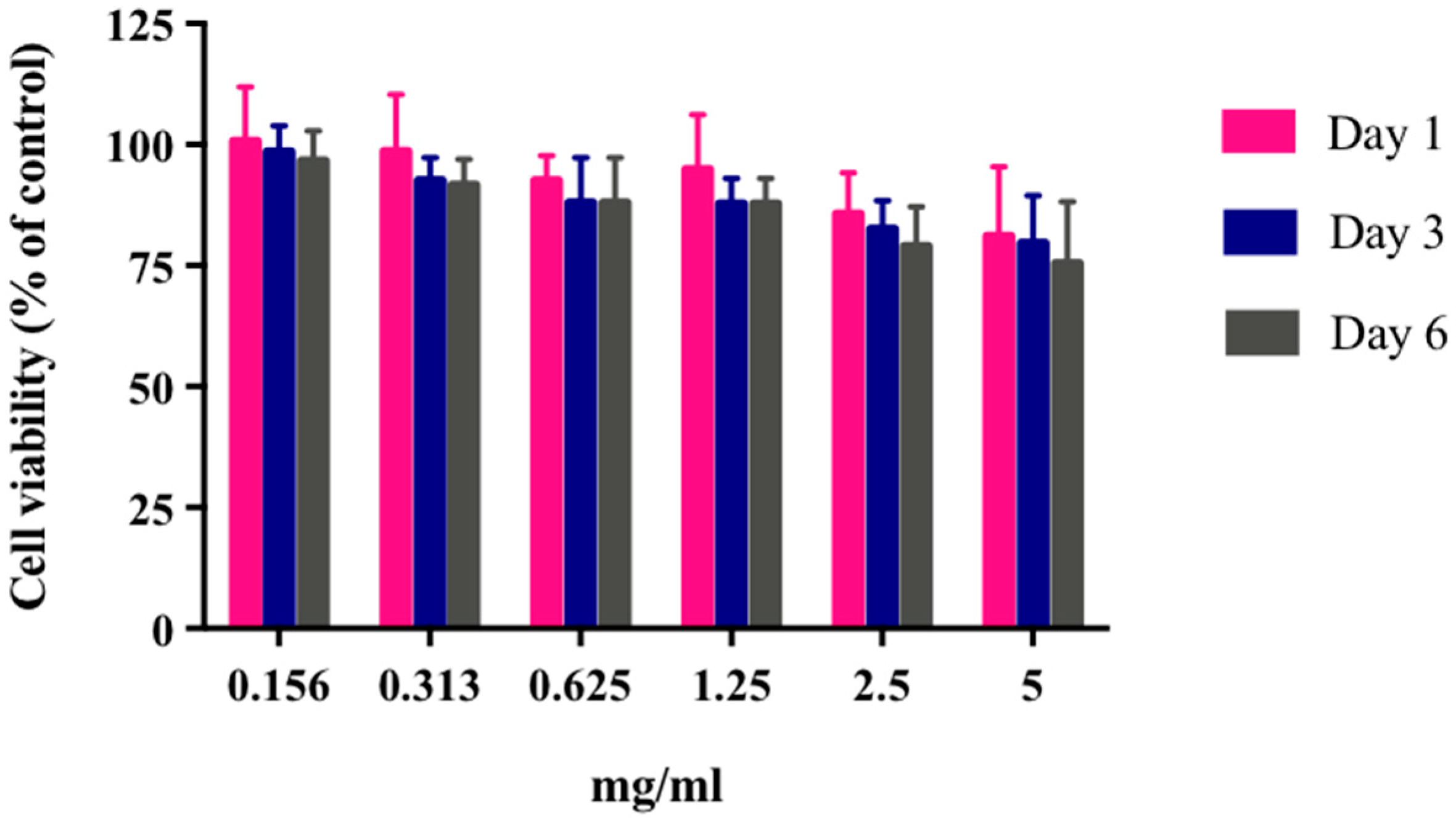
There is no possibility that the produced NPs can be toxic to the mammalian cells used in this work. The obtained results showed that HEK293 cells continued to grow despite the presence of these particles even with extreme NP concentration (5 mg/mL) (
Figure 12). The results were not surprising, as γ-PGA is known to be a nontoxic material. Besides γ-PGA, CH and its derivatives are widely used for drug delivery; this has been attributed to their biodegradability and biocompatibility. CH can also be degraded into nontoxic products in vivo [
40]. As the fabrication of γ-PGA NPs started only in the last decade, there are very few data regarding their effect on mammalian cells. However, a study by Hsieh et al. (2005) demonstrated that the addition of γ-PGA and CH into the medium enhances NP hydrophilic properties. It can also increase serum protein intake as well as the attachment and growth of rat osteosarcoma cells [
40].
4. Materials and Methods
4.1. γ-PGA Production, Purification, and Characterization
Bacterial γ-PGA was produced by
B. licheniformis ATCC 9455a in a 5 L fermenter (Electrolab, Tewkesbury, UK) as reported earlier [
49]. Infrared spectra were recorded on a Nicolet 380 FT-IR (Thermo Fisher Scientific Inc., Wilmington, DE, USA) with 32 scans and 4 cm
−1 resolution. Samples were ground with KBr (Sigma Aldrich, Milano, Italy) (1 mg sample/100 mg KBr), and pelletized under pressure.
TGA measurements were carried out using a TA-TGA2950 (TA Instruments, New Castle, DE, USA). Analyses were performed at 10 °C/min from room temperature to 600 °C both under air and under nitrogen flow. DSC measurements were performed using a TA-DSC-Q100 (TA Instruments, New Castle, DE, USA) apparatus equipped with a liquid nitrogen cooling system (LNCS) accessory. Heating scans were run at 20 °C/min, from −100 °C to 200 °C in a helium atmosphere. Between heating scans, quench cooling was applied. Melting temperature (Tm) was taken at the peak maximum of endotherm, while the glass-transition temperature (Tg) was taken as the midpoint of the stepwise increase of the specific heat associated with the transition.
Wide-angle X-ray diffraction measurements (WAXS) were carried out at room temperature with a PANalytical X’Pert PRO diffractometer equipped with an X’Celerator detector for ultrafast data collection (PANalytical B.V., Almelo, The Netherlands). A Cu anode was used as X-ray source (KR radiation: λ = 0.154 18 nm, 40 kV, 40 mA), and ¼ divergence slit was used to collect the data in 2θ range from 2° to 60°. The degree of crystallinity (χc) was evaluated as the ratio of the crystalline peak areas to the total area under the scattering curve.
Dynamic mechanical measurements (DMTA) were carried out in dual cantilever mode, at 3 °C/min and 3 Hz from −120 °C to 280 °C using a DMTA MkII (Polymer Laboratories Ltd., Church Stretton, UK). A strip (8 mm wide) cut from a glass fiber nonwoven mat (Whatman GF/C, Sigma Aldrich, Milano, Italy) was used to support the polymer. Polymer deposition was carried out by means of multiple immersions of the glass-fiber support in a 1% aqueous solution of Sample 2. A solvent-evaporation step (in oven under vacuum at 80 °C) was performed between consecutive immersions. The final glass-fiber strip coated with the polymer was subjected to DMTA measurement.
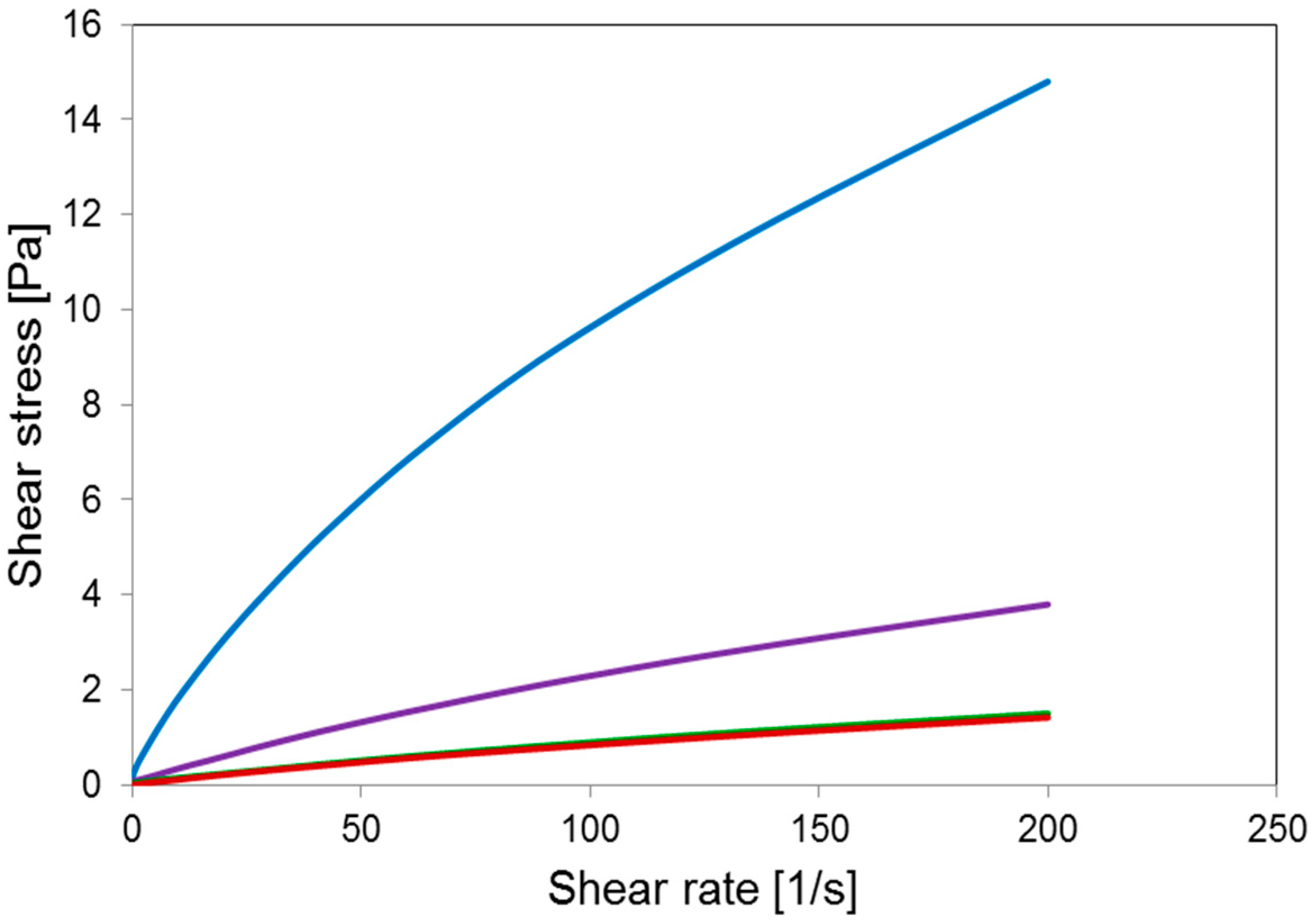
Rheological measurements were carried out at 25 °C using an Anton Paar MCR102 Rheometer (Anton Paar GmbH, Graz, Austria) with cone-plate CP50-1-SN19392 geometry (d = 0.102 mm, acquisition time 100 s, shear rate from 0.01 s−1 to 200 s−1).
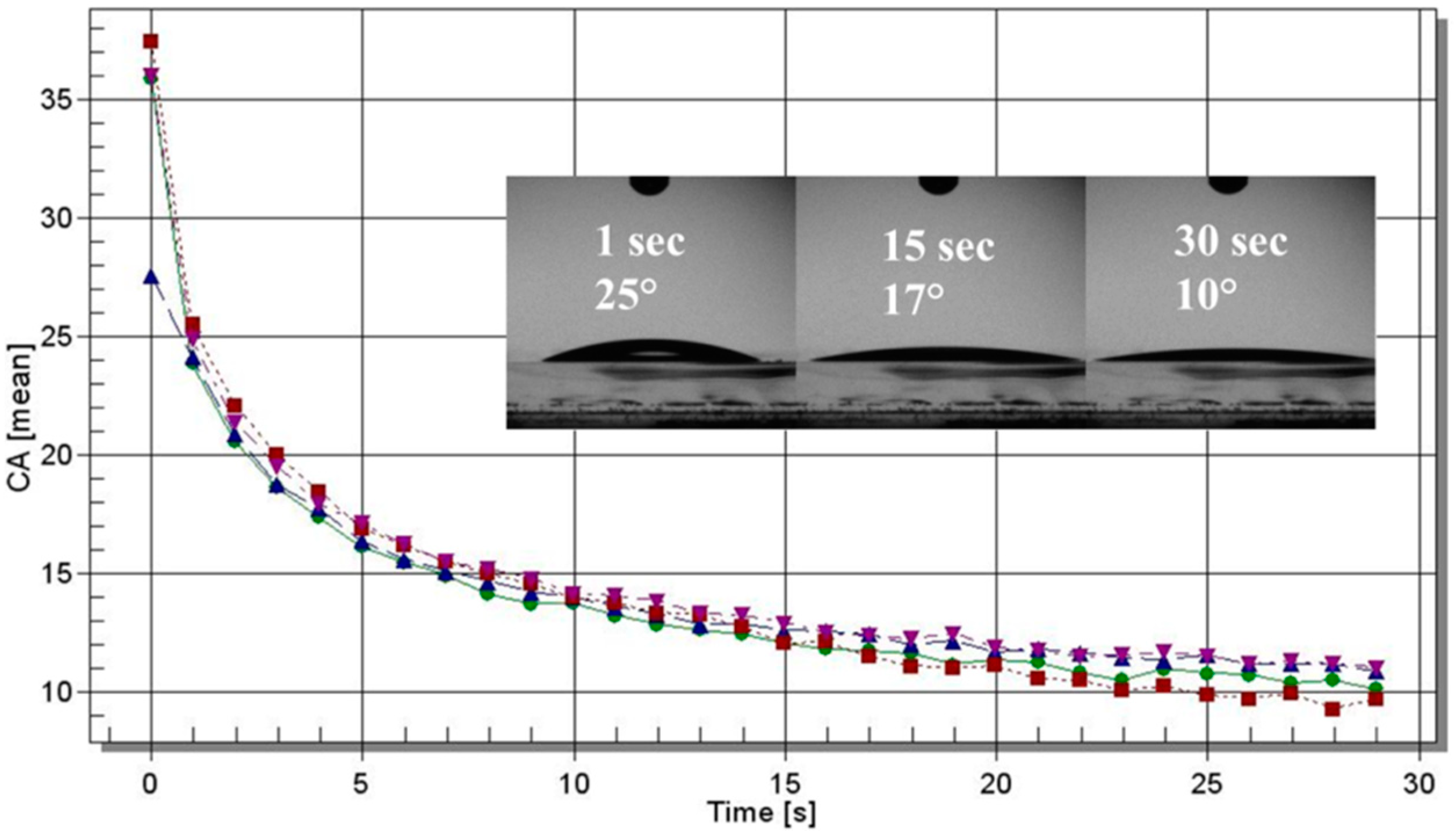
Static water-contact angle measurements were performed on samples coated on glass slides (from a 0.8% aqueous solution) by means of a Laurell (WS-650-23NPP) spin coater (Laurell Technologies Corporation, North Wales, PA, USA) under ambient conditions. The contact-angle experiments were run with an optical contact angle and surface-tension meter KSV’s CAM 100 (KSV, Espoo, Finland) using distilled water. The water-drop profile images were collected every second in a time range of 0–30 s. Four drops per sample were analyzed.
The enantiomeric composition of the produced γ-PGA was investigated using HPLC (Thermo Scientific, Schwerte, Germany). The hydrolysis of γ-PGA was performed according to many studies [
50,
51]. Initially, the lyophilized polymer was resuspended in 6 M HCl (Sigma, Irvine, UK) at the concentration of 1 mg/mL, and hydrolyzed at 110 °C for 24 h. The hydrolyzed samples were then lyophilized again and dissolved in water and methanol (30:70) to the final concentration of 300 μg/mL. After that, 10 μL of the samples was injected into a Chirobiotic T column, 25 cm × 4.6 mm I.D., 5 μm particles (Astec, Sigma-Aldrich, Irvine, UK) at 25 °C using a mobile phase: water; methanol (Thermo Fisher Scientific, Altrincham, UK); and formic acid (Thermo Fisher Scientific, Altrincham, UK) (30:70:0.02, respectively) at a flow rate of 1 mL/min according to the manufacturer’s instructions. Chromatography was performed using a Dionex Ultimate 3000 series HPLC (Thermo Scientific, Schwerte, Germany) connected to an ultraviolet (UV) spectrometer (Thermo Scientific, Schwerte, Germany). The analytical software used was Chromeleon 7 (Thermo Scientific, Schwerte, Germany). In order to determine retention time, standards of
d- and
l-form of glutamic acid were first tested. After that, the mass of glutamic acid was used to determine the amounts of
d- and
l-glutamic acid by peak integration. Number average molar mass M
n was determined by conventional aqueous-based GPC at Smithers Rapra in Shrewsbury, United Kingdom. An MZ Hema guard plus 2× Hema Linear column (Cognis Performance Chemicals Ltd., Southampton, UK) was used for analysis. The GPC experiments were carried out in 0.2 M NaNO
3, 0.01 M NaH
2PO
4, at pH = 7, with a flow rate of 1 mL/min at 30 °C. The GPC system used was calibrated with polyacrylate standards. The data were collected and analyzed using Polymer Laboratories’ “Cirrus 3.0” software Supplied by Polymer Laboratories (Salop, UK).
4.2. Adenovirus Preparation
The HEK293 cell line was used to amplify Adenovirus-GFP (purchased from Applied Biological Materials, Inc., Richmond, BC, Canada). The adenovirus was purified using an Adenovirus mini purification viraKit (Virapur LLC, San Diego, CA, USA). Then, the adenovirus was titered using an Adeno-X Rapid Titer Kit (Clontech Laboratories, Inc., Mountain View, CA, USA). Initially, 0.5 mL of healthy, log-phase HEK 293 cells (2.5 × 105 cells/mL) suspended in standard growth medium (DMEM + 10% FBS + antibiotics) was seeded in each well of a 24-well plate. Using PBS as diluent, 10-fold serial dilutions of the viral sample were prepared from 10−2 to 10−7 mL. After that, 50 µL of viral dilution was added to each well. Subsequent to incubation for 48 h at 37 °C with 5% CO2, cell medium was aspirated. Then, cells were fixed by adding 0.5 mL ice-cold 100% methanol to each well, and the plate was incubated at −20 °C for 10 min. Subsequently, methanol was aspirated and the wells were rinsed 3 times with PBS + 1% BSA. The final rinse was aspirated from the wells and 0.25 mL of (1:1000) diluted mouse antihexon antibody was added to each well, then incubated for 1 h at 37 °C. The antihexon antibody was aspirated, and cells were washed with 1 mL PBS + 1% BSA. Then, 0.25 mL of (1:500) diluted rat antimouse antibody (conjugate) was added to each well, and incubated for 1 h at 37 °C. Prior to removing rat antimouse antibody, a diaminobenzidine (DAB) working solution was prepared by diluting 10X DAB substrate 1:10 with 1X stable peroxidase buffer. The rat antimouse antibody dilution was subsequently aspirated and again rinsed with 1 mL PBS + 1% BSA. After removing the final PBS + 1% BSA rinse, 500 µL of DAB working solution was added to each well, and incubated at room temperature for 10 min. Finally, the DAB solution was aspirated and 1 mL PBS was added to each well. The mean of a minimum of 3 fields of brown positive cells were counted using a microscope with a 20× objective. Calculations of infectious units (ifu)/mL for each well were performed using the manufacturer’s manual.
4.3. Adenovirus Encapsulation
Chitosan (50–190 kDa) with 75–85% deacetylation (purchased from Sigma, UK), was dissolved in a 0.1 M glacial acetic acid (Acros, UK) solution (pH = 5.5) to a final concentration of 10 mg/mL. The prepared solution was left to stir overnight, followed by a purification process in a dialysis tube (Specturm, Irving, TX, USA) with a 10 kDa cut-off. After 3 days of dialysing, CH was precipitated by increasing the pH to 10. The precipitant was washed 3 times and freeze-dried.
Solutions of CH and γ-PGA were prepared before forming NPs. Purified CH was dissolved in 0.1 M glacial acetic acid at concentration of 10 mg/mL and left to stir overnight on a magnetic stirrer. The following day, the CH solution was autoclaved under pressure of 10 psi for 20 min at 121 °C. Subsequently, the solution was passed through a filter (0.45 μm) to eliminate nondissolved particles. Finally, 0.05% of CH solution was obtained with pH = 5.5. To prepare the γ-PGA solution, 10 mg/mL of γ-PGA in deionized water was thermally depolymerized at 100 °C for 180 min. Then, 0.15% of γ-PGA in 0.1% sodium tripolyphosphate (TPP) solution was prepared at pH = 6.8. Multi-ion-crosslinked Ad-loaded NPs were fabricated by a simple ionic-gelation method as described previously [
52], with some modifications. In brief, 3.0 × 10
8 PFU/mL of Ad in 200 μL PBS solution was added to 7.8 mL of CH and left to stir on ice for 30 min. Subsequently, 2 mL of γ-PGA was added drop-wisely using a digital pipette (Thermo Fisher Scientific, Waltham, MA, USA) with a flow rate of 2 mL/min. The solution was left to mix on a magnetic stirrer. Suspension was spontaneously formed after 1 h of crosslinking. The obtained particles were transferred to a sterile centrifuged tube containing a bed of glycerol (Sigma, UK), and kept in the fridge to set for 30 min. After that the particles were collected by centrifugation at 8000×
g for 80 min at 4 °C. To remove the Ad that was adsorbed to the surface of the NPs or free in the supernatant, the obtained particles were collected by a washing and centrifugation (Sigma, UK) step at 8000×
g for 80 min at 4 °C. The pellet was then resuspended in 2.5% of trehalose (Sigma, UK), which can be used directly or freeze-dried. Another sample was prepared under the same circumstances without γ-PGA as a control. To determine the EE of Ad in γ-PGA-CH NPs, the amount of free Ad in supernatants was assayed by the Adeno-X Rapid Titer Kit as shown below:
SEM (Zeiss EVO50, Carl Zeiss Ltd., Cambridge, UK) analysis was performed to determine the morphological surface structure of freeze-dried NPs. Samples were prepared by freeze-drying. The obtained fine dry powder samples were placed on an aluminum stub with a double-sided carbon tape, then coated with gold. Analyses were conducted using an SEM and microphotographs were analyzed using SmartSEM software V05.06 (Carl Zeiss Ltd., Cambridge, UK). The size and zeta potential of γ-PGA nanoparticles synthesized during this project were continuously measured using dynamic light scattering from Malvern (HeNe laser, Malvern, UK).
4.4. In Vitro Virus Release from Chitosan-γ-PGA NPs
The Ad release profile from produced NPs (0.05:0.15 CH:γ-PGA ratio) was obtained in a DMEM medium (pH = 7.0), supplemented with 10% fetal bovine serum (FBS), 1% l-glutamine, and 1% antibiotic antimycotic solution (Sigma, UK). The pellet after the centrifugation and washing step was resuspended in the medium and incubated in an orbital shaker at 37 °C under agitation (150 rpm). At predetermined time intervals, NP suspension was centrifuged 8000× g for 80 min, and supernatant was removed and assayed by plaque-forming assay to determine the amount of the released viruses. Then, the NP pellet was again resuspended in the fresh medium and agitated; the amount of viruses released from the produced NPs was expressed as a percentage with respect to the total amount of viruses encapsulated into the NPs. Release results were obtained daily in triplicate for a time course of 10 days.
4.5. Fluorescent Labeled NPs
To investigate NP cellular uptake, it was vital to label the particle with fluorescent dye. To label NPs with FITC, a method described in a previous study [
53] was followed. In brief, 20 mg of FITC (Sigma, UK) in 20 mL dehydrated methanol was added to 20 mL 1%
w/
v CH in 0.1 M acetic acid solution. Then, it was left for 3 h to react in the dark at room temperature. The FITC-labeled chitosan was precipitated by raising the pH to 10 with 1 M NaOH. The unreacted FITC was washed away with deionized water and separated by centrifugation until no fluorescence was detected in the supernatant. The FITC-CH was dissolved in acetic acid and dialyzed in 5 L of deionized water for 3 days under darkness, with water being replaced every day. The nanoparticles were then fabricated using the same method as described above.
4.6. Adenovirus Immunogenicity
An indirect ELISA test was carried out to check whether NPs have the potential to shield Ad from neutralizing antibodies or not. In brief, flat-bottomed 96-well plates were coated for 1 h at 37 °C with 50 µL of (1:1000) diluted antihexon antibody (primary antibody). Following 3 washes with PBS, plates were blocked with 100 μL of PBS + 2% BSA at 37 °C for 1 h. Plates were then washed 3 times with PBS. Naked Ad for a standard curve was prepared by a 10-fold serial dilution using PBS from 9.8 × 106, to 9.8 × 102 ifu/mL, and each dilution was added at 50 µL per well. The same amount of encapsulated Ad was also diluted down and applied to coat the 96-well plate, as above. After 1 h at room temperature, viruses were washed 3 times, followed by the addition of 50 µL more of (1:1000) diluted antihexon antibody (primary antibody) and incubated for 1 h at 37 °C. Subsequently, wells were rinsed 3 times with PBS, and 50 µL of (1:500) diluted rat antimouse antibody (conjugate antibody) was added. Plates were incubated for 1 h at room temperature and protected from direct light. After washing 3 times with PBS, 50 µL per well of tetramethylbenzidine (TMB) (Sigma, UK) substrate was added. After 4 min of incubation in the dark, the reaction was stopped upon the addition of 2 M HCl. The absorbance of the resulting solution was measured at 450 nm in a Multiskan Ascent (Thermo Labsystems, Altrincham, UK) plate spectrophotometer.
4.7. Cytotoxicity Assay
The stock MTT solution was prepared by dissolving 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma, Gillingham, UK) in PBS to a final concentration of 5 mg/mL. Cells were cultured in 96-well plates (5.0 × 103/well) and were left for 24 h to be attached to the bottom of the incubator at 37 °C with 5% CO2. Cells were exposed to different concentrations of NPs prepared by a 2-fold serial dilution using a fresh culture medium, i.e., 5, 2.5, 1.25, 0.625, 0.313, and 0.156 mg/mL. There was also a positive control of cells that was not exposed to NPs. After 24 h, the medium in each well was replaced with 200 μL of fresh medium containing different concentrations of NPs and incubated at 37 °C with 5% CO2. NP cytotoxicity was investigated at day 1, 3, and 6 by adding 50 μL of MTT (5 mg/mL) to the wells; the plates were protected from direct light. The solution was removed after incubation for 4 h at 37 °C. Subsequently, 80 μL of 99.9% DMSO and 20 μL glycine buffer (0.1 M glycine and 0.1 M NaCl equilibrated to pH = 10.5) (Sigma) were added. The absorbance of the resulting solution was measured at 540 nm in a Multiskan Ascent (ThermoLabsystems, Altrincham, UK) plate spectrophotometer.
4.8. Statistical Analysis
All results were statistically analyzed using the Student’s t-test and a one-way ANOVA in statistical package GraphPad Prism version 6.03 (GraphPad Software, Inc., La Jolla, CA, USA). p ≤ 0.05 was considered to be statistically significant.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












