Suppression of Hepatitis C Virus Genome Replication and Particle Production by a Novel Diacylglycerol Acyltransferases Inhibitor

 ,
,
Abstract
:
1. Introduction
2. Results
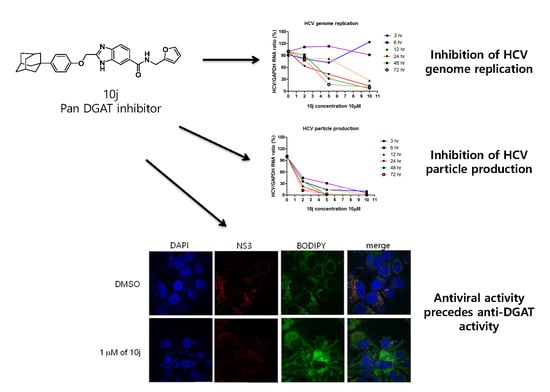
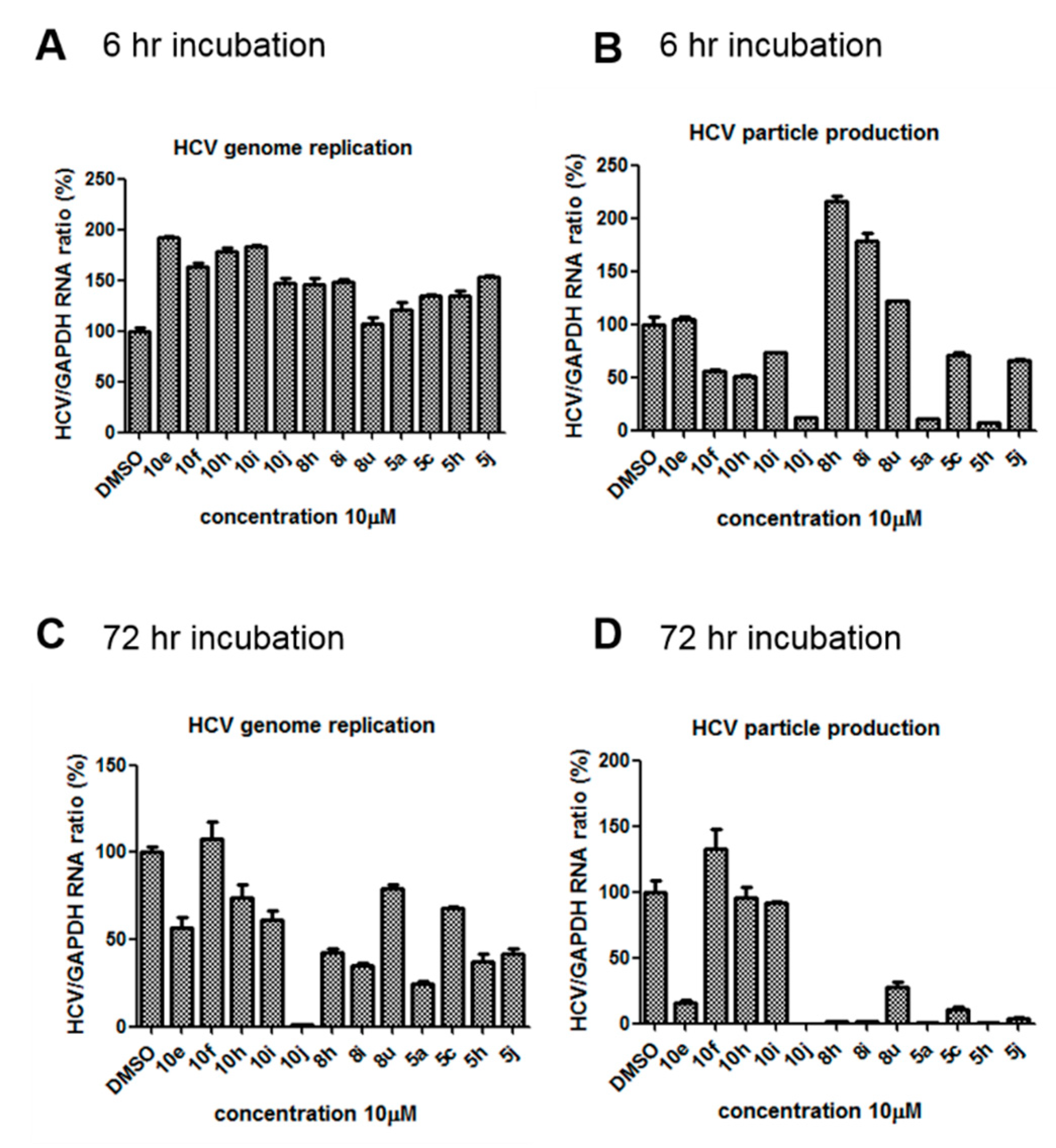
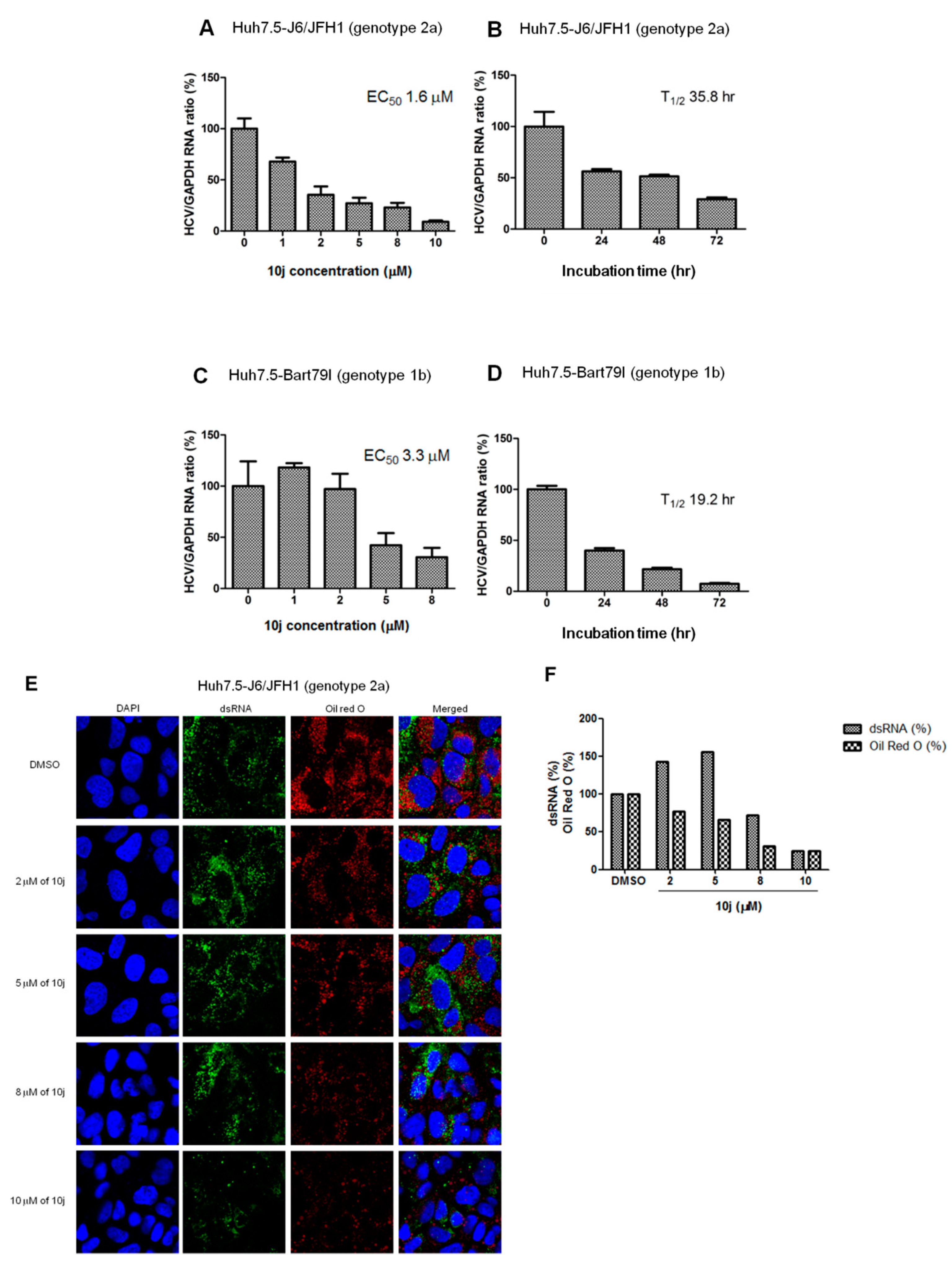
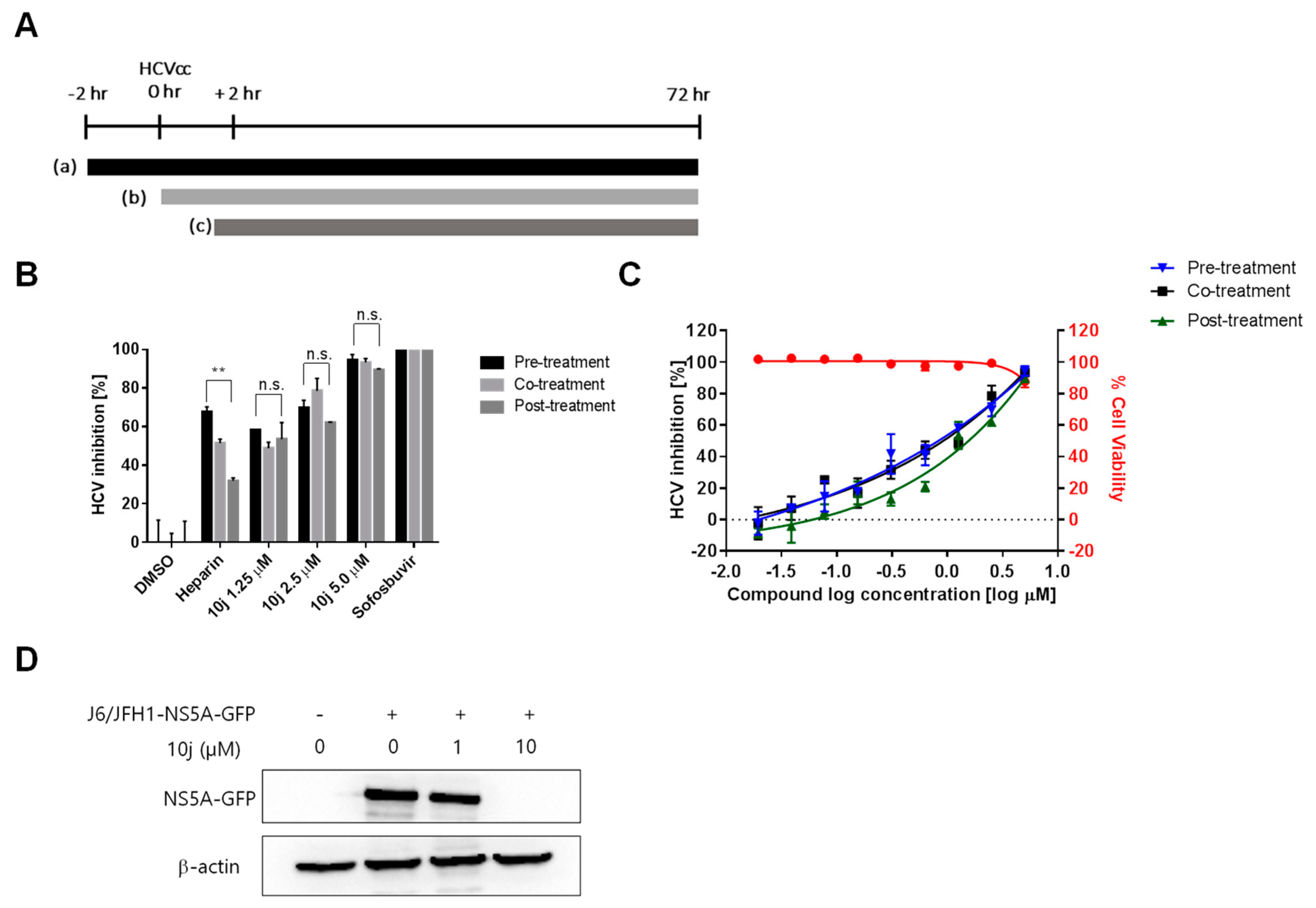
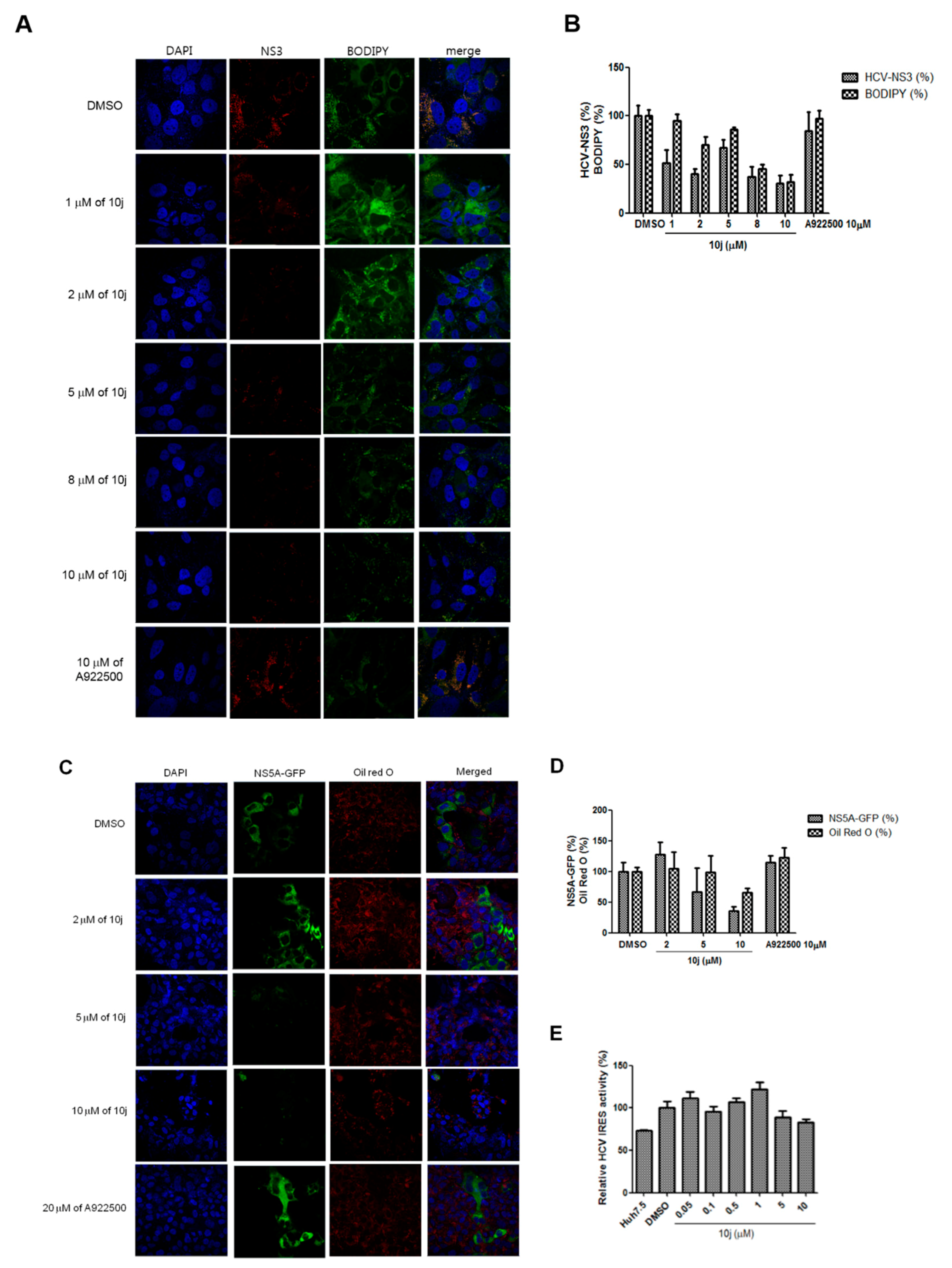
2.1. 10j Suppresses Both HCV Genome Replication and Particle Production
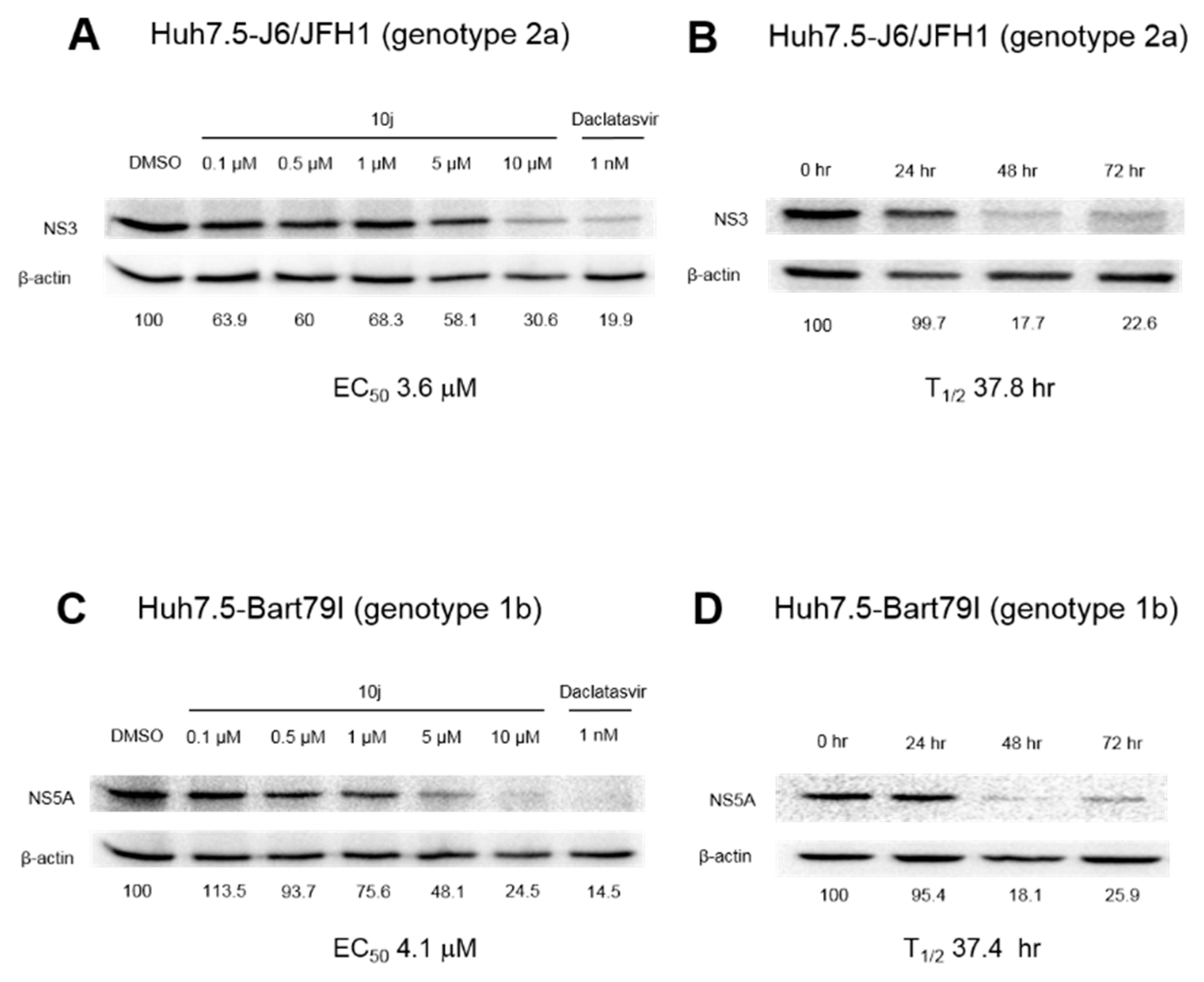
2.2. 10j Reduces Expression Levels of HCV Proteins
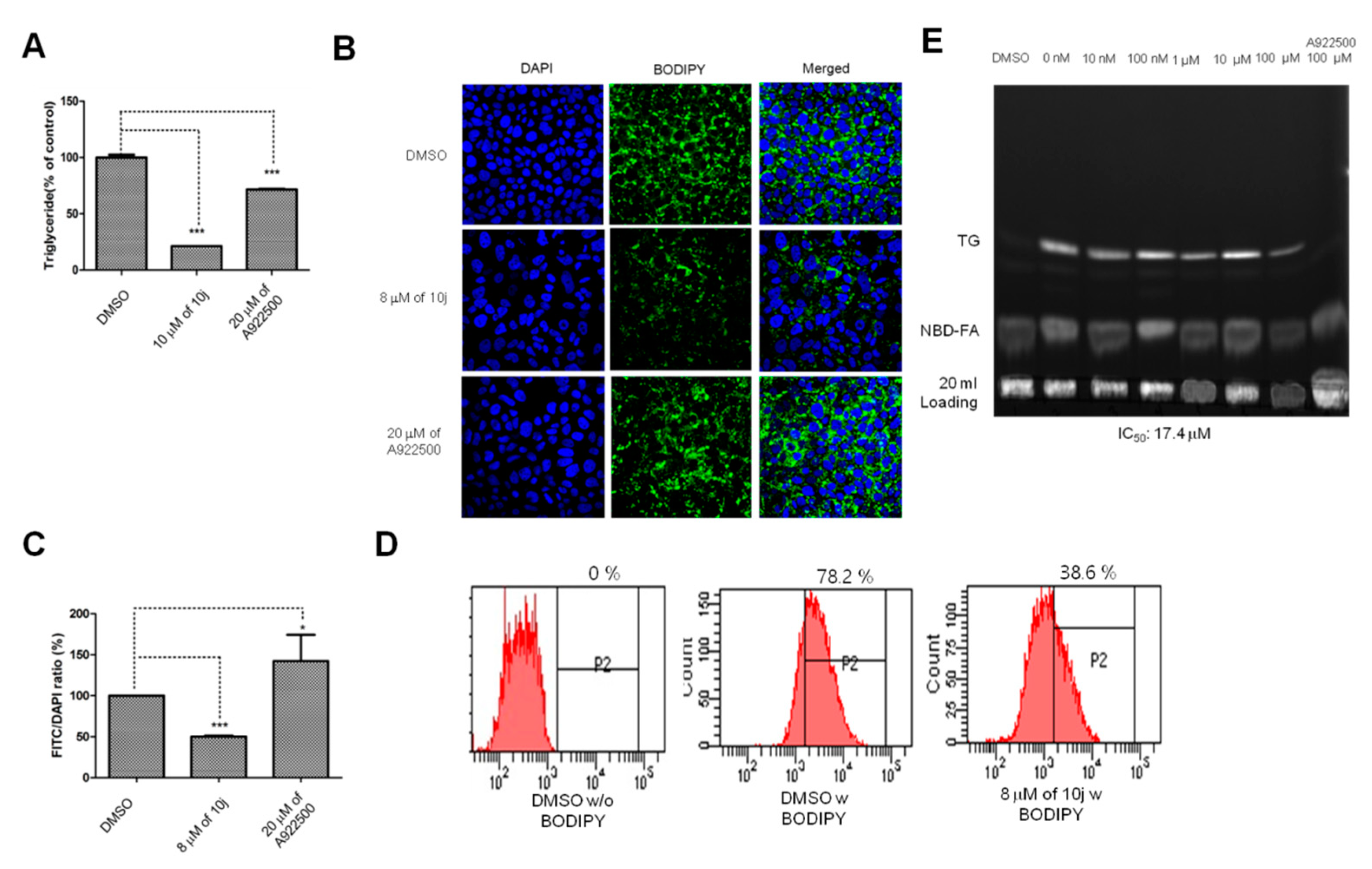
2.3. 10j Suppresses the Biosynthesis of TG
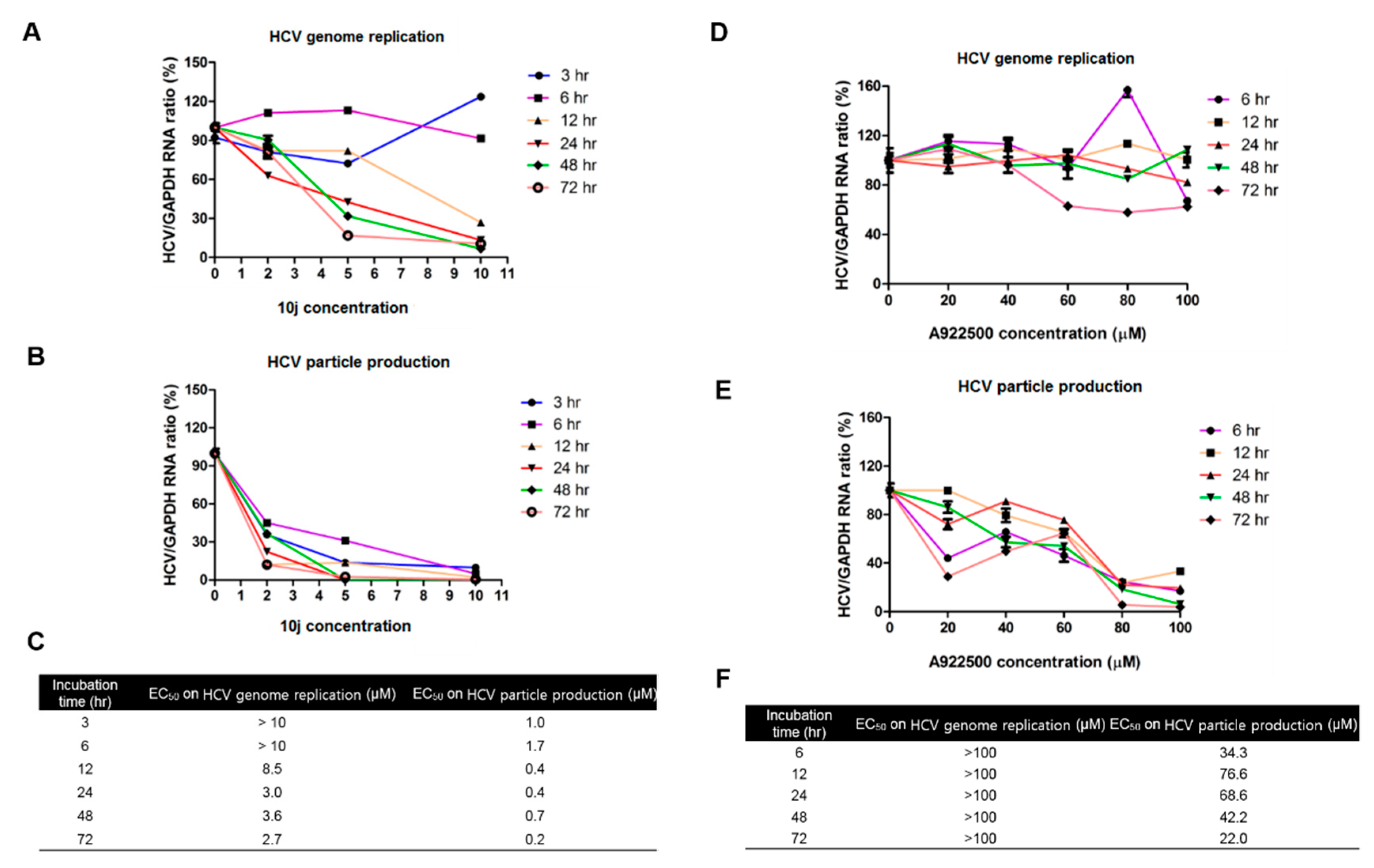
2.4. Inhibition of HCV Genome Replication by 10j Precedes Its Suppression of TG Synthesis
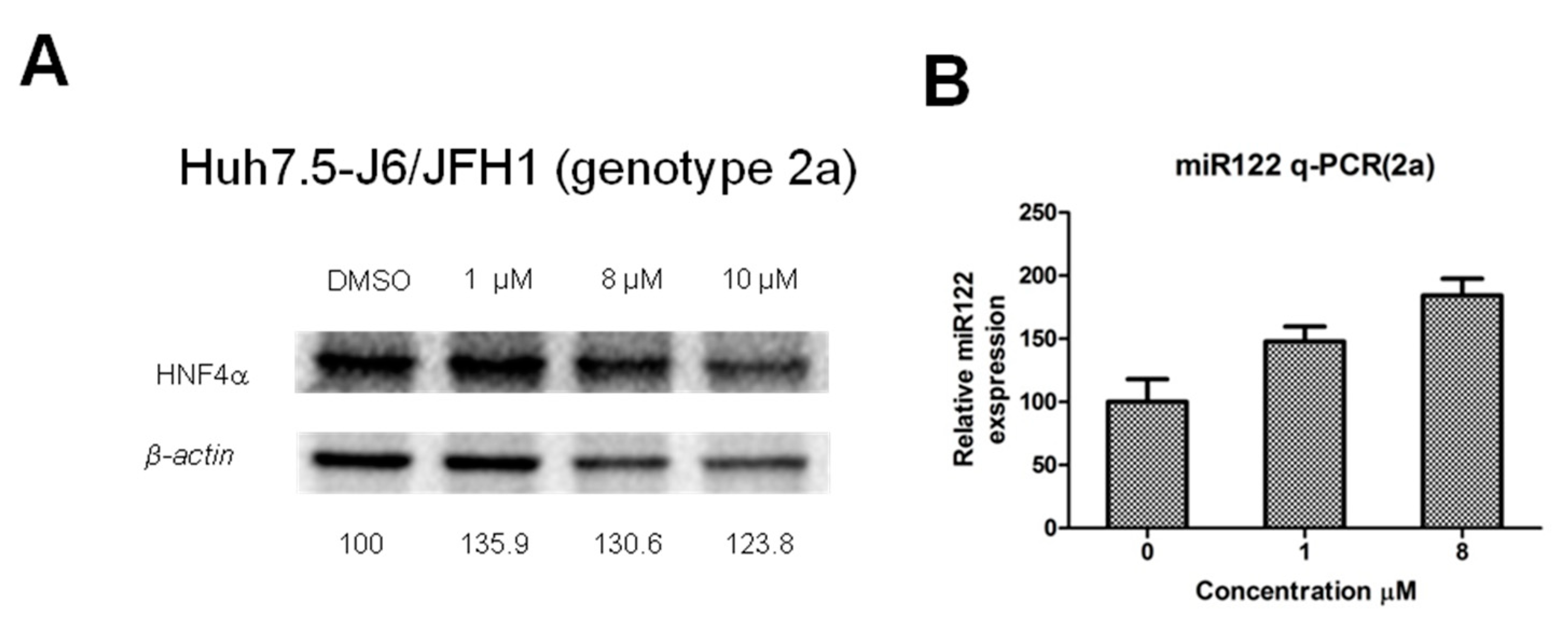
2.5. Effects of 10j on Levels of a Liver-Specific Marker Such as HNF4α and miR122
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids
4.3. In-Vitro Transcription for Production of HCV RNA Genomes
4.4. Generation of Stable HCV-Positive Cell Lines
4.5. Cell Viability and Anti-HCV Replication Analysis Using a Luciferase Assay
4.6. TG Quantification Assay
4.7. Visualization of Intracellular LD
4.8. FACS Analysis
4.9. Quantitative Real-Time RT-PCR Analysis
4.10. Immunofluorescence Analysis
4.11. Western Blot Analysis
4.12. HCV Genome Replication and Particle Production Assay
4.13. HCVcc Infectivity and the Time-of-Addition Assay
4.14. Total Membrane Isolation and DGAT-1 In Vitro Assay
4.15. HCV IRES-Dependent Translation
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alter, M.J. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.R. Clinical practice. Chronic hepatitis C infection. N. Engl. J. Med. 2011, 364, 2429–2438. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; McCourt, D.W.; Wychowski, C.; Feinstone, S.M.; Rice, C.M. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. USA 1993, 90, 10583–10587. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Wychowski, C.; Lin, C.; Feinstone, S.M.; Rice, C.M. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 1993, 67, 1385–1395. [Google Scholar] [PubMed]
- Wilby, K.J.; Partovi, N.; Ford, J.A.; Greanya, E.; Yoshida, E.M. Review of boceprevir and telaprevir for the treatment of chronic hepatitis C. Can. J. Gastroenterol. 2012, 26, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Daclatasvir: Potential role in hepatitis C. Drug Des. Dev. Ther. 2013, 7, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Everson, G.T.; Sims, K.D.; Rodriguez-Torres, M.; Hezode, C.; Lawitz, E.; Bourliere, M.; Loustaud-Ratti, V.; Rustgi, V.; Schwartz, H.; Tatum, H.; et al. Efficacy of an interferon- and ribavirin-free regimen of daclatasvir, asunaprevir and bms-791325 in treatment-naive patients with HCV genotype 1 infection. Gastroenterology 2014, 146, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl coa:Diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.; Obiang-Obounou, B.W.; Kim, M.; Choi, Y.; Lee, H.S.; Lee, K. Therapeutic strategies for metabolic diseases: Small-molecule diacylglycerol acyltransferase (dgat) inhibitors. ChemMedChem 2014, 9, 2410–2424. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Herker, E.; Modi, A.A.; Haas, J.T.; Ramage, H.R.; Farese, R.V., Jr.; Ott, M. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 2013, 288, 9915–9923. [Google Scholar] [CrossRef] [PubMed]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V., Jr.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gane, E.; Stedman, C.; Dole, K.; Chen, J.; Meyers, C.D.; Wiedmann, B.; Zhang, J.; Raman, P.; Colvin, R.A. A diacylglycerol transferase 1 inhibitor is a potent hepatitis C antiviral in vitro but not in patients in a randomized clinical trial. ACS Infect. Dis. 2017, 3, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kwon, J.; Kim, M.O.; Singh, S.; Kim, S.K.; Lee, K.; Lee, K.; Lee, H.S.; Choi, Y. Discovery of a novel series of indolyl hydrazide derivatives as diacylglycerol acyltransferase-1 inhibitors. Bull. Korean Chem. Soc. 2015, 36, 628–635. [Google Scholar]
- Lee, K.; Goo, J.I.; Jung, H.Y.; Kim, M.; Boovanahalli, S.K.; Park, H.R.; Kim, M.O.; Kim, D.H.; Lee, H.S.; Choi, Y. Discovery of a novel series of benzimidazole derivatives as diacylglycerol acyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7456–7460. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, M.; Lee, B.; Goo, J.; Kim, J.; Naik, R.; Seo, J.H.; Kim, M.O.; Byun, Y.; Song, G.Y.; et al. Discovery of indolyl acrylamide derivatives as human diacylglycerol acyltransferase-2 selective inhibitors. Org. Biomol. Chem. 2013, 11, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-ph-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., 2nd; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Parviz, F.; Matullo, C.; Garrison, W.D.; Savatski, L.; Adamson, J.W.; Ning, G.; Kaestner, K.H.; Rossi, J.M.; Zaret, K.S.; Duncan, S.A. Hepatocyte nuclear factor 4α controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 2003, 34, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kuroda, K.; Ikeda, M.; Wakita, T.; Kato, N.; Makishima, M. Hepatitis C virus NS4B targets lipid droplets through hydrophobic residues in the amphipathic helices. J. Lipid Res. 2013, 54, 881–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.M.; Zeuzem, S. Diacylglycerol acyltransferase-1: A critical host factor for hepatitis C virus assembly and potential new drug target. Gastroenterology 2011, 140, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.S.; Murayama, A.; Kang, W.; Kim, M.S.; Yoon, S.K.; Fukasawa, M.; Kondoh, M.; Kim, J.S.; Kim, H.; Kato, T.; et al. Hepatitis C virus entry is impaired by claudin-1 downregulation in diacylglycerol acyltransferase-1-deficient cells. J. Virol. 2014, 88, 9233–9244. [Google Scholar] [CrossRef] [PubMed]
- Dibrov, S.M.; Ding, K.; Brunn, N.D.; Parker, M.A.; Bergdahl, B.M.; Wyles, D.L.; Hermann, T. Structure of a hepatitis C virus rna domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proc. Natl. Acad. Sci. USA 2012, 109, 5223–5228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Liu, N.; Zhan, P.; Jiang, X.; Liu, X. Discovery of HCV NS5B thumb site i inhibitors: Core-refining from benzimidazole to indole scaffold. Eur. J. Med. Chem. 2015, 94, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Suzuki, T.; Hirashima, S.; Mizutani, K.; Yoshida, A.; Ando, I.; Ikeda, S.; Adachi, T.; Hashimoto, H. Benzimidazole inhibitors of hepatitis C virus NS5B polymerase: Identification of 2-[(4-diarylmethoxy)phenyl]-benzimidazole. Bioorg. Med. Chem. Lett. 2006, 16, 1859–1863. [Google Scholar] [CrossRef] [PubMed]
- El Diwani, H.I.; Abdel-Mohsen, H.T.; Salama, I.; Ragab, F.A.; Ramla, M.M.; Galal, S.A.; Abdalla, M.M.; Abdel-Wahab, A.; El Demellawy, M.A. Synthesis, molecular modeling and biological evaluation of novel benzimidazole derivatives as inhibitors of hepatitis C virus RNA replication. Chem. Pharm. Bull. 2014, 62, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Asthana, S.; Shukla, S.; Vargiu, A.V.; Ceccarelli, M.; Ruggerone, P.; Paglietti, G.; Marongiu, M.E.; Blois, S.; Giliberti, G.; La Colla, P. Different molecular mechanisms of inhibition of bovine viral diarrhea virus and hepatitis C virus RNA-dependent RNA polymerases by a novel benzimidazole. Biochemistry 2013, 52, 3752–3764. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumpter, R., Jr.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Sklan, E.H.; Staschke, K.; Oakes, T.M.; Elazar, M.; Winters, M.; Aroeti, B.; Danieli, T.; Glenn, J.S. A rab-gap tbc domain protein binds hepatitis C virus NS5A and mediates viral replication. J. Virol. 2007, 81, 11096–11105. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science (N. Y.) 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Kim, H.Y.; Jo, S.; Park, E.; Choi, J.; Kong, S.; Park, D.S.; Heo, J.M.; Lee, J.S.; Ko, Y.; et al. Synthesis and evaluation of hexahydropyrimidines and diamines as novel hepatitis C virus inhibitors. Eur. J. Med. Chem. 2013, 70, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Interaction of hepatitis C virus core protein with janus kinase is required for efficient production of infectious viruses. Biomol. Ther. 2013, 21, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.J.; Dvory-Sobol, H.; Lee, C.; Cho, S.J.; Bryson, P.; Masek, M.; Elazar, M.; Frank, C.W.; Glenn, J.S. Identification of a class of hcv inhibitors directed against the nonstructural protein NS4B. Sci. Transl. Med. 2010, 2, 15ra16. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yoon, K.D.; Lee, M.; Cho, Y.; Choi, G.; Jang, H.; Kim, B.; Jung, D.H.; Oh, J.G.; Kim, G.W.; et al. Identification of a resveratrol tetramer as a potent inhibitor of hepatitis C virus helicase. Br. J. Pharmacol. 2016, 173, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Madduma Hewage, S.R.; Piao, M.J.; Kang, K.A.; Ryu, Y.S.; Han, X.; Oh, M.C.; Jung, U.; Kim, I.G.; Hyun, J.W. Hesperidin attenuates ultraviolet b-induced apoptosis by mitigating oxidative stress in human keratinocytes. Biomol. Ther. 2016, 24, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, N.; Kalliampakou, K.I.; Kotta-Loizou, I.; Befani, C.; Liakos, P.; Simos, G.; Mentis, A.F.; Kalliaropoulos, A.; Doumba, P.P.; Smirlis, D.; et al. Low oxygen tension enhances hepatitis C virus replication. J. Virol. 2013, 87, 2935–2948. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Yang, J.; Jo, E.; Lee, J.Y.; Kim, H.Y.; Bartenschlager, R.; Shin, E.C.; Bae, Y.S.; Windisch, M.P. A Novel Inhibitor IDPP Interferes with Entry and Egress of HCV by Targeting Glycoprotein E1 in a Genotype-Specific Manner. Sci. Rep. 2017, 7, 44676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFie, P.J.; Stone, S.J. A fluorescent assay to quantitatively measure in vitro acyl CoA: Diacylglycerol acyltransferase activity. J. Lipid Res. 2011, 52, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lee, J.H.; Myung, H.J. An Interferon Resistance Induced by the Interaction between HCV NS5B and Host p48. Korean J. Microbiol. Biotechnol. 2008, 4, 353–359. [Google Scholar]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Code | Chemical Stucture |
|---|---|---|
| Benzimidazoles (pan DGATs inhibitors) | 10e |  |
| 10f |  | |
| 10h |  | |
| 10i |  | |
| 10j |  | |
| Indolyl hydrazides (DGAT-1 inhibitors) | 8h |  |
| 8i |  | |
| 8u |  | |
| Indolyl acrylamides (DGAT-2 inhibitors) | 5a |  |
| 5c |  | |
| 5h |  | |
| 5j |  | |
| DGAT-1 inhibitor | A922500 |  |
| Species | Code | IC50 (RM) (μM) | IC50 (DGAT-1) (μM) | IC50 (DGAT-2) (μM) | TG % (10 μM in HepG2) |
|---|---|---|---|---|---|
| Benzimidazoles (pan DGATs inhibitors) | 10e | 27.5 | N/D | N/D | N/D |
| 10f | >50 | N/D | N/D | N/D | |
| 10h | >50 | N/D | N/D | N/D | |
| 10i | 20.0 | N/D | N/D | N/D | |
| 10j | 4.4 | 9.0 | 17.3 | 45.0 | |
| Indolyl hydrazides (DGAT-1 inhibitors) | 8h | N/D | 20.4 | >10 | 71.2 |
| 8i | N/D | 2.1 | >10 | 94.8 | |
| 8u | N/D | 1.5 | >10 | 55.7 | |
| Indolyl acrylamides (DGAT-2 inhibitors) | 5a | 8.8 | >100 | 6.9 | N/D |
| 5c | 13.2 | 89.1 | 6.8 | N/D | |
| 5h | 2.5 | >100 | 6.9 | 33.2 | |
| 5j | 9.4 | >100 | 7.4 | N/D | |
| DGAT-1 inhibitor | A922500 | N/D | 0.007 | N/D | N/D |
| Species | Code | EC50 (μM) | CC50 (μM) | TI |
|---|---|---|---|---|
| Benzimidazoles (pan DGATs inhibitors) | 10e | >10 | >10 | >1.0 |
| 10f | >10 | >10 | >1.0 | |
| 10h | 7.7 | >10 | >1.3 | |
| 10i | 0.9 | 3.9 | 4.4 | |
| 10j | 1.5 | 38.9 | 24.6 | |
| Indolyl hydrazides (DGAT-1 inhibitors) | 8h | 6.7 | >10 | >1.5 |
| 8i | 5.1 | >10 | >2.0 | |
| 8u | >10 | >10 | >1.0 | |
| Indolyl acrylamides (DGAT-2 inhibitors) | 5a | 1.9 | 3.7 | 1.9 |
| 5c | >10 | >10 | >1.0 | |
| 5h | 2.1 | 5.1 | 2.2 | |
| 5j | 1.1 | 3.4 | 3.0 | |
| DGAT-1 inhibitor | A922500 | 50.8 | 71.9 | 1.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Goo, J.-I.; Kim, M.I.; Lee, S.-J.; Choi, M.; Than, T.T.; Nguyen, P.H.; Windisch, M.P.; Lee, K.; Choi, Y.; et al. Suppression of Hepatitis C Virus Genome Replication and Particle Production by a Novel Diacylglycerol Acyltransferases Inhibitor. Molecules 2018, 23, 2083. https://doi.org/10.3390/molecules23082083
Kim D, Goo J-I, Kim MI, Lee S-J, Choi M, Than TT, Nguyen PH, Windisch MP, Lee K, Choi Y, et al. Suppression of Hepatitis C Virus Genome Replication and Particle Production by a Novel Diacylglycerol Acyltransferases Inhibitor. Molecules. 2018; 23(8):2083. https://doi.org/10.3390/molecules23082083
Chicago/Turabian StyleKim, Dahee, Ja-Il Goo, Mi Il Kim, Sung-Jin Lee, Moonju Choi, Thoa Thi Than, Phuong Hong Nguyen, Marc P. Windisch, Kyeong Lee, Yongseok Choi, and et al. 2018. "Suppression of Hepatitis C Virus Genome Replication and Particle Production by a Novel Diacylglycerol Acyltransferases Inhibitor" Molecules 23, no. 8: 2083. https://doi.org/10.3390/molecules23082083