First-Row Late Transition Metals for Catalytic Alkene Hydrofunctionalisation: Recent Advances in C-N, C-O and C-P Bond Formation


Abstract
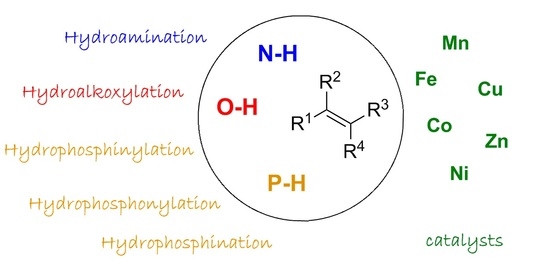
:
{kind=link}
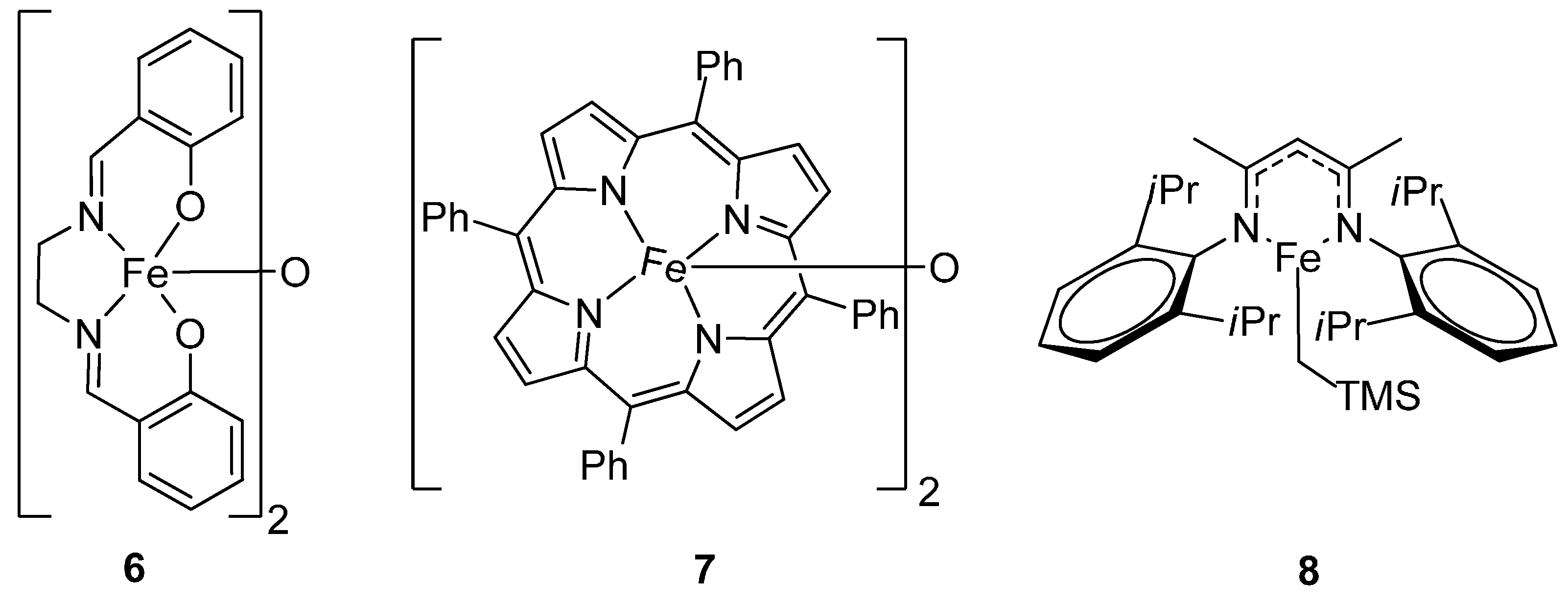{kind=link}
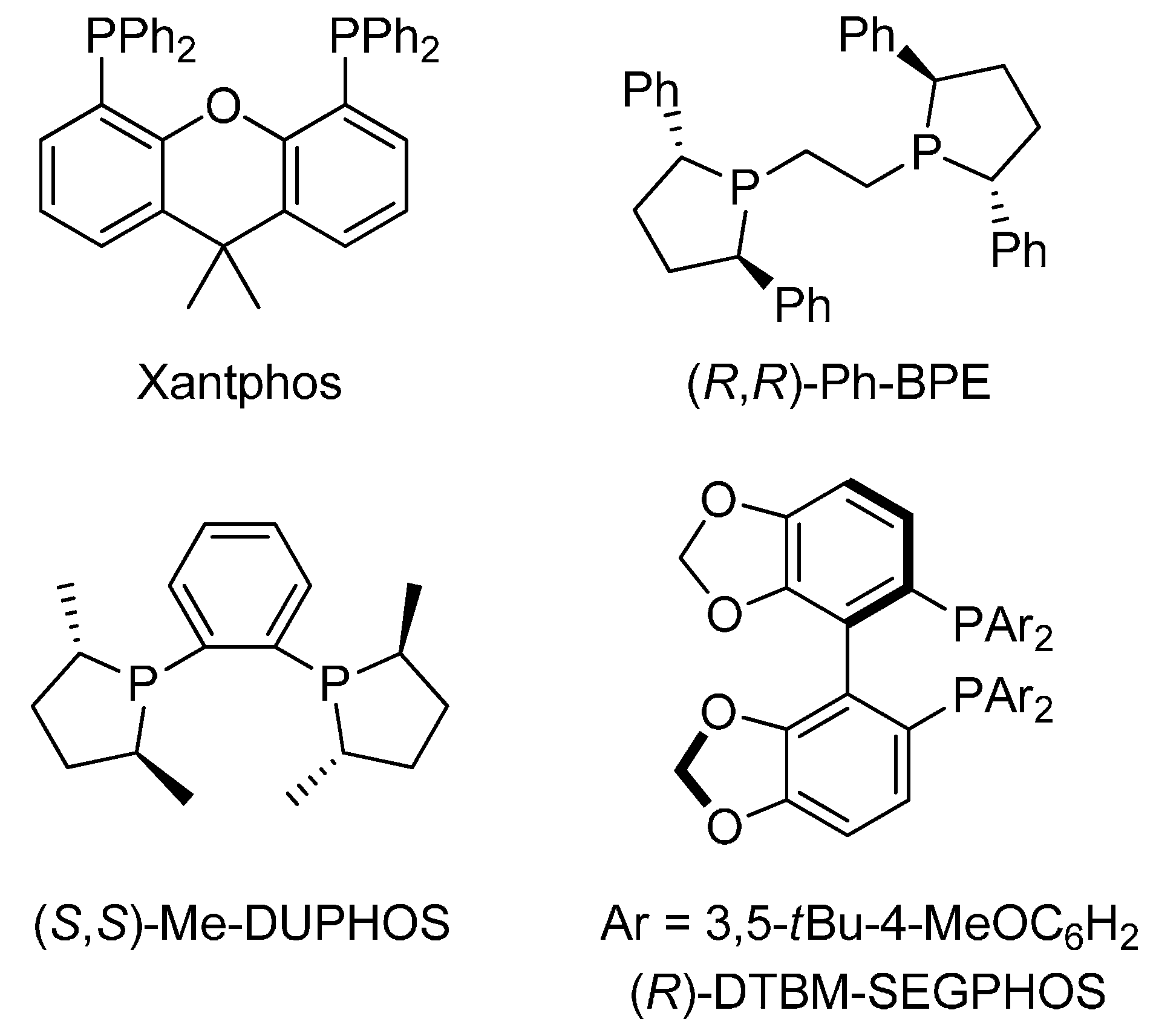{kind=link}
{kind=link}
{kind=link}
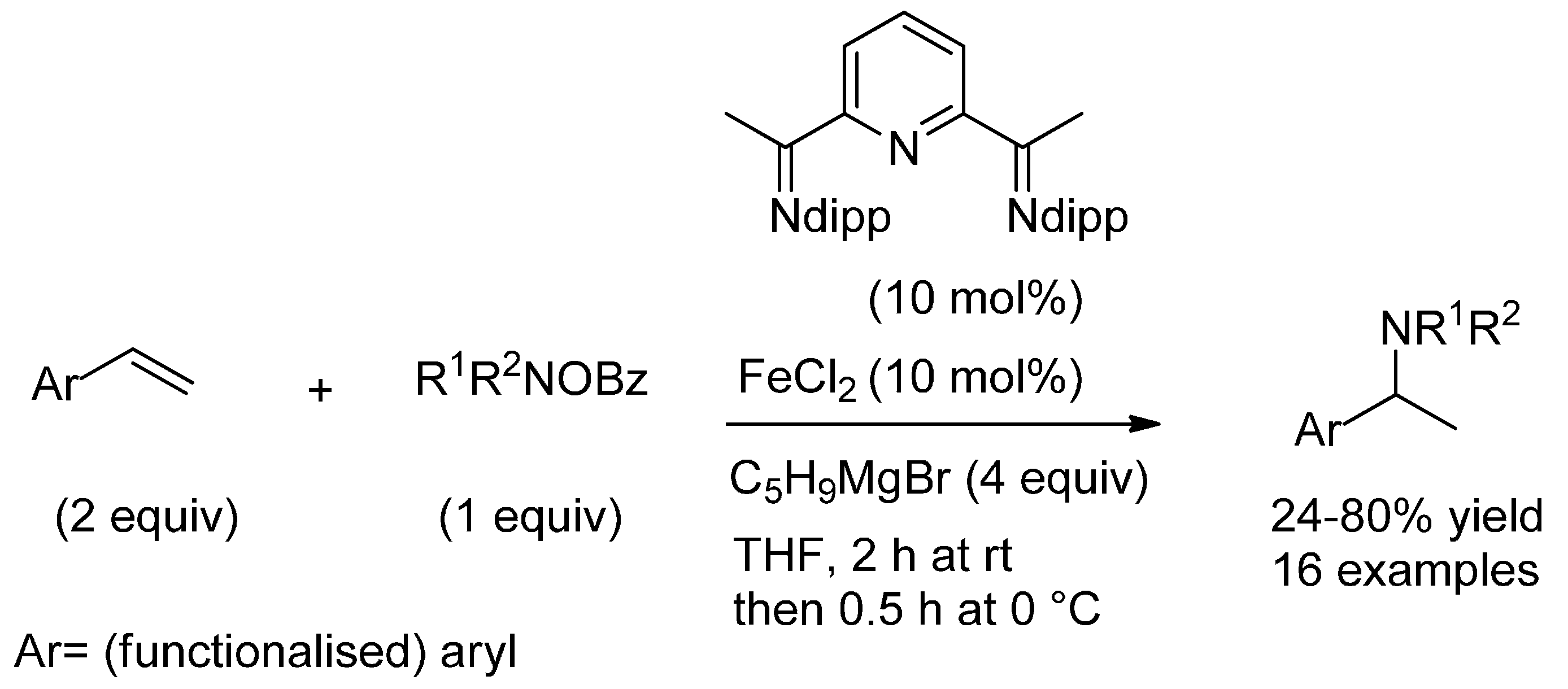{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
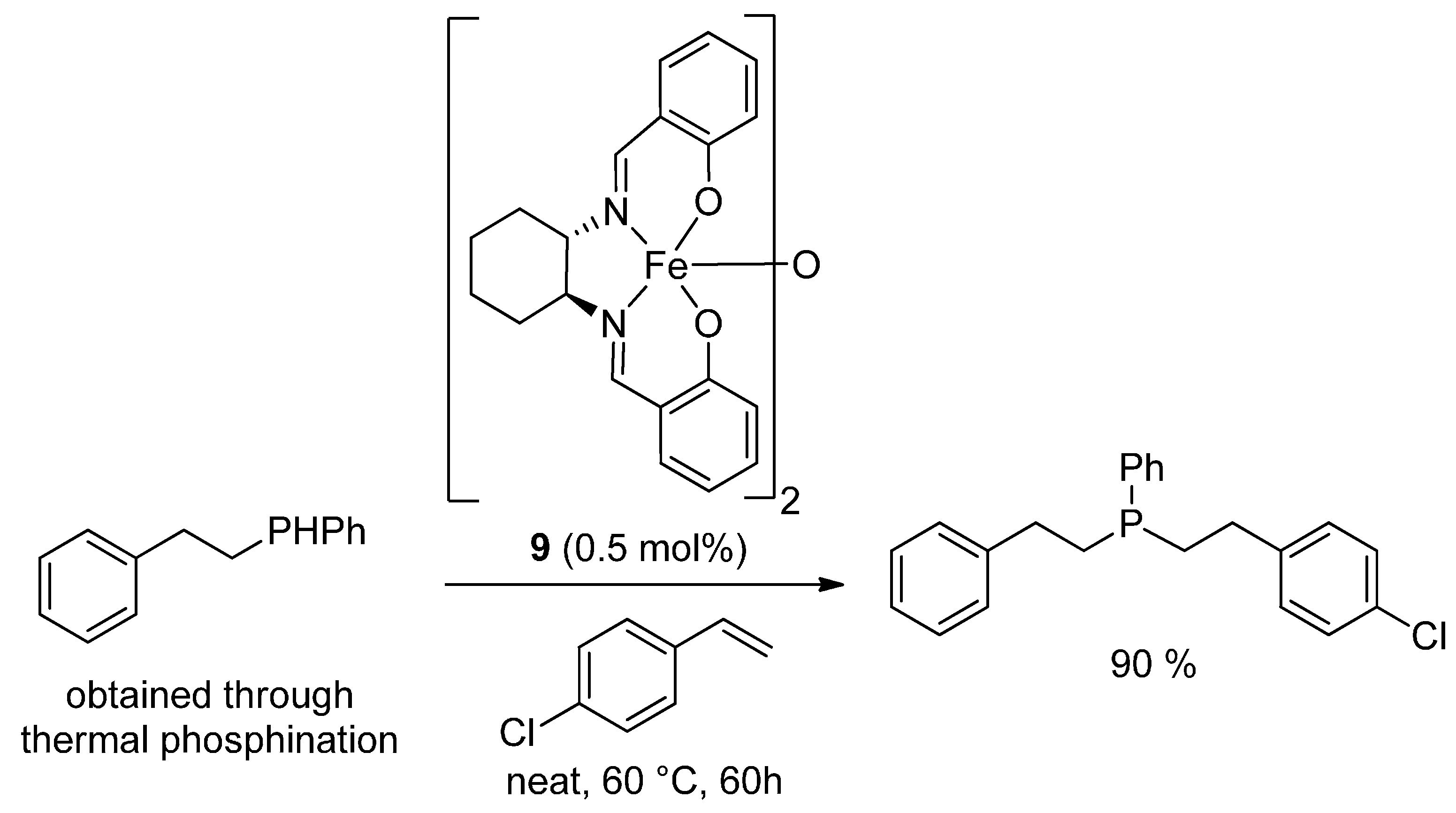{kind=link}
{kind=link}
{kind=link}
{kind=link}
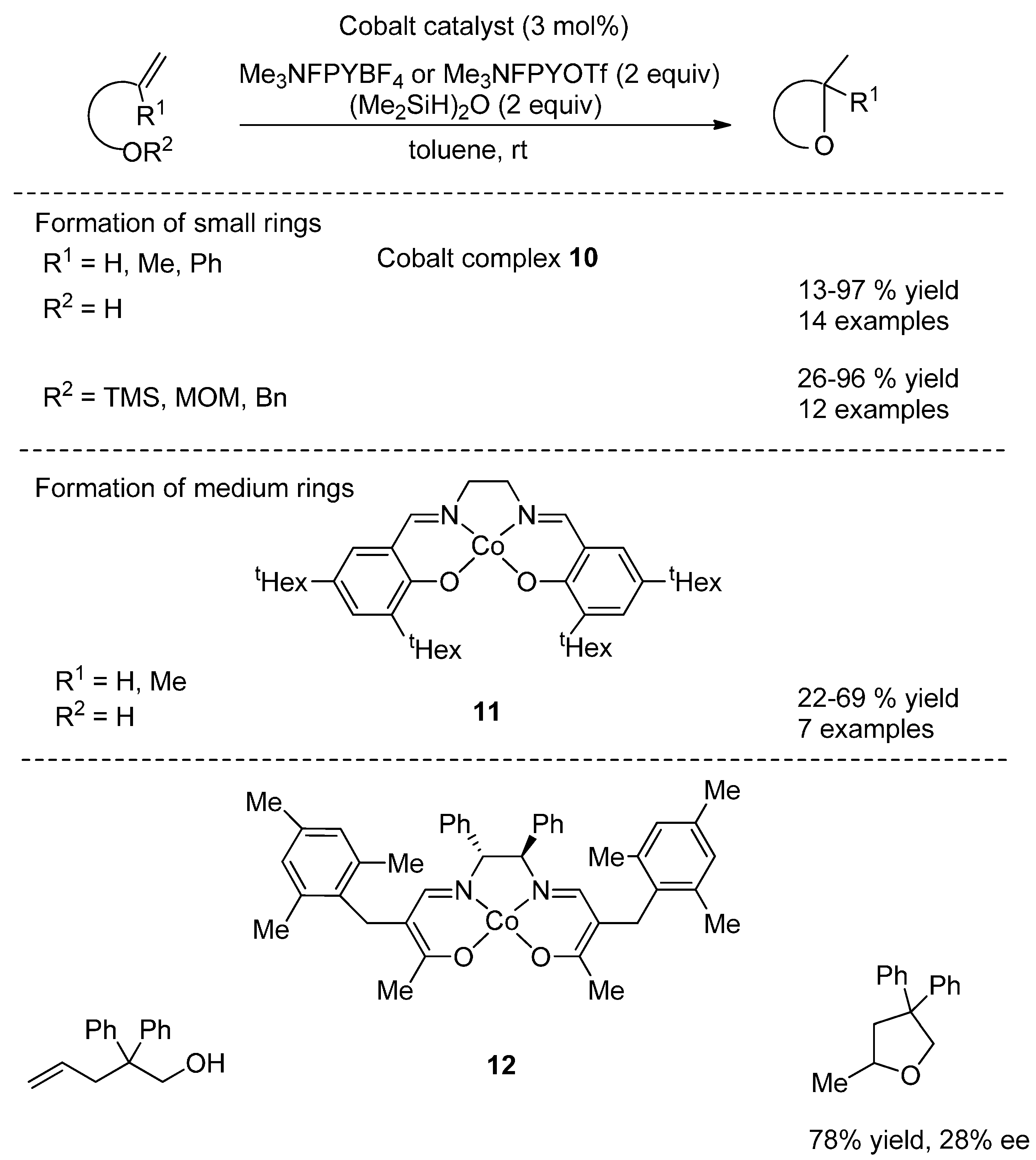{kind=link}
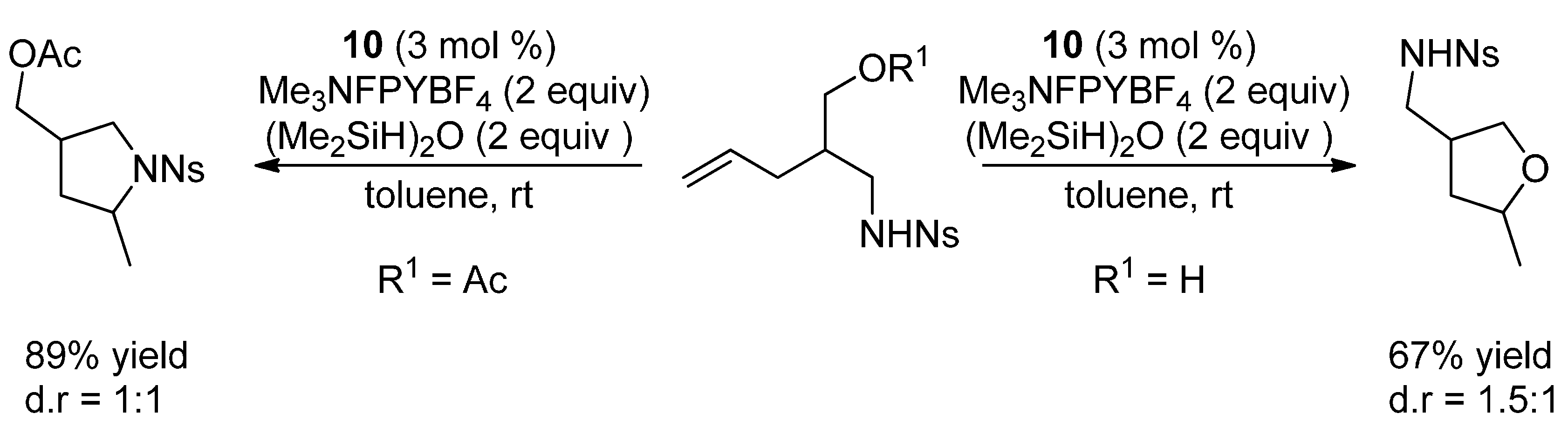{kind=link}
{kind=link}
{kind=link}
{kind=link}
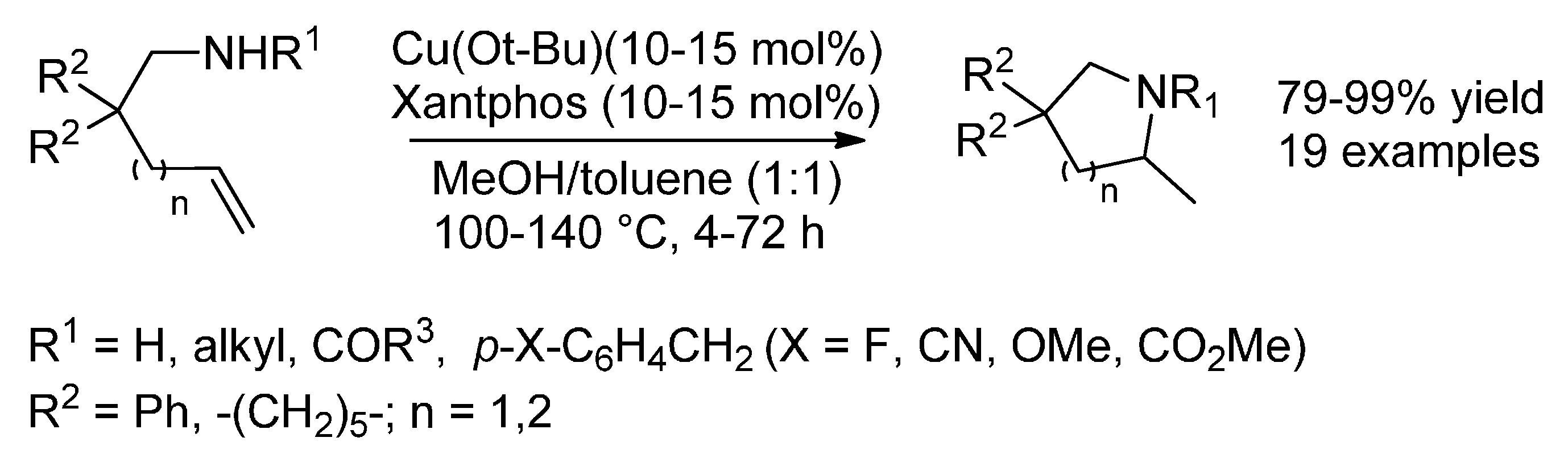{kind=link}
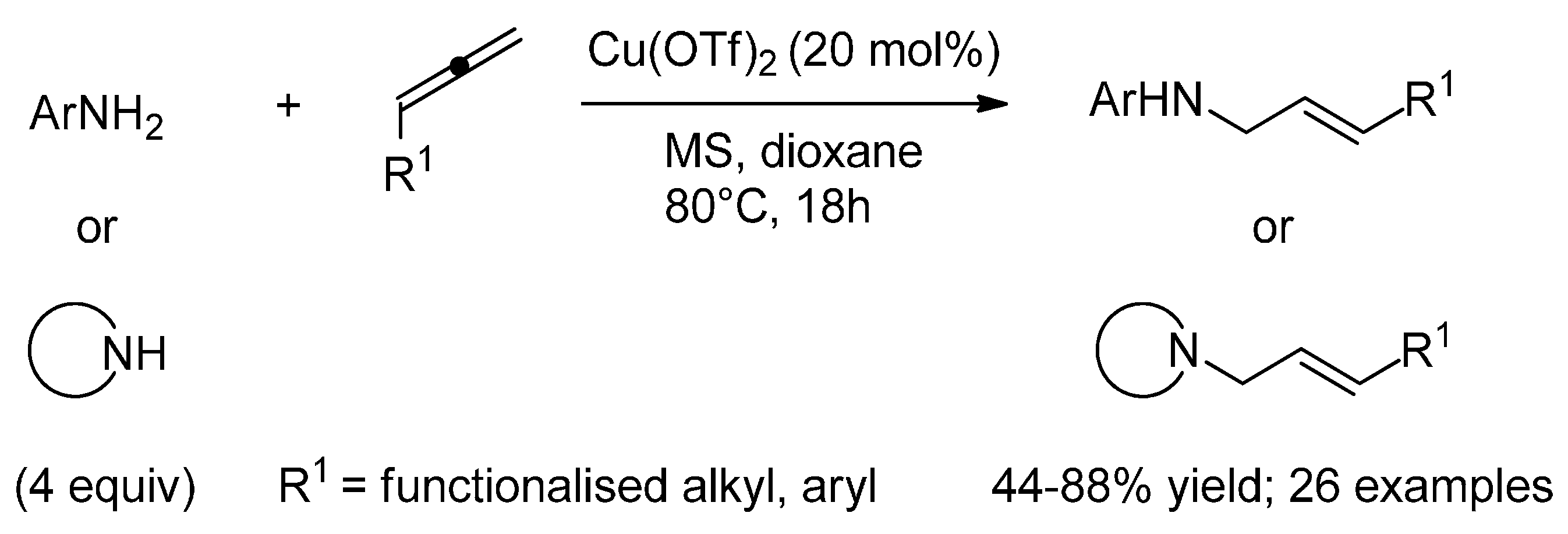{kind=link}
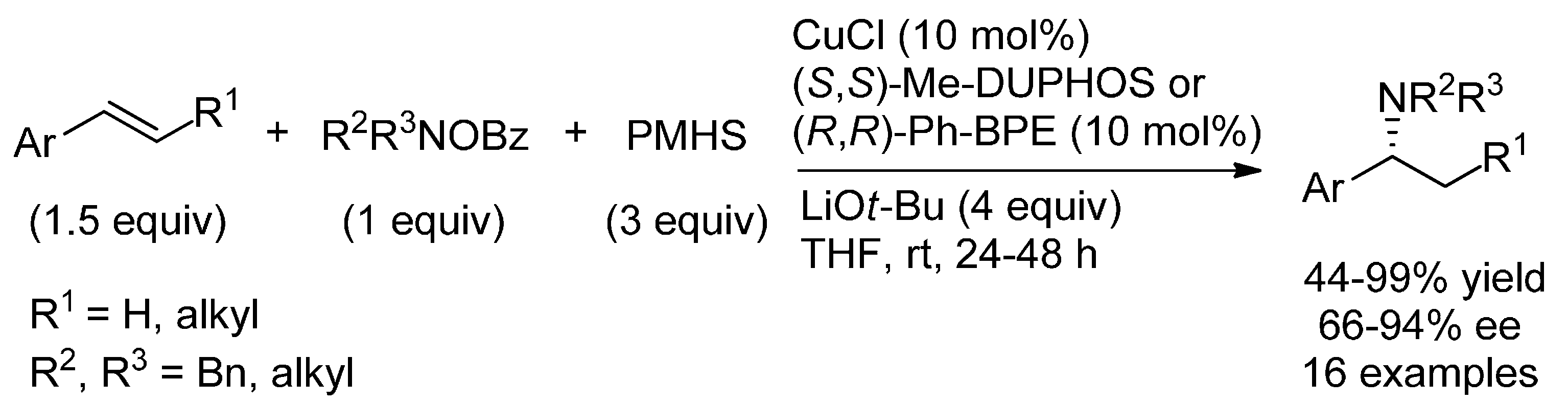{kind=link}
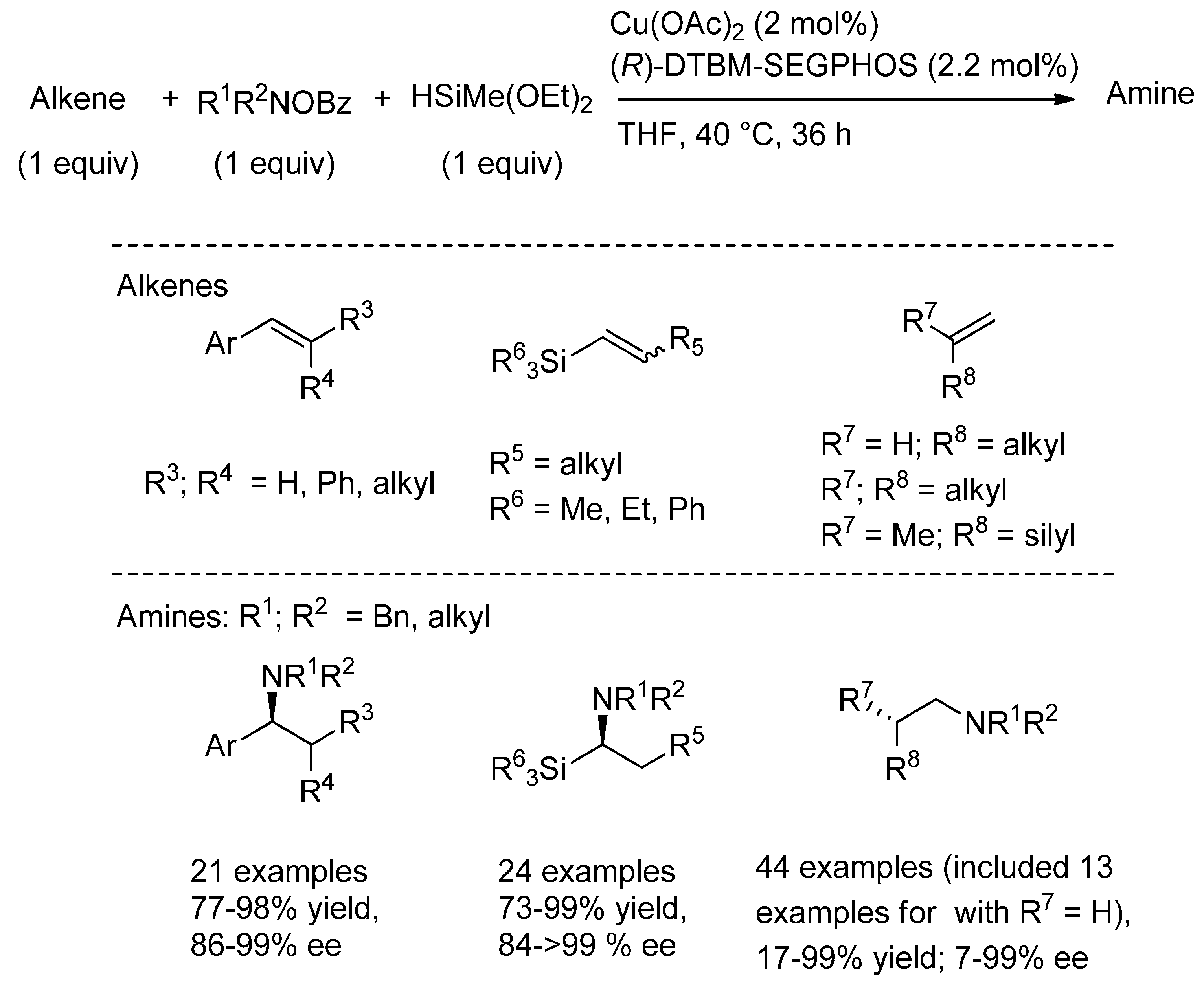{kind=link}
{kind=link}
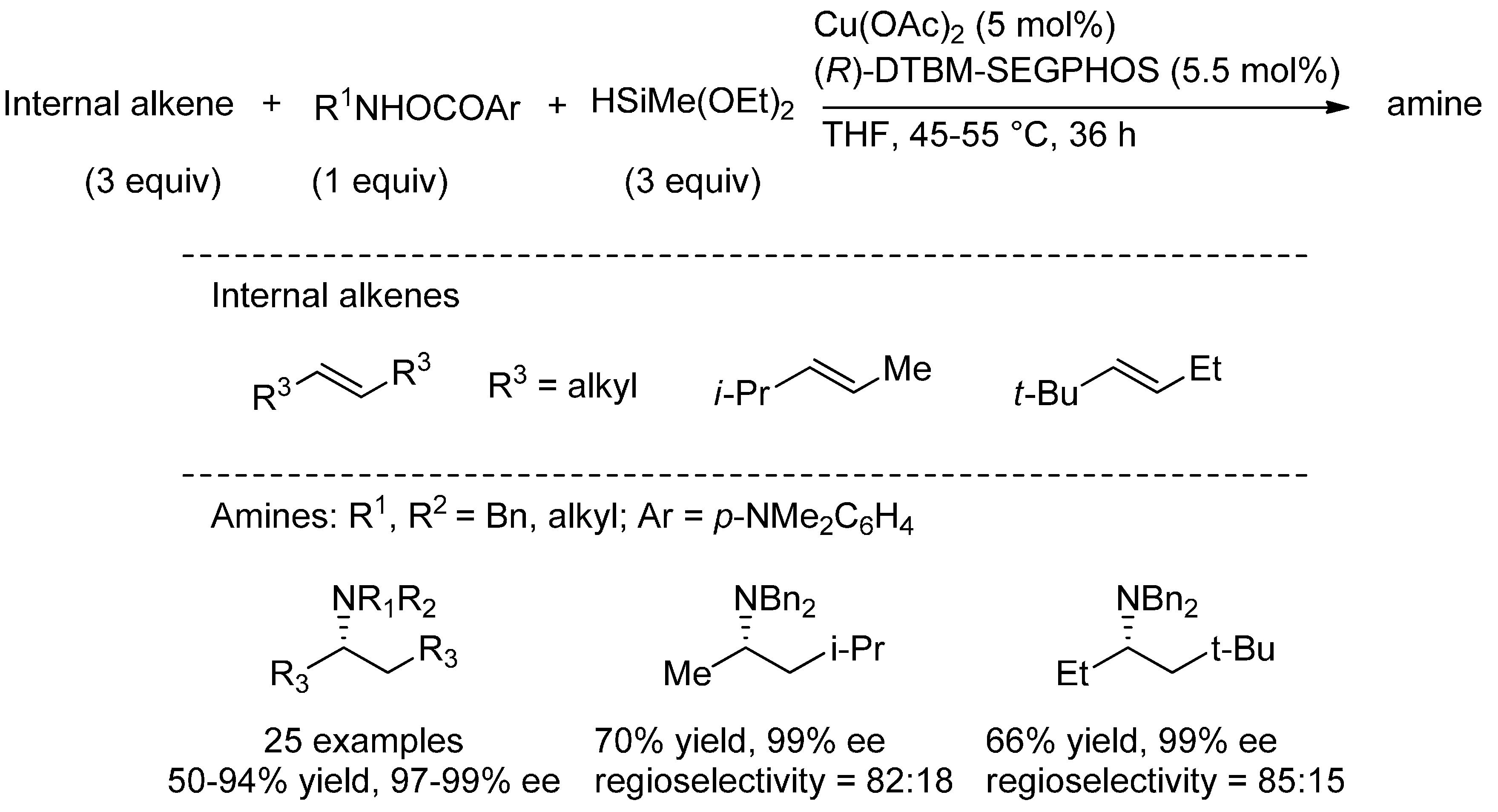{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Manganese
2.1. Hydroamination and Hydroalkoxylation
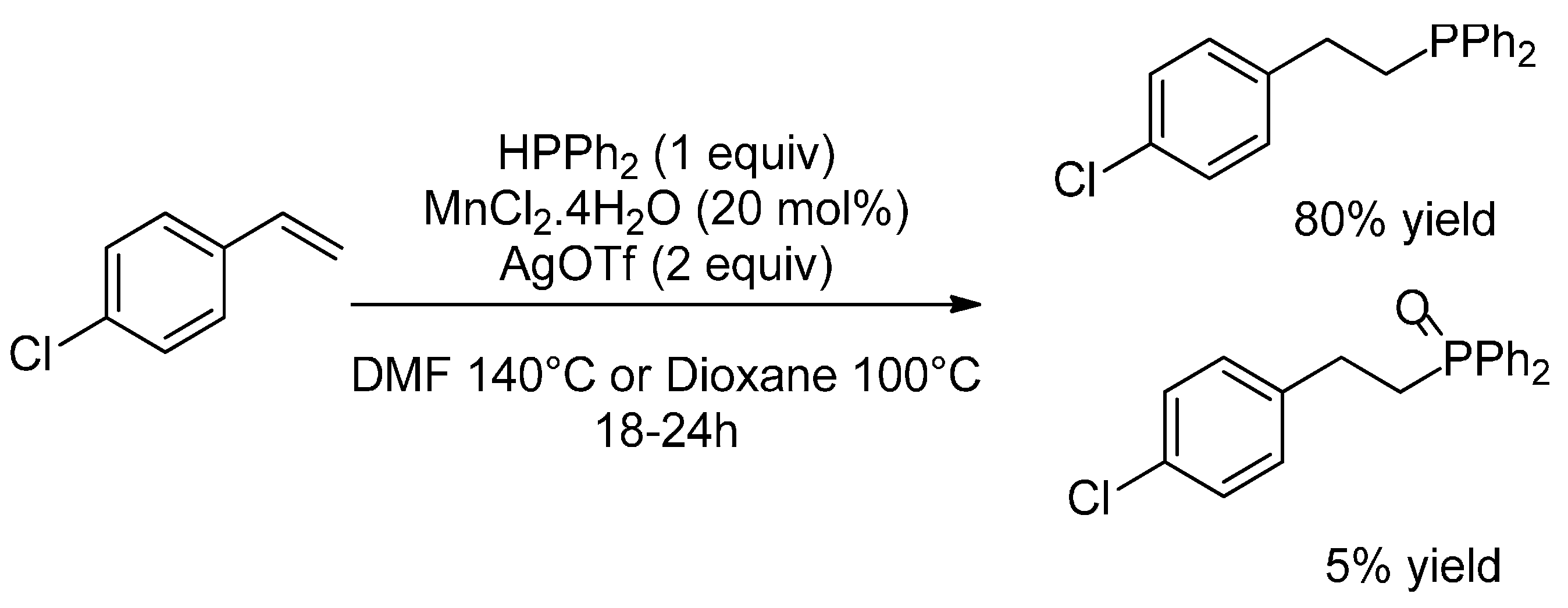
2.2. Hydrophosphination
3. Iron
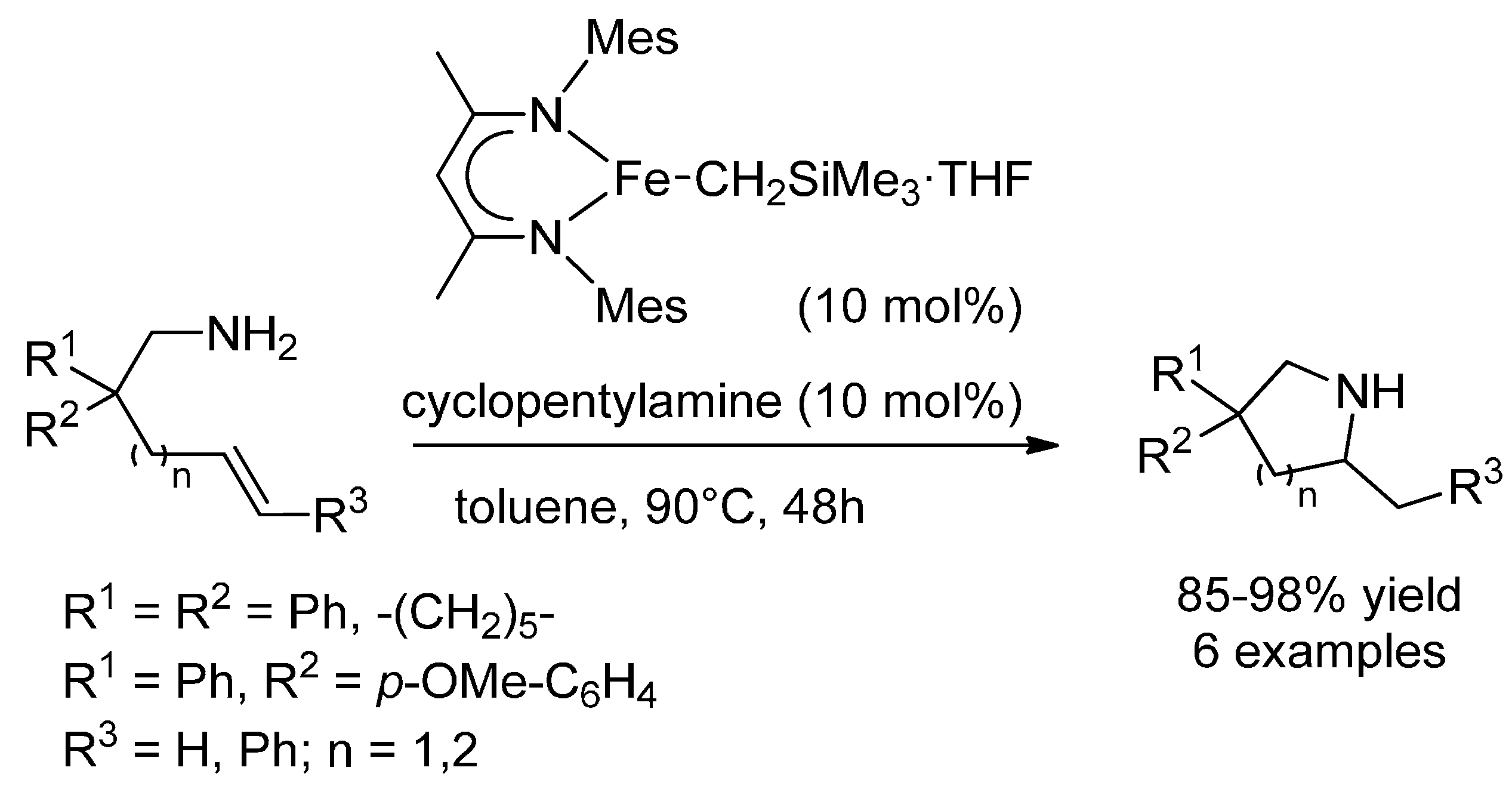
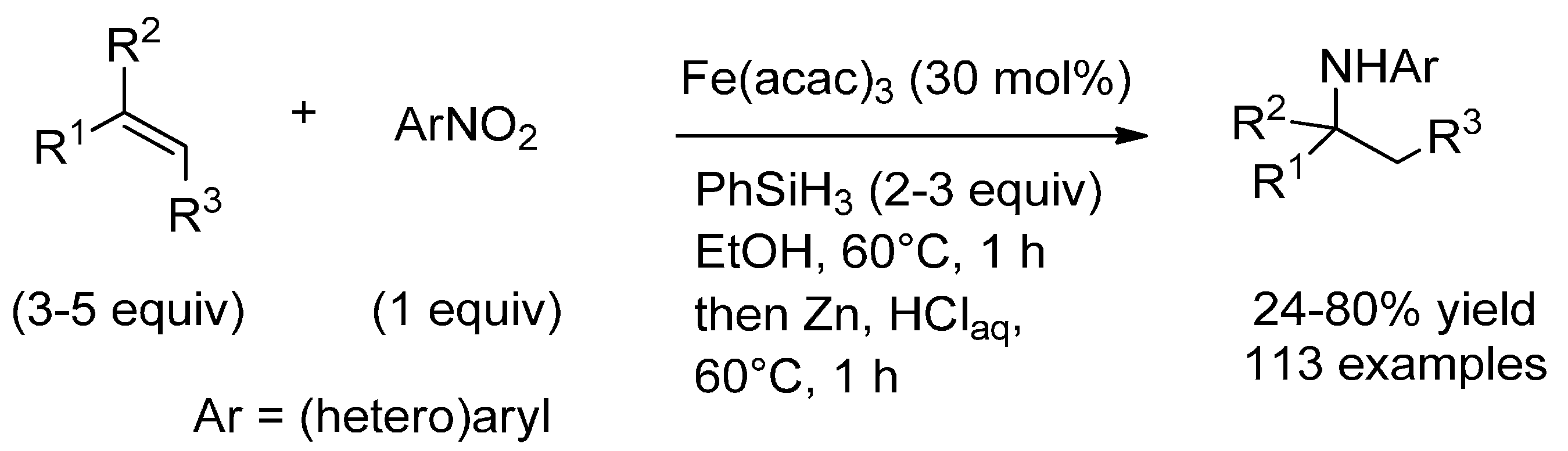
3.1. (Formal) Hydroamination
3.2. Hydroalkoxylation
3.3. Hydrophosphination
4. Cobalt
4.1. Formal Hydroamidation
4.2. Hydroalkoxylation
5. Nickel
5.1. Hydroamination
5.2. Hydroalkoxylation
5.3. Hydrophosphinylation
6. Copper
6.1. (Formal) Hydroamination
6.2. Hydroalkoxylation
6.3. Hydrophosphination
7. Zinc
7.1. Hydroamination
7.2. Hydroalkoxylation
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct addition of amines to alkenes and ALKYNES. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.; Stephan, D.W. Stoichiometric and catalytic activation of P-H and P-P bonds. Chem. Soc. Rev. 2008, 37, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.J.; Marks, T.J. Organo-f-element catalysts for efficient and highly selective hydroalkoxylation and hydrothiolation. Dalton Trans. 2010, 39, 6576–6588. [Google Scholar] [CrossRef] [PubMed]
- Ananikov, V.P.; Tanaka, M. (Eds.) Hydrofunctionalization; Springer: Berlin/Heidelberg, Germany, 2013; Volume 43, pp. 1–325. [Google Scholar]
- Rodriguez-Ruiz, V.; Carlino, R.; Bezzenine-Lafollée, S.; Gil, R.; Prim, D.; Schulz, E.; Hannedouche, J. Recent developments in alkene hydrofunctionalisation promoted by homogeneous catalysts based on earth abundant elements: Formation of C-N, C-O and C-P bond. Dalton Trans. 2015, 44, 12029–12059. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L.J. Late transition metal-catalyzed hydroamination and hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [Google Scholar] [CrossRef] [PubMed]
- Bernoud, E.; Lepori, C.; Mellah, M.; Schulz, E.; Hannedouche, J. Recent advances in metal free- and late transition metal-catalysed hydroamination of unactivated alkenes. Catal. Sci. Technol. 2015, 5, 2017–2037. [Google Scholar] [CrossRef]
- Musacchio, A.J.; Lainhart, B.C.; Zhang, X.; Naguib, S.G.; Sherwood, T.C.; Knowles, R.R. Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines. Science 2017, 355, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Ensign, S.C.; Vanable, E.P.; Kortman, G.D.; Weir, L.J.; Hull, K.L. Anti-markovnikov hydroamination of homoallylic amines. J. Am. Chem. Soc. 2015, 137, 13748–13751. [Google Scholar] [CrossRef] [PubMed]
- Takemiya, A.; Hartwig, J.F. Rhodium-catalyzed intramolecular, anti-markovnikov hydroamination. Synthesis of 3-arylpiperidines. J. Am. Chem. Soc. 2006, 128, 6042–6043. [Google Scholar] [CrossRef] [PubMed]
- Munro-Leighton, C.; Delp, S.A.; Alsop, N.M.; Blue, E.D.; Gunnoe, T.B. Anti-markovnikov hydroamination and hydrothiolation of electron-deficient vinylarenes catalyzed by well-defined monomeric copper (I) amido and thiolate complexes. Chem. Commun. 2008, 111–113. [Google Scholar] [CrossRef]
- Beller, M.; Trauthwein, H.; Eichberger, M.; Breindl, C.; Herwig, J.; Müller, T.E.; Thiel, O.R. The first rhodium-catalyzed anti-markovnikov hydroamination: Studies on hydroamination and oxidative amination of aromatic olefins. Chem. Eur. J. 1999, 5, 1306–1319. [Google Scholar] [CrossRef]
- Hamilton, D.S.; Nicewicz, D.A. Direct catalytic anti-markovnikov hydroetherification of alkenols. J. Am. Chem. Soc. 2012, 134, 18577–18580. [Google Scholar] [CrossRef] [PubMed]
- Asaoka, S.; Kitazawa, T.; Wada, T.; Inoue, Y. Enantiodifferentiating anti-markovnikov photoaddition of alcohols to 1,1-diphenylalkenes sensitized by chiral naphthalenecarboxylates. J. Am. Chem. Soc. 1999, 121, 8486–8498. [Google Scholar] [CrossRef]
- Douglass, M.R.; Marks, T.J. Organolanthanide-catalyzed intramolecular hydrophosphination/cyclization of phosphinoalkenes and phosphinoalkynes. J. Am. Chem. Soc. 2000, 122, 1824–1825. [Google Scholar] [CrossRef]
- Routaboul, L.; Toulgoat, F.; Gatignol, J.; Lohier, J.F.; Norah, B.; Delacroix, O.; Alayrac, C.; Taillefer, M.; Gaumont, A.C. Iron-salt-promoted highly regioselective α and β hydrophosphination of alkenyl arenes. Chem. Eur. J. 2013, 19, 8760–8764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Du Lee, S.; Widenhoefer, R.A. Intermolecular hydroamination of ethylene and 1-alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral gold (I) complexes. J. Am. Chem. Soc. 2009, 131, 5372–5373. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, A.L.; Nguyen, H.N.; Hultzsch, K.C. Asymmetric intermolecular hydroamination of unactivated alkenes with simple amines. Angew. Chem. Int. Ed. 2010, 49, 8984–8987. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Miao, L.; Hosseini, A.S.; Chemler, S.R. Copper-catalyzed intramolecular alkene carboetherification: Synthesis of fused-ring and bridged-ring tetrahydrofurans. J. Am. Chem. Soc. 2012, 134, 12149–12156. [Google Scholar] [CrossRef] [PubMed]
- Guoa, J.; Teo, P. Anti-markovnikov oxidation and hydration of terminal olefins. Dalton Trans. 2014, 43, 6952–6964. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, L. Anti-markovnikov hydration of terminal alkenes: A coupled catalytic cycle Approach. ChemCatChem 2012, 4, 321–322. [Google Scholar] [CrossRef]
- Carney, J.R.; Dillon, B.R.; Thomas, S.P. Recent advances of manganese catalysis for organic synthesis. Eur. J. Org. Chem. 2016, 3912–3929. [Google Scholar] [CrossRef]
- Crossley, S.W.M.; Obradors, C.; Martinez, R.M.; Shenvi, R.A. Mn-, Fe-, and Co-catalyzed radical hydrofunctionalizations of olefins. Chem. Rev. 2016, 116, 8912–9000. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Perez, A.; Vidal-Moya, J.A.; Cabrero-Antonino, J.R.; Al-Deyab, S.S.; Al-Resayes, S.I.; Corma, A. Copper (I)-catalyzed hydrophosphination of styrenes. J. Organomet. Chem. 2011, 696, 362–367. [Google Scholar] [CrossRef]
- Komeyama, K.; Morimoto, T.; Takaki, K. A simple and efficient iron-catalyzed intramolecular hydroamination of unactivated olefins. Angew. Chem. Int. Ed. 2006, 45, 2938–2941. [Google Scholar] [CrossRef] [PubMed]
- Michaux, J.; Terrasson, V.; Marque, S.; Wehbe, J.; Prim, D.; Campagne, J.M. Intermolecular FeCl3-catalyzed hydroamination of styrenes. Eur. J. Org. Chem. 2007, 2601–2603. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, Y.; Wei, H.; Xu, B.; Zhang, C.; Li, Y.; Qian, G.; Zhang, X.; Li, K.; Li, W. Lewis acid catalyzed intermolecular olefin hydroamination: Scope, limitation, and mechanism. Eur. J. Org. Chem. 2008, 1929–1936. [Google Scholar] [CrossRef]
- Dal Zotto, C.; Michaux, J.; Zarate-Ruiz, A.; Gayon, E.; Virieux, D.; Campagne, J.M.; Terrasson, V.; Pieters, G.; Gaucher, A.; Prim, D. FeCl3-catalyzed addition of nitrogen and 1,3-dicarbonyl nucleophiles to olefins. J. Organomet. Chem. 2011, 696, 296–304. [Google Scholar] [CrossRef]
- Jung, M.S.; Kim, W.S.; Shin, Y.H.; Jin, H.J.; Kim, Y.S.; Kang, E.J. Chemoselective activities of Fe(III) catalysts in the hydrofunctionalization of allenes. Org. Lett. 2012, 14, 6262–6265. [Google Scholar] [CrossRef] [PubMed]
- Bernoud, E.; Oulié, P.; Guillot, R.; Mellah, M.; Hannedouche, J. Well-defined four-coordinate iron(II) complexes for intramolecular hydroamination of primary aliphatic alkenylamines. Angew. Chem. Int. Ed. 2014, 53, 4930–4934. [Google Scholar] [CrossRef] [PubMed]
- Huehls, C.B.; Lin, A.; Yang, J. Iron-catalyzed intermolecular hydroamination of styrenes. Org. Lett. 2014, 16, 3620–3623. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.C.; Yabe, Y.; Baran, P.S. A practical and catalytic reductive olefin coupling. J. Am. Chem. Soc. 2014, 136, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.C.; Gui, J.; Yabe, Y.; Pan, C.M.; Baran, P.S. Functionalized olefin cross-coupling to construct carbon–carbon bonds. Nature 2014, 516, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Pan, C.M.; Jin, Y.; Qin, T.; Lo, J.C.; Lee, B.J.; Spergel, S.H.; Mertzman, M.E.; Pitts, W.J.; La Cruz, T.E.; et al. Practical olefin hydroamination with nitroarenes. Science 2015, 348, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.; Von Wangelin, A.J. Hydroaminations of alkenes: A radical, revised, and expanded version. Angew. Chem. Int. Ed. 2015, 54, 11906–11908. [Google Scholar] [CrossRef] [PubMed]
- Obradors, C.; Martinez, R.M.; Shenvi, R.A. Ph(i-PrO)SiH2: An exceptional reductant for metal-catalyzed hydrogen atom transfers. J. Am. Chem. Soc. 2016, 138, 4962–4971. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Shaver, M.P.; Thomas, S.P. Amine-bis(phenolate) iron(III)-catalyzed formal hydroamination of Olefins. Chem. Asian J. 2016, 11, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Shaver, M.P.; Thomas, S.P. Chemoselective nitro reduction and hydroamination using a single iron catalyst. Chem. Sci. 2016, 7, 3031–3035. [Google Scholar] [CrossRef]
- Greenhalgh, M.D.; Jones, A.S.; Thomas, S.P. Iron-catalysed hydrofunctionalisation of alkenes and alkynes. ChemCatChem 2015, 7, 190–222. [Google Scholar] [CrossRef]
- Komeyama, K.; Morimoto, T.; Nakayama, Y.; Takaki, K. Cationic iron-catalyzed intramolecular hydroalkoxylation of unactivated olefins. Tetrahedron Lett. 2007, 48, 3259–3261. [Google Scholar] [CrossRef]
- Ke, F.; Li, Z.; Xiang, H.; Zhou, X. Catalytic hydroalkoxylation of alkenes by iron(III) Catalyst. Tetrahedron Lett. 2011, 52, 318–320. [Google Scholar] [CrossRef]
- Notar Francesco, I.; Cacciuttolo, B.; Pascu, O.; Aymonier, C.; Pucheault, M.; Antoniotti, S. Simple salts of abundant metals (Fe, Bi, and Ti) supported on montmorillonite as efficient and recyclable catalysts for regioselective intramolecular and intermolecular hydroalkoxylation reactions of double bonds and tandem processes. RCS Adv. 2016, 6, 19807–19818. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Martínez del Campo, T.; Redondo, M.C.; Fernández, I. Striking Alkenol versus allenol reactivity: Metal-catalyzed chemodifferentiating oxycyclization of enallenols. Chem. Eur. J. 2011, 17, 15005–15013. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Almendros, P.; Quirós, M.T. Accessing skeletal diversity under iron catalysis using substrate control: Formation of pyrroles versus lactones. Adv. Synth. Catal. 2011, 353, 585–594. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.W.; Jung, M.S.; Ahn, K.H.; Kang, E.J. Regioselectivities in Fe(III)-catalyzed cycloisomerization reactions of γ-allenyl alcohols. Bull. Korean Chem. Soc. 2015, 36, 2846–2850. [Google Scholar] [CrossRef]
- Wagner, A.; Hampel, N.; Zipse, H.; Ofial, A.R. Sequential oxidative α-cyanation/anti-markovnikov hydroalkoxylation of allylamines. Org. Lett. 2015, 17, 4770–4773. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.J.; Webster, R.L. Room temperature hydrophosphination using a simple iron salen pre-catalyst. Chem. Commun. 2014, 50, 12109–12111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, K.J.; Espinal-Viguri, M.; Mahon, M.F.; Webster, R.L. A study of two highly active, air-stable iron(III)-µ-oxo precatalysts: Synthetic scope of hydrophosphination using phenyl- and diphenylphosphine. Adv. Synth. Catal. 2016, 358, 2460–2468. [Google Scholar] [CrossRef] [Green Version]
- King, A.K.; Buchard, A.; Mahon, M.F.; Webster, R.L. Facile, Catalytic Dehydrocoupling of phosphines using β-diketiminate iron(II) complexes. Chem. Eur. J. 2015, 21, 15960–15963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinal-Viguri, M.; King, A.K.; Lowe, J.P.; Mahon, M.F.; Webster, R.L. Hydrophosphination of unactivated alkenes and alkynes using iron(II): catalysis and mechanistic insight. ACS Catal. 2016, 6, 7892–7897. [Google Scholar] [CrossRef]
- Shigehisa, H.; Aoki, T.; Yamaguchi, S.; Shimizu, N.; Hiroya, K. Hydroalkoxylation of unactivated olefins with carbon radicals and carbocation species as key intermediates. J. Am. Chem. Soc. 2013, 135, 10306–10309. [Google Scholar] [CrossRef] [PubMed]
- Shigehisa, H.; Koseki, N.; Shimizu, N.; Fujisawa, M.; Niitsu, M.; Hiroya, K. Catalytic hydroamination of unactivated olefins using a co catalyst for complex molecule synthesis. J. Am. Chem. Soc. 2014, 136, 13534–13537. [Google Scholar] [CrossRef] [PubMed]
- Shigehisa, H.; Hayashi, M.; Ohkawa, H.; Suzuki, T.; Okayasu, H.; Mukai, M.; Yamazaki, A.; Kawai, R.; Kikuchi, H.; Satoh, Y.; et al. Catalytic synthesis of saturated oxygen heterocycles by hydrofunctionalization of unactivated olefins : Unprotected and protected strategies. J. Am. Chem. Soc. 2016, 10597–10604. [Google Scholar] [CrossRef] [PubMed]
- Shepard, S.M.; Diaconescu, P.L. Redox-switchable hydroelementation of a cobalt complex supported by a ferrocene-based ligand. Organometallics 2016, 35, 2446–2453. [Google Scholar] [CrossRef]
- Baker, R.; Cook, A.H.; Halliday, D.E.; Smith, T.N. Reaction of amines with 1,3-dienes catalysed by nickel complexes. J. Chem. Soc. Perkin Trans. 2 1974, 1511–1517. [Google Scholar] [CrossRef]
- Baker, R.; Onions, A.; Popplestone, R.J.; Smith, T.N. Reactions of amines and active methylene compounds with buta-1,3-diene and isoprene: Catalysis by nickel, cobalt, rhodium, and iridium complexes. J. Chem. Soc. Perkin Trans. 2 1975, 1133–1138. [Google Scholar] [CrossRef]
- Pawlas, J.; Nakao, Y.; Kawatsura, M.; Hartwig, J.F. A general nickel-catalyzed hydroamination of 1,3-Dienes by Alkylamines: Catalyst Selection, Scope, and mechanism. J. Am. Chem. Soc. 2002, 124, 3369–3679. [Google Scholar] [CrossRef]
- Bigot, S.; El Alami, M.S.I.; Mifleur, A.; Castanet, Y.; Suisse, I.; Mortreux, A.; Sauthier, M. Nickel-catalysed hydroalkoxylation reaction of 1,3-butadiene: Ligand controlled selectivity for the efficient and atom-economical synthesis of alkylbutenyl ethers. Chem. Eur. J. 2013, 19, 9785–9788. [Google Scholar] [CrossRef] [PubMed]
- Mifleur, A.; Ledru, H.; Lopes, A.; Suisse, I.; Mortreux, A.; Sauthier, M. Synthesis of short-chain alkenyl ethers from primary and biosourced alcohols via the nickel-catalyzed hydroalkoxylation reaction of butadiene and derivatives. Adv. Synth. Catal. 2016, 358, 110–121. [Google Scholar] [CrossRef]
- Mifleur, A.; Mortreux, A.; Suisse, I.; Sauthier, M. Synthesis of C4 chain glyceryl ethers via nickel-catalyzed butadiene hydroalkoxylation reaction. J. Mol. Catal. A Chem. 2017, 427, 25–30. [Google Scholar] [CrossRef]
- Ortial, S.; Fisher, H.C.; Montchamp, J.L. Hydrophosphinylation of unactivated terminal alkenes catalyzed by nickel chloride. J. Org. Chem. 2013, 78, 6599–6608. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.G.; Whittall, N.; Hii, K.K. Copper-catalyzed intermolecular hydroamination of alkenes. Org. Lett. 2006, 8, 3561–3564. [Google Scholar] [CrossRef] [PubMed]
- Turnpenny, B.W.; Hyman, K.L.; Chemler, S.R. Chiral indoline synthesis via enantioselective intramolecular copper-catalyzed alkene hydroamination. Organometallics 2012, 31, 7819–7822. [Google Scholar] [CrossRef] [PubMed]
- Michon, C.; Medina, F.; Capet, F.; Roussel, P.; Agbossou-Niedercorn, F. Inter- and intramolecular hydroamination of unactivated alkenes catalysed by a combination of copper and silver salts: The unveiling of a brønstedt acid catalysis. Adv. Synth. Catal. 2010, 35, 3293–3305. [Google Scholar] [CrossRef]
- Ohmiya, H.; Moriya, T.; Sawamura, M. Cu (I)-catalyzed intramolecular hydroamination of unactivated alkenes bearing a primary or secondary amino group in alcoholic solvents. Org. Lett. 2009, 11, 2145–2147. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, H.; Yoshida, M.; Sawamura, M. Protecting-group-free route to hydroxylated pyrrolidine and piperidine derivatives through Cu (I)-catalyzed intramolecular hydroamination of alkenes. Synlett 2010, 14, 2136–2140. [Google Scholar]
- Blieck, R.; Bahri, J.; Taillefer, M.; Monnier, F. Copper-catalyzed hydroamination of terminal allenes. Org. Lett. 2016, 18, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Hirano, K.; Satoh, T.; Miura, M. Copper-catalyzed intermolecular regioselective hydroamination of styrenes with polymethylhydrosiloxane and hydroxylamines. Angew. Chem. Int. Ed. 2013, 52, 10830–10834. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Niljianskul, N.; Buchwald, S.L. Enantio- and regioselective CuH-catalyzed hydroamination of alkenes. J. Am. Chem. Soc. 2013, 135, 15746–15749. [Google Scholar] [CrossRef] [PubMed]
- Pirnot, M.T.; Wang, Y.M.; Buchwald, S.L. Copper hydride catalyzed hydroamination of alkenes and alkynes. Angew. Chem. Int. Ed. 2016, 55, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, C.; Krause, N.; Lipshutz, B.H. CuH-catalyzed reactions. Chem. Rev. 2008, 108, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.J.; Lalic, G.; Sadighi, J.P. Coinage metal hydrides: Synthesis, characterization, and reactivity. Chem. Rev. 2016, 116, 8318–8372. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, Q.; Dong, Y.; Liu, H. Transition-metal-catalyzed electrophilic amination: Application of O-benzoylhydroxylamines in the construction of the C-N bond. Chem. Eur. J. 2017, 11, 2481–2511. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Hirano, K.; Satoh, T.; Miura, M. Copper-catalyzed enantioselective formal hydroamination of oxa- and azabicyclic alkenes with hydrosilanes and hydroxylamines. Org. Lett. 2014, 16, 1498–1501. [Google Scholar] [CrossRef] [PubMed]
- Niljianskul, N.; Zhu, S.; Buchwald, S.L. Enantioselective synthesis of α-aminosilanes by copper-catalyzed hydroamination of vinylsilanes. Angew. Chem. Int. Ed. 2015, 54, 1638–1641. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Buchwald, S.L. Enantioselective CuH-catalyzed anti-markovnikov hydroamination of 1,1-disubstituted alkenes. J. Am. Chem. Soc. 2014, 136, 15913–15916. [Google Scholar] [CrossRef] [PubMed]
- Bandar, J.S.; Pirnot, M.T.; Buchwald, S.L. Mechanistic Studies lead to dramatically improved reaction conditions for the Cu-catalyzed asymmetric hydroamination of olefins. J. Am. Chem. Soc. 2015, 137, 14812–14818. [Google Scholar] [CrossRef] [PubMed]
- Tobisch, S. CuH-catalysed hydroamination of styrene with hydroxylamine esters: A coupled cluster scrutiny of mechanistic pathways. Chem. Eur. J. 2016, 22, 8290–8300. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.; Buchwald, S.L. Design of modified amine transfer reagents allows the synthesis of α-chiral secondary amines via CuH-catalyzed hydroamination. J. Am. Chem. Soc. 2015, 137, 9716–9721. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, S.L.; Niu, D.; Liu, P.; Buchwald, S.L. Catalytic Asymmetric Hydroamination of Unactivated Internal Olefins to Aliphatic amines. Science 2015, 349, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Butcher, T.W.; Zhang, J.; Hartwig, J.F. Regioselective, asymmetric formal hydroamination of unactivated internal alkenes. Angew. Chem. Int. Ed. 2016, 55, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Fogg, D.E.; Dos Santos, E.N. Tandem catalysis: A taxonomy and illustrative review. Coord. Chem. Rev. 2004, 248, 2365–2379. [Google Scholar] [CrossRef]
- Shi, S.L.; Buchwald, S.L. Copper-catalysed selective Hydroamination Reactions of alkynes. Nat. Chem. 2015, 7, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Niljianskul, N.; Buchwald, S.L. A Direct approach to amines with remote stereocentres by enantioselective CuH-catalysed reductive relay hydroamination. Nat. Chem. 2016, 8, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.L.; Wong, Z.L.; Buchwald, S.L. Copper-catalysed enantioselective stereodivergent synthesis of amino alcohols. Nature 2016, 532, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sortais, J.B.; Darcel, C. Amine synthesis via transition metal homogeneous catalysed hydrosilylation. RSC Adv. 2016, 6, 57603–57625. [Google Scholar] [CrossRef]
- Taylor, J.G.; Whittall, N.; Hii, K.K. (Mimi). Copper(II)-catalysed addition of O-H bonds to norbornene. Chem. Commun. 2005, 5103–5105. [Google Scholar] [CrossRef] [PubMed]
- Adrio, L.A.; Hii, K.K. (Mimi). A Recyclable Copper(II) Catalyst for the Annulation of Phenols with 1,3-Dienes. Chem. Commun. 2008, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Tschan, M.J.L.; Thomas, C.M.; Strub, H.; Carpentier, J.F. Copper(II) triflate as a source of triflic acid: Effective, green catalysis of hydroalkoxylation reactions. Adv. Synth. Catal. 2009, 351, 2496–2504. [Google Scholar] [CrossRef]
- Murayama, H.; Nagao, K.; Ohmiya, H.; Sawamura, M. Copper (I)-catalyzed intramolecular hydroalkoxylation of unactivated alkenes. Org. Lett. 2015, 17, 2039–2041. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, P.H. Copper-catalyzed intramolecular hydroalkoxylation of α-(1-hydroxy-1-alkyl- and -aryl)methylallenoates by a 5-endo Mode for Preparation of 2-alkyl- and 2-aryl-2,5-dihydrofurans. J. Org. Chem. 2012, 77, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Zulys, A.; Dochnahl, M.; Hollmann, D.; Löhnwitz, K.; Herrmann, J.S.; Roesky, P.W.; Blechert, S. Intramolecular hydroamination of functionalized alkenes and alkynes with a homogenous zinc catalyst. Angew. Chem. Int. Ed. 2005, 44, 7794–7798. [Google Scholar] [CrossRef] [PubMed]
- Dochnahl, M.; Pissarek, J.W.; Blechert, S.; Löhnwitz, K.; Roesky, P.W. A new homogeneous zinc complex with increased reactivity for the intramolecular hydroamination of alkenes. Chem. Commun. 2006, 3405–3407. [Google Scholar] [CrossRef] [PubMed]
- Löhnwitz, K.; Molski, M.J.; Lühl, A.; Roesky, P.W.; Dochnahl, M.; Blechert, S. Aminotroponiminate zinc complexes with different leaving groups as catalysts for the intramolecular hydroamination of alkenes. Eur. J. Inorg. Chem. 2009, 10, 1369–1375. [Google Scholar] [CrossRef]
- Dochnahl, M.; Löhnwitz, K.; Lühl, A.; Pissarek, J.W.; Biyikal, M.; Roesky, P.W.; Blechert, S. Functionalized aminotroponiminate zinc complexes as catalysts for the intramolecular hydroamination of alkenes. Organometallics 2010, 29, 2637–2645. [Google Scholar] [CrossRef]
- Jenter, J.; Lühl, A.; Roesky, P.W.; Blechert, S. Aminotroponiminate zinc complexes as catalysts for the intramolecular hydroamination. J. Organomet. Chem. 2011, 696, 406–418. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sen, T.K.; Ghorai, P.K.; Samuel, P.P.; Schulzke, C.; Mandal, S.K. Phenalenyl-based organozinc catalysts for intramolecular hydroamination reactions: A combined catalytic, kinetic, and mechanistic investigation of the catalytic cycle. Chem. Eur. J. 2012, 18, 10530–10545. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Sen, T.K.; Ghorai, P. K.; Mandal, S.K. Organozinc catalyst on a phenalenyl scaffold for intramolecular hydroamination of aminoalkenes. Organometallics 2013, 32, 7213–7224. [Google Scholar] [CrossRef]
- Horrillo-Martinez, P.; Hultzsch, K.C. Intramolecular hydroamination/cyclization of aminoalkenes catalysed by diamidobinaphthyl magnesium- and zinc-complexes. Tetrahedron Lett. 2009, 50, 2054–2056. [Google Scholar] [CrossRef]
- Arbour, J.L.; Rzepa, H.S.; White, A.J.P.; Hii, K.K. (Mimi). Unusual regiodivergence in metal-catalysed intramolecular cyclisation of γ-allenols. Chem. Commun. 2009, 7125–7127. [Google Scholar] [CrossRef] [PubMed]







































© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezzenine-Lafollée, S.; Gil, R.; Prim, D.; Hannedouche, J. First-Row Late Transition Metals for Catalytic Alkene Hydrofunctionalisation: Recent Advances in C-N, C-O and C-P Bond Formation. Molecules 2017, 22, 1901. https://doi.org/10.3390/molecules22111901
Bezzenine-Lafollée S, Gil R, Prim D, Hannedouche J. First-Row Late Transition Metals for Catalytic Alkene Hydrofunctionalisation: Recent Advances in C-N, C-O and C-P Bond Formation. Molecules. 2017; 22(11):1901. https://doi.org/10.3390/molecules22111901
Chicago/Turabian StyleBezzenine-Lafollée, Sophie, Richard Gil, Damien Prim, and Jérôme Hannedouche. 2017. "First-Row Late Transition Metals for Catalytic Alkene Hydrofunctionalisation: Recent Advances in C-N, C-O and C-P Bond Formation" Molecules 22, no. 11: 1901. https://doi.org/10.3390/molecules22111901