
Mixed-Sequence Recognition of Double-Stranded DNA Using Enzymatically Stable Phosphorothioate Invader Probes

Abstract
:
1. Introduction

2. Results and Discussion
2.1. Synthesis of Modified ONs and Experimental Design

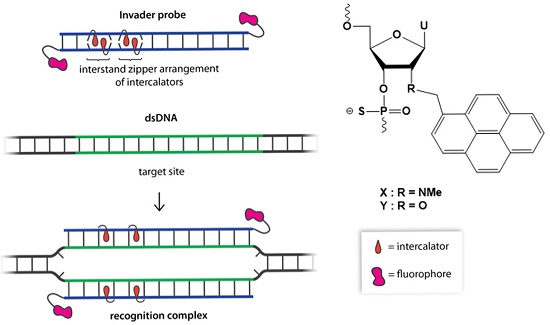{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ON | PS-DNA Sequence | B = | ΔTm (°C) | |||
|---|---|---|---|---|---|---|
| +cDNA | +cRNA | |||||
| X | Y | X | Y | |||
| B1 | 5′-GBG ATA TGC | +5.0 | +4.5 | −4.0 | −4.0 | |
| B2 | 5′-GTG ABA TGC | +13.0 | +12.0 | ±0.0 | ±0.0 | |
| B3 | 5′-GTG ATA BGC | +8.0 | +5.5 | −4.0 | −4.0 | |
| B4 | 3′-CAC BAT ACG | +4.0 | +2.0 | −5.0 | −5.5 | |
| B5 | 3′-CAC TAB ACG | +12.5 | +11.0 | +3.0 | +1.5 | |
| B6 | 3′-CAC BAB ACG | +11.0 | +10.5 | <−8.0 | −6.5 | |
| B7 | 5′-GGB ATA TAT AGG C | +6.0 | +5.5 | - | - | |
| B8 | 3′-CCA BAT ATA TCC G | +9.5 | +9.0 | - | - | |
| B9 | 5′-GGB ABA TAT AGG C | +12.0 | +12.5 | - | - | |
| B10 | 3′-CCA BA BATA TCC G | +16.0 | +16.0 | - | - | |
| B11 | 5′- GGT ABA BAT AGG C | +16.5 | +16.0 | - | - | |
| B12 | 3′- CCA TAB ABA TCC G | +18.0 | +17.5 | - | - | |
| B13 | 5′-GGB ATA TAB AGG C | +15.5 | +13.5 | - | - | |
| B14 | 3′-CCA BAT ATA BCC G | +18.0 | +16.0 | - | - | |
2.2. Thermal Denaturation Properties of X-/Y-Modified PS-DNA
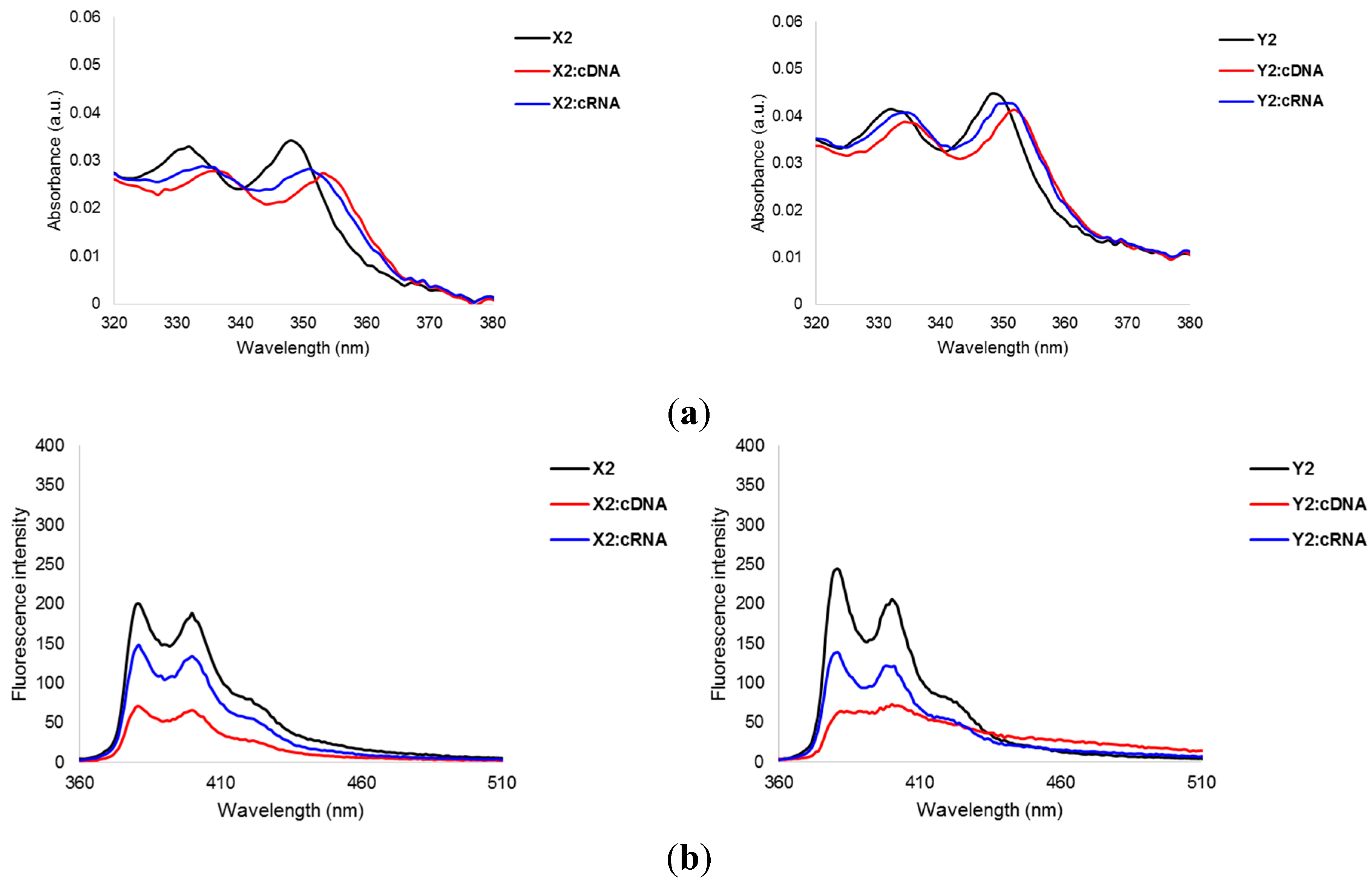
2.3. Photophysical Properties of X-/Y-Modified PS-DNA

2.4. Thermal Denaturation Properties of PS-DNA Duplexes with Interstrand Zipper Arrangements of X or Y Monomers
| ON | ZP | PS-DNA Duplex | B = | Tm (ΔTm)/°C | |
|---|---|---|---|---|---|
| X | Y | ||||
| B1 | +4 | 5′-GBG ATA TGC | 29.5 | 29.5 | |
| B5 | 3′-CAC TAB ACG | (>+19.5) | (>+19.5) | ||
| B1 | +2 | 5′-GBG ATA TGC | <10.0 | <10.0 | |
| B4 | 3′-CAC BAT ACG | ||||
| B2 | +1 | 5′-GTG ABA TGC | <10.0 | <10.0 | |
| B5 | 3′-CAC TAB ACG | ||||
| B2 | −1 | 5′-GTG ABA TGC | 22.0 | 19.0 | |
| B4 | 3′-CAC BAT ACG | (>+12.0) | (>+9.0) | ||
| B7 | +1 | 5′-GGB ATA TAT AGG C | 13.5 | 12.5 | |
| B8 | 3′-CCA BAT ATA TCC G | (−4.0) | (−5.0) | ||
| B9 | +1 | 5′-GGB ABA TAT AGG C | 17.5 | 23.5 | |
| B10 | 3′-CCA BA BATA TCC G | (±0.0) | (+6.0) | ||
| B11 | +1 | 5′- GGT ABA BAT AGG C | 18.0 | 26.0 | |
| B12 | 3′- CCA TAB ABA TCC G | (+0.5) | (+8.5) | ||
| B13 | +1 | 5′-GGB ATA TAB AGG C | 24.5 | 24.0 | |
| B14 | 3′-CCA BAT ATA BCC G | (+7.0) | (+6.5) | ||
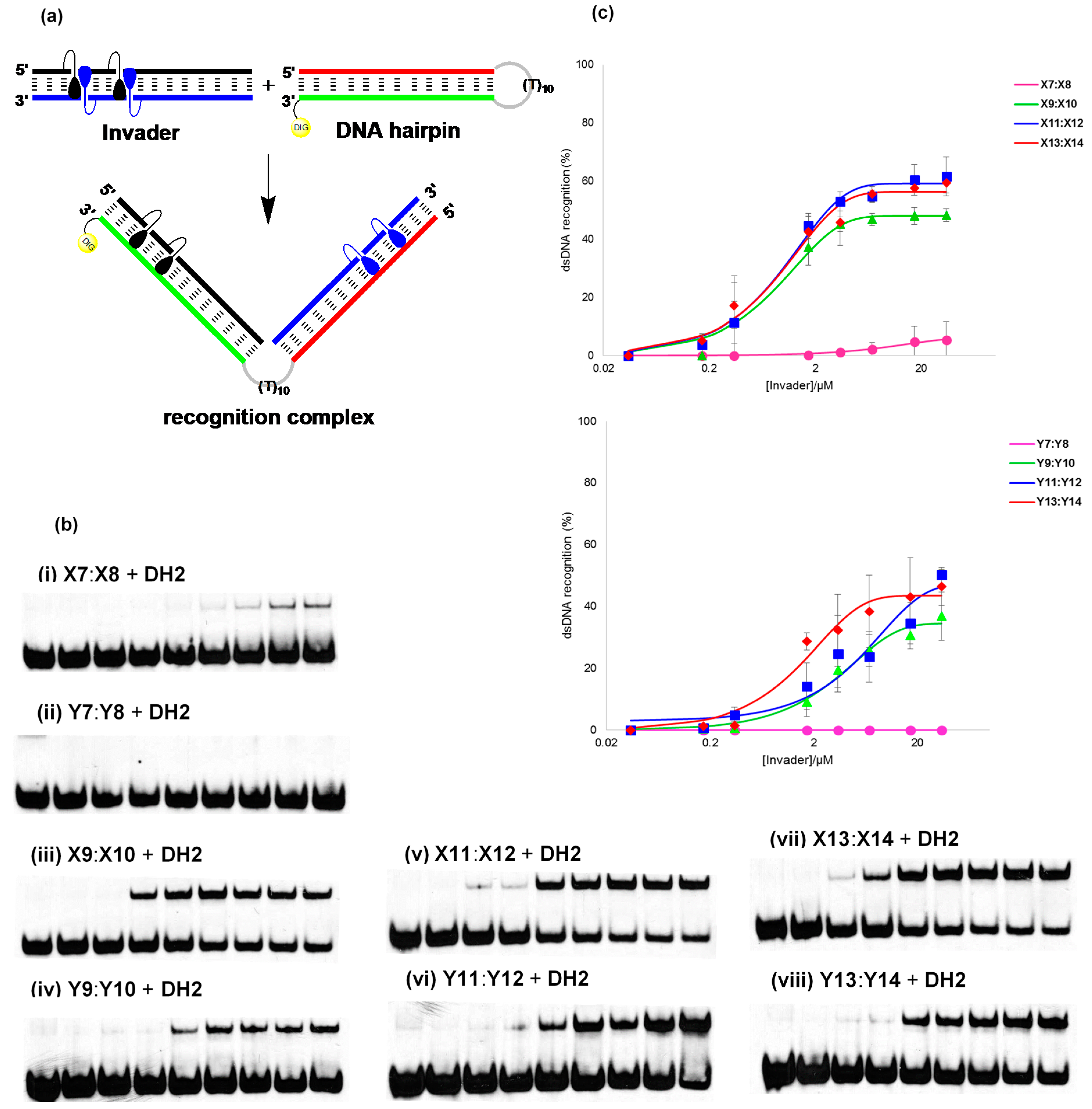
2.5. Recognition of DNA Hairpins Using Energetically Activated Probe Duplexes
2.6. Enzymatic Stability of Individual Invader Strands


3. Experimental Section
3.1. Protocol—Synthesis and Purification of ONs
3.2. Protocol—Thermal Denaturation Studies
3.3. Protocol—Absorption Spectra
3.4. Protocol—Steady-State Fluorescence Emission Spectra
3.5. Protocol—Electrophoretic Mobility Shift Assay
3.6. Protocol—3′-Exonuclease Stability Assay
3.7. Definition of Zipper Nomenclature
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Rogers, F.A.; Lloyd, J.A.; Glazer, P.M. Triplex-forming oligonucleotides as potential tools for modulation of gene expression. Curr. Med. Chem. Anti- Cancer Agents 2005, 5, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Stains, C.I.; Ooi, A.T.; Segal, D.J. Direct detection of double-stranded DNA: Molecular methods and applications for DNA diagnostics. Mol. Biol. Syst. 2006, 2, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Peptide Nucleic Acids (PNA) in chemical biology and drug discovery. Chem. Biodivers. 2010, 7, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Aiba, Y.; Sumaoka, J.; Komiyama, M. Artificial DNA cutters for DNA manipulation and genome engineering. Chem. Soc. Rev. 2011, 40, 5657–5668. [Google Scholar] [CrossRef] [PubMed]
- Vaijayanthi, T.; Bando, T.; Pandian, G.N.; Sugiyama, H. Progress and prospects of pyrrole-imidazole polyamide-fluorophore conjugates as sequence-selective DNA probes. Chem. Biol. Chem. 2012, 13, 2170–2185. [Google Scholar] [CrossRef] [PubMed]
- Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. The triple helix: 50 Years later, the outcome. Nucleic Acids Res. 2008, 36, 5123–5138. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Dervan, P.B.; Edelson, B.S. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol. 2003, 13, 284–299. [Google Scholar] [CrossRef]
- Blackledge, M.S.; Melander, C. Programmable DNA-binding small molecules. Bioorg. Med. Chem. 2013, 21, 6101–6114. [Google Scholar] [CrossRef] [PubMed]
- Rusling, D.A.; Powers, V.E.C.; Ranasinghe, R.T.; Wang, Y.; Osborne, S.D.; Brown, T.; Fox, K. Four base recognition by triplex-forming oligonucleotides at physiological pH. Nucleic Acids Res. 2005, 33, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- Hari, Y.; Obika, S.; Imanishi, T. Towards the sequence-selective recognition of double-stranded DNA containing pyrimidine-purine interruptions by triplex-forming oligonucleotides. Eur. J. Org. Chem. 2012, 2875–2887. [Google Scholar] [CrossRef]
- Kutyavin, I.V.; Rhinehart, R.L.; Lukhtanov, E.A.; Gorn, V.V.; Meyer, R.B., Jr.; Gamper, H.B., Jr. Oligonucleotides containing 2-aminoadenine and 2-thiothymine act as selectively binding complementary agents. Biochemistry 1996, 35, 11170–11176. [Google Scholar] [CrossRef] [PubMed]
- Lohse, J.; Dahl, O.; Nielsen, P.E. Double duplex invasion by peptide nucleic acid: A general principle for sequence-specific targeting of double-stranded DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 11804–11808. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, T.; Yoshida, J.; Yamamoto, Y.; Sumaoka, J.; Tedeschi, T.; Corradini, R.; Sforza, S.; Komiyama, M. Chiral introduction of positive charges to PNA for double-duplex invasion to versatile sequences. Nucleic Acids Res. 2008, 36, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Rapireddy, S.; Bahal, R.; Ly, D.H. Strand invasion of mixed-sequence, double-helical B-DNA by gamma-peptide nucleic acids containing g-clamp nucleobases under physiological conditions. Biochemistry 2011, 50, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Bahal, R.; Sahu, B.; Rapireddy, S.; Lee, C.M.; Ly, D.H. Sequence-unrestricted, Watson-Crick recognition of double helical B-DNA by (R)-MiniPEG-γPNAs. Chem. Bio. Chem. 2012, 13, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Didion, B.A.; Karmakar, S.; Guenther, D.C.; Sau, S.P.; Verstegen, J.P.; Hrdlicka, P.J. Invaders: Recognition of double-stranded DNA using duplexes modified with interstrand zippers of 2′-O-(pyren-1-yl)methylribonucleotides. Chem. Biol. Chem. 2013, 4, 3447–3454. [Google Scholar] [CrossRef] [PubMed]
- Guenther, D.C.; Anderson, G.H.; Karmakar, S.; Anderson, B.A.; Didion, B.A.; Guo, W.; Verstegen, J.P.; Hrdlicka, P.J. Invader probes: Harnessing the energy of intercalation to facilitate recognition of chromosomal DNA for diagnostic applications. Chem. Sci. 2015, 6, 5006–5015. [Google Scholar] [CrossRef]
- Sau, S.P.; Madsen, A.S.; Podbevsek, P.; Andersen, N.K.; Kumar, T.S.; Andersen, S.; Rathje, R.L.; Anderson, B.A.; Guenther, D.C.; Karmakar, S.; et al. Identification and characterization of 2nd generation Invader LNAs for mixed-sequence recognition of double-stranded DNA. J. Org. Chem. 2013, 78, 9560–9570. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Madsen, A.S.; Guenther, D.C.; Gibbons, B.C.; Hrdlicka, P.J. Recognition of double-stranded DNA using energetically activated duplexes with interstrand zippers of 1-, 2- or 4-pyrenyl-functionalized O2ʹ-alkylated RNA monomers. Org. Biomol. Chem. 2014, 12, 7758–7773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crothers, D.M. Calculation of binding isotherms for heterogeneous polymers. Biopolymers 1968, 6, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Persil, O.; Hud, N.V. Harnessing DNA intercalation. Trends Biotechnol. 2007, 25, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Jain, S.C.; Sobell, H.M. Visualization of drug-nucleic acid interactions at atomic resolution. Structure of an ethidium-dinucleoside monophosphate crystalline complex ethidium—5-iodouridylyl (3′-5′) adenosine. J. Mol. Biol. 1977, 114, 301–315. [Google Scholar] [CrossRef]
- Williams, L.D.; Egli, M.; Gao, Q.; Rich, A. Structure and Function, Volume 1: Nucleic Acids; Sarma, R.H., Sarma, M.H., Eds.; Adenine Press: Albany, NY, USA, 1992; pp. 107–125. [Google Scholar]
- Sau, S.P.; Kumar, T.S.; Hrdlicka, P.J. Invader LNA—Efficient targeting of short DNA duplexes. Org. Biomol. Chem. 2010, 8, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.A.; Onley, J.J.; Hrdlicka, P.J. Recognition of double-stranded DNA using energetically activated duplexes modified with N2′-pyrene-, perylene-, or coronene-functionalized 2′-N-methyl-2′-amino-DNA monomers. J. Org. Chem. 2015, 80, 5395–5406. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Guenther, D.C.; Hrdlicka, P.J. Recognition of mixed-sequence DNA duplexes: Design guidelines for Invaders based on 2′-O-(pyren-1-yl)methyl-RNA monomers. J. Org. Chem. 2013, 78, 12040–12048. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Bacon, T.A.; Wickstrom, E. Oligodeoxynucleoside phosphotothioate stability in subcellular extracts, culture media, sera and cerebrospinal fluid. J. Biochem. Biophys. Methods 1990, 20, 259–267. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Anderson, B.A.; Rathje, R.L.; Andersen, S.; Jensen, T.; Nielsen, P.; Hrdlicka, P.J. High-affinity DNA-targeting using readily accessible mimics of N2′-functionalized 2′-amino-α-l-LNA. J. Org. Chem. 2011, 76, 7119–7131. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Fukunaga, Y.; Sasa, K.; Ohtoshi, Y.; Kanaori, K.; Hayashi, H.; Nakano, H.; Yamana, K. Pyrene is highly emissive when attached to the RNA duplex but not to the DNA duplex: The structural basis of this difference. Nucleic Acids Res. 2005, 33, 5887–5895. [Google Scholar] [CrossRef] [PubMed]
- For a discussion of binding specificities of X-/Y-modified PS-DNA, see the Supporting Information (Tables S3 and S4).
- Asanuma, H.; Fujii, T.; Kato, T.; Kashida, H. Coherent interactions of dyes assembled on DNA. J. Photochem. Photobiol. C 2012, 13, 124–135. [Google Scholar] [CrossRef]
- Dougherty, G.; Pilbrow, J.R. Physicochemical probes of intercalation. Int. J. Biochem. 1984, 16, 1179–1192. [Google Scholar] [CrossRef]
- Manoharan, M.; Tivel, K.L.; Zhao, M.; Nafisi, K.; Netzel, T.L. Base-sequence dependence of emission lifetimes for dna oligomers and duplexes covalently labeled with pyrene—Relative electron-transfer quenching efficiencies of A-nucleoside, G-nucleoside, C-nucleoside, and T-nucleoside toward pyrene. J. Phys. Chem. 1995, 99, 17461–17472. [Google Scholar] [CrossRef]
- Wilson, J.N.; Cho, Y.; Tan, S.; Cuppoletti, A.; Kool, E.T. Quenching of fluorescent nucleobases by neighboring DNA: The “Insulator” concept. Chem. Biol. Chem. 2008, 9, 279–285. [Google Scholar] [CrossRef] [PubMed]
- For data on DNAse I stability, see Figure S7 in the Supporting Information.
- Dioubankova, N.N.; Malakhov, A.D.; Stetsenko, D.A.; Gait, M.J.; Volynsky, P.E.; Efremov, R.G.; Korshun, V.A. Pyrenemethyl ara-uridine-2ʹ-carbamate: A strong interstrand excimer in the major groove of a DNA duplex. Chem. Biol. Chem. 2003, 4, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M. A step-by-step guide to non-linear regression analysis of experimental data using a Microsoft Excel spreadsheet. Comput. Meth. Prog. Biomed. 2001, 65, 181–200. [Google Scholar] [CrossRef]
- Sample Availability: Select Invader probe samples are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, B.A.; Karmakar, S.; Hrdlicka, P.J. Mixed-Sequence Recognition of Double-Stranded DNA Using Enzymatically Stable Phosphorothioate Invader Probes. Molecules 2015, 20, 13780-13793. https://doi.org/10.3390/molecules200813780
Anderson BA, Karmakar S, Hrdlicka PJ. Mixed-Sequence Recognition of Double-Stranded DNA Using Enzymatically Stable Phosphorothioate Invader Probes. Molecules. 2015; 20(8):13780-13793. https://doi.org/10.3390/molecules200813780
Chicago/Turabian StyleAnderson, Brooke A., Saswata Karmakar, and Patrick J. Hrdlicka. 2015. "Mixed-Sequence Recognition of Double-Stranded DNA Using Enzymatically Stable Phosphorothioate Invader Probes" Molecules 20, no. 8: 13780-13793. https://doi.org/10.3390/molecules200813780