
New Lignans and Iridoid Glycosides from Dipsacus asper Wall

Abstract
:1. Introduction

2. Results and Discussion

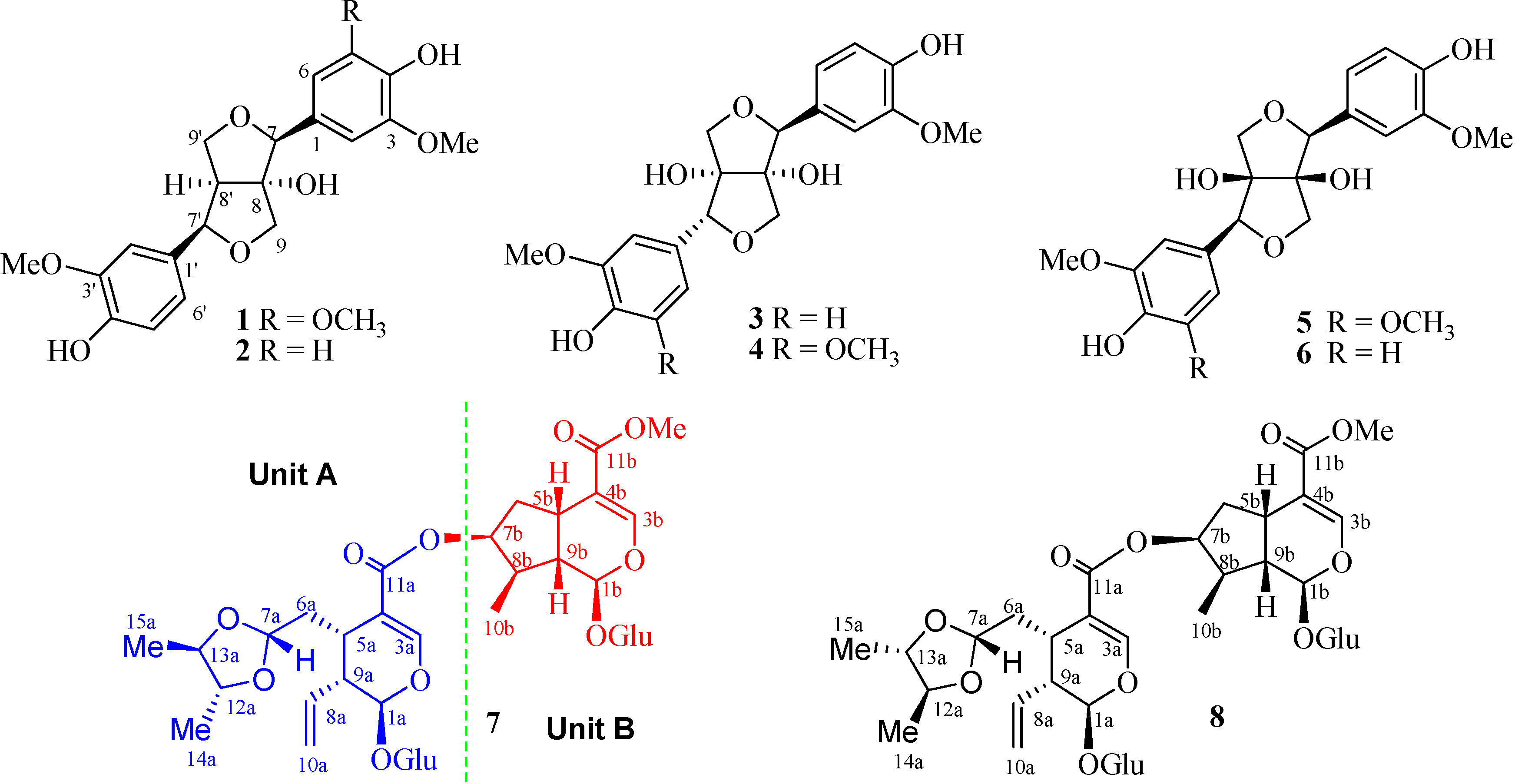{kind=link}
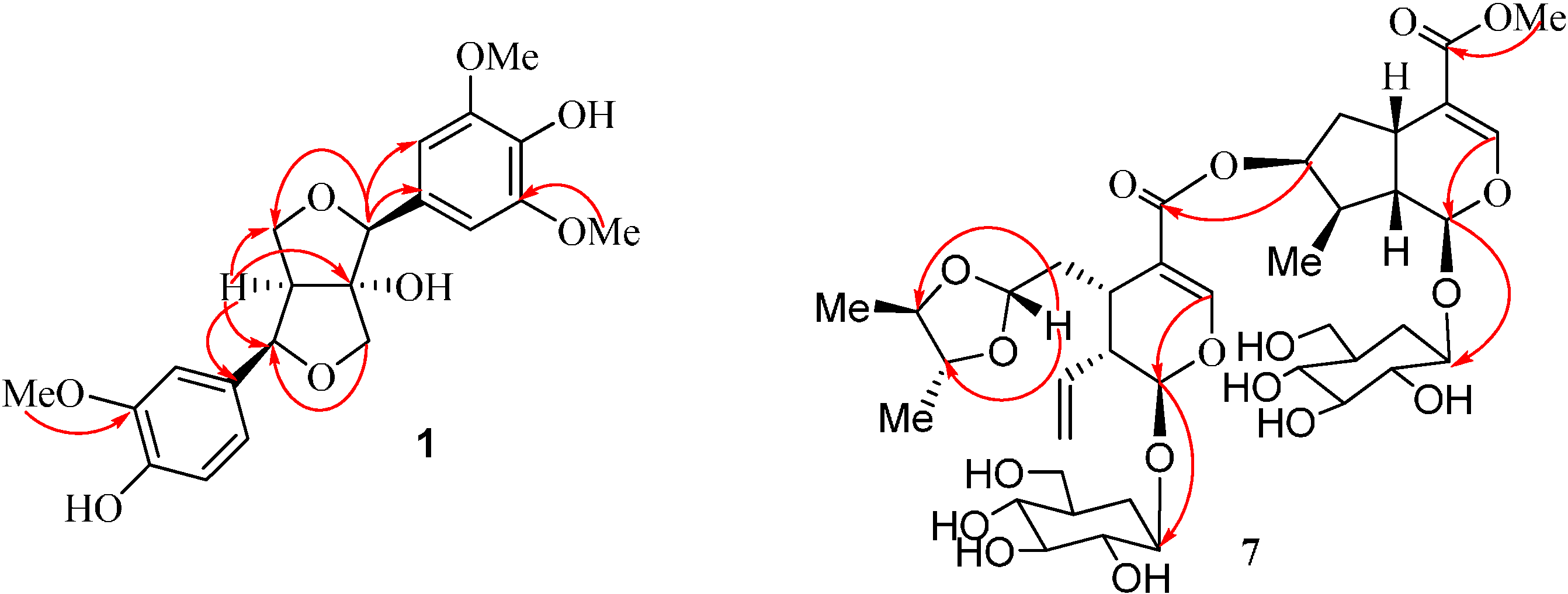{kind=link}
{kind=link}
| NO. | 1 (CD3OD) | 3 (CDCl3) | 4 (CDCl3) | 5 (CDCl3) | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J) | δC | δH (J) | δC | δH (J) | δC | δH (J) | δC | |
| 1 | — | 129.6 | — | 128.6 | — | 129.3 | — | 129.3 |
| 2 | 6.75 (s) | 103.9 | 6.92 (d, 1.8) | 109.2 | 6.97 (d, 1.8) | 112.3 | 6.93 (d, 1.8) | 109.9 |
| 3 | — | 149.3 | — | 147.2 | — | 146.9 | — | 147.1 |
| 4 | — | 136.2 | — | 145.7 | — | 145.7 | — | 145.7 |
| 5 | — | 149.3 | 6.76 (dd, 7.8, 1.8) | 115.2 | 6.69 (dd, 7.8, 1.8) | 114.7 | 6.77 (dd, 7.8, 1.8) | 115.1 |
| 6 | 6.75 (s) | 103.9 | 6.81 (d, 7.8) | 117.5 | 6.77 (d, 7.8) | 120.2 | 6.80 (d, 7.8) | 118.1 |
| 7 | 4,65 (s) | 87.4 | 4.53 (s) | 85.3 | 4.53 (s) | 85.4 | 4.66 (s) | 84.6 |
| 8 | — | 94.4 | — | 88.2 | — | 88.2 | — | 84.7 |
| 9α | 3.42 (d, 9.0) | 77.4 | 3.51 (d,9.0) | 74.3 | 3.54 (d, 9.0) | 74.5 | 3.39 (d,9.0) | 72.9 |
| 9β | 3.65 (d, 9.0) | 3.19 (d,9.0) | 3.20 (d, 9.0) | 3.68 (d,9.0) | ||||
| 1' | — | 133.5 | — | 127.7 | — | 128.4 | — | 128.4 |
| 2' | 7.07 (d, 1.8) | 111.3 | 6.97 (d, 1.8) | 112.2 | 6.93 (d, 1.8) | 102.5 | 6.64 (d, 1.8) | 103.2 |
| 3' | — | 149.5 | — | 146.8 | — | 147.8 | — | 147.7 |
| 4' | — | 148.0 | — | 145.9 | — | 134.7 | — | 134.7 |
| 5' | 6.77 (d, 7.8) | 116.2 | — | 115.2 | — | 147.8 | — | 147.7 |
| 6' | 6.85 (dd, 7.8, 1.8) | 120.8 | 6.78 (d, 1.8) | 120.2 | 6.93 (d, 1.8) | 102.5 | 6.64 d, 1.8) | 103.2 |
| 7' | 4.43 (d, 7.8) | 90.7 | 4.30 (s) | 88.9 | 4.30 (s) | 88.9 | 4.66 (s) | 84.5 |
| 8' | 2.64 (dd, 7.8, 6.6) | 64.1 | — | 85.3 | — | 85.4 | — | 84.7 |
| 9'α | 4.01 (dd, 9.0, 1.2) | 70.7 | 4.18 (d,9.0) | 75.5 | 4.18 (d, 9.0) | 74.5 | 3.68 (d, 9.0) | 73.0 |
| 9'β | 4.07 (dd, 9.0, 1.2) | 3.43 (d,9.0) | 3.44 (d, 9.0) | 3.39 (d, 9.0) | ||||
| 3-OMe | 3.85 (s) | 56.9 | 3.76 (s) | 55.6 | 3.76 (s) | 55.6 | 3.76 (s) | 55.5 |
| 5-OMe | 3.85 (s) | 56.9 | — | — | — | — | — | — |
| 3'-OMe | 3.87 (s) | 56.6 | 3.76 (s) | 55.6 | 3.76 (s) | 56.0 | 3.76 (s) | 55.9 |
| 5'-OMe | — | — | — | — | 3.76 (s) | 56.0 | 3.76 (s) | 55.9 |


| NO. | 7 | 8 | ||
|---|---|---|---|---|
| δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | |
| 1a | 5.54 (d, 6.0) | 98.0 | 5.55 (d, 6.0) | 98.0 |
| 3a | 7.44 (s) | 153.5 | 7.44 (s) | 153.6 |
| 4a | — | 113.6 | — | 113.6 |
| 5a | 3.02 (br q, 6.0) | 30.2 | 3.01 (br q, 6.0) | 30.1 |
| 6a | 1.73–1.80 (m) 2.02 (ddd, 6.0, 6.0, 14.0) | 35.9 | 1.73–1.82 (m) 1.99 (ddd, 6.0, 6.0, 14.0) | 36.0 |
| 7a | 5.13 (dd, 4.2, 6.0) | 103.4 | 5.13 (dd, 4.2, 6.0) | 103.3 |
| 8a | 5.75 (ddd, 8.4, 10.2, 18.0) | 136.0 | 5.75 (ddd, 8.4, 10.2, 18.0) | 136.0 |
| 9a | 2.75 (ddd,5.4, 6.0, 8.4) | 45.5 | 2.75 (ddd, 5.4, 6.0, 8.4) | 45.6 |
| 10a | 5.29 (d, 18.0) 5.26 (d, 10.2) | 120.0 | 5.29 (d, 18.0) 5.25 (d, 10.2) | 120.0 |
| 11a | — | 168.6 | — | 168.6 |
| 12a | 3.56 (d,6.0) | 61.4 | 3.56 (d, 3.5) | 81.2 |
| 13a | 3.56 (d,6.0) | 79.4 | 3.56 (d, 3.5) | 79.6 |
| 14a | 1.19 (d,6.0) | 17.5 | 1.20 (d, 6.0) | 17.6 |
| 15a | 1.25 (d,6.0) | 17.6 | 1.25 (d, 6.0) | 17.7 |
| 1'a | 4.69 (d,7.8) | 100.4 | 4.69 (d, 7.8) | 100.4 |
| 2'a | 3.20 (dd,7.8,9.0) | 75.0 | 3.20 (dd, 7.8, 9.0) | 75.0 |
| 3'a | 3.25–3.39 (m) | 78.2 | 3.25–3.39 (m) | 78.2 |
| 4'a | 3.25–3.39 (m) | 71.9 | 3.25–3.39 (m) | 71.9 |
| 5'a | 3.25–3.39 (m) | 78.6 | 3.25–3.39 (m) | 78.6 |
| 6'a | 3.66 (dd, 6.0, 12.0) 3.88–3.92 (m) | 63.0 | 3.67 (dd, 6.0, 12.0) 3.91 (br d, 12.0) | 63.0 |
| 1b | 5.27 (d, 4.2) | 97.7 | 5.27 (d, 4.2) | 97.6 |
| 3b | 7.42 (s) | 152.6 | 7.42 (s) | 152.6 |
| 4b | — | 112.2 | — | 112.2 |
| 5b | 3.13 (br q, 7.8) | 32.8 | 3.12 (br q, 7.8) | 32.7 |
| 6b | 1.73–1.80 (m) 2.32 (br dd, 7.8, 14.4) | 40.6 | 1.74–1.82 (m) 2.32 (br dd, 7.8, 14.4) | 40.5 |
| 7b | 5.20 (dd, 4.2, 7.8) | 78.6 | 5.19 (dd, 4.2, 7.8) | 78.6 |
| 8b | 2.09–2.16 (m) | 41.2 | 2.10–2.16 (m) | 41.1 |
| 9b | 2.09–2.16 (m) | 47.4 | 2.10–2.16 (m) | 47.4 |
| 10b | 1.07 (d, 6.5) | 14.0 | 1.07 (d, 6.0) | 13.9 |
| 11b | — | 169.5 | — | 169.5 |
| 12b | 3.69 (s) | 51.9 | 3.69 (s) | 51.9 |
| 1'b | 4.67 (d, 7.8) | 100.3 | 4.67 (d, 7.8) | 100.3 |
| 2'b | 3.20 (dd, 7.8, 9.0) | 74.9 | 3.20 (dd, 7.8, 9.0) | 74.9 |
| 3'b | 3.25–3.39 (m) | 78.2 | 3.25–3.39 (m) | 78.2 |
| 4'b | 3.25–3.39 (m) | 71.8 | 3.25–3.39 (m) | 71.8 |
| 5'b | 3.25–3.39 (m) | 78.6 | 3.25–3.39 (m) | 78.6 |
| 6'b | 3.66 (dd, 6.0, 12.0) 3.88–3.92 (m) | 63.0 | 3.66 (dd,6.0,12.0) 3.89 (br d,12.0) | 63.0 |
3. Experimental Section
3.1. General
3.2. Plant Material
3.3. Extration and Isolation
3.4. Spectroscopic Data
3.5. Acid Hydrolysis of Compounds 8 and 9
3.6. HIV-1 Integrase Strand Transfer Inhibition Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jung, H.W.; Jung, J.K.; Son, K.H.; Lee, D.H.; Kang, T.M.; Kim, Y.S.; Park, Y.K. Inhibitory effects of the root extract of Dipsacus asperoides C.Y. Cheng et al T.M.Ai on collagen-induced arthritis in mice. J. Ethnopharmacol. 2012, 139, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.M.; Na, M.K.; Thong, P.T.; Su, N.D.; Sok, D.; Song, K.S.; Seong, Y.H.; Bae, K. Antioxidant activity of caffeoyl quinic acid derivatives from the roots of Dipsacus asper Wall. J. Ethnopharmacol. 2006, 108, 188–192. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Kiyohara, H.; Matsumoto, T.; Yamada, H. Fractionation and Chemical Properties of Immunomodulating Polysaccharides from Roots of Dipsacus asperoides. Planta Med. 1997, 63, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Zhang, R.; Li, C.; Ma, X.; Liu, L.; Wang, J.P.; Mei, Q.B. The osteoprotective effect of Radix Dipsaci extract in ovariectomized rats. J. Ethnopharmacol. 2009, 123, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Wang, X.L.; Guo, B.L.; Huang, W.H.; Xiao, P.G.; Huang, C.Q.; Zheng, L.Z.; Zhang, G.; Qin, L.; Tu, G.Z. Triterpenoid saponins from Dipsacus asper and their activities in vitro. J. Asian Nat. Prod. Res. 2011, 13, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.Q.; Wang, Y.; Franzblau, S.G.; Montenegro, G.; Yang, D.; Timmermann, B.N. Antitubercular constituents of Valeriana laxiflora. Planta Med. 2004, 70, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Cuadra, P.; Fajardo, V. A new lignan from the Patagonian Valeriana Carnosa Sm. Bol. Soc. Chil. Quím. 2002, 47, 361–366. [Google Scholar] [CrossRef]
- Takahashi, K.; Nakagawa, T. Studies on constituents of medicinal plants. VIII. The stereochemistry of paulownin and isopaulownin. Chem. Pharm. Bull. 1966, 14, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Cowan, S.; Stewart, M.; Abbiw, D.K.; Latif, Z.; Sarker, S.D.; Nash, R.J. Lignans from Strophanthus gratus. Fitoterapia 2001, 72, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Scholle, S.; Hölzl, J.; Khudeir, N.; Hess, S.; Müller, C.E. Lignans isolated from valerian: Identification and characterization of a new olivil derivative with partial agonistic activity at A1 adenosine receptors. J. Nat. Prod. 2002, 65, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Ishikawa, Y.; Kasahara, H.; Yamanaka, J.I.; Kameoka, H. An insect growth inhibitory lignan from flower buds of Magnolia fargesii. Phytochemistry 1994, 35, 611–613. [Google Scholar] [CrossRef]
- MacRae, W.D.; Towers, G.H. Non-alkaloidal constituents of Virola elongate bark. Phytochemistry 1985, 24, 561–566. [Google Scholar] [CrossRef]
- Gréger, H.; Hofer, O. New unsymmetrically substituted tetrahydrofurofuran lignans from Artemisia absinthium: Assignment of the relative stereochemistry by lanthanide induced chemical shifts. Tetrahedron 1980, 36, 3551–3558. [Google Scholar] [CrossRef]
- Pan, W.; Liu, K.; Guan, Y.; Tan, G.T.; Hung, N.V.; Cuong, N.M.; Soejarto, D.D.; Pezzuto, J.M.; Fong, H.H.; Zhang, H.J. Bioactive compounds from Vitex leptobotrys. J. Nat. Prod. 2014, 77, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; Li, Z.L.; Chen, H.; Liu, X.Q.; Zhou, W.; Hua, H.M. Four new cytotoxic tetrahydrofuranoid lignans from Sinopodophyllum emodi. Planta Med. 2012, 78, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Wei, H.B.; Xu, Y.Z.; Zeng, J.; Gao, K. Antioxidant lignans from the roots of Vladimiria muliensis. Planta Med. 2013, 79, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, Á.; Szabó, L.F.; Podányi, B. New bis-iridoid glucosides from Dipsacus laciniatus. J. Nat. Prod. 1993, 56, 1486–1499. [Google Scholar] [CrossRef]
- Podányi, B.; Reid, R.S.; Kocsis, A.; Szabó, L. Laciniatoside V: A new bis-iridoid glucoside. isolation and structure elucidation by 2D NMR Spectroscopy. J. Nat. Prod. 1989, 52, 135–142. [Google Scholar] [CrossRef]
- Tian, X.Y.; Wang, Y.H.; Liu, H.Y.; Liu, H.Y.; Yu, S.S.; Fang, W.S. On the chemical constituents of Dipsacus asper. Chem. Pharm. Bull. 2007, 55, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Ma, X.H.; Liu, B.; Chen, W.Z.; Wang, C.X.; Cheng, S.H. A novel high-throughput format assay for HIV-1 integrase strand transfer reaction using magnetic beads. Acta Pharmacol. Sin. 2008, 29, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Mitaine-Offer, A.C.; Penez, N.; Miyamoto, T.; Delaude, C.; Mirjolet, J.F.; Duchamp, O.; Lacaille-Dubois, M.A. Acylated triterpene saponins from the roots of Securidaca longepedunculata. Phytochemistry 2010, 71, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1–8 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Ma, G.; Zhang, D.; Huang, W.; Ding, G.; Hu, H.; Tu, G.; Guo, B. New Lignans and Iridoid Glycosides from Dipsacus asper Wall. Molecules 2015, 20, 2165-2175. https://doi.org/10.3390/molecules20022165
Sun X, Ma G, Zhang D, Huang W, Ding G, Hu H, Tu G, Guo B. New Lignans and Iridoid Glycosides from Dipsacus asper Wall. Molecules. 2015; 20(2):2165-2175. https://doi.org/10.3390/molecules20022165
Chicago/Turabian StyleSun, Xinguang, Guoxu Ma, Dawei Zhang, Wenhua Huang, Gang Ding, Huagang Hu, Guangzhong Tu, and Baolin Guo. 2015. "New Lignans and Iridoid Glycosides from Dipsacus asper Wall" Molecules 20, no. 2: 2165-2175. https://doi.org/10.3390/molecules20022165