EOP, a Newly Synthesized Ethyl Pyruvate Derivative, Attenuates the Production of Inflammatory Mediators via p38, ERK and NF-κB Pathways in Lipopolysaccharide-Activated BV-2 Microglial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
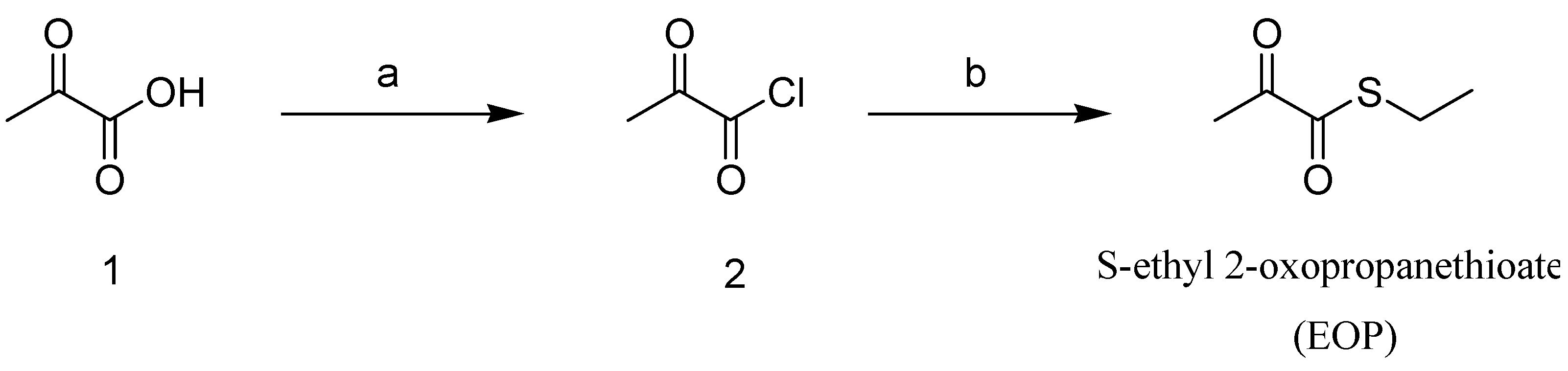
2.1. Synthesis of the Novel Ethyl Pyruvate Derivative, EOP

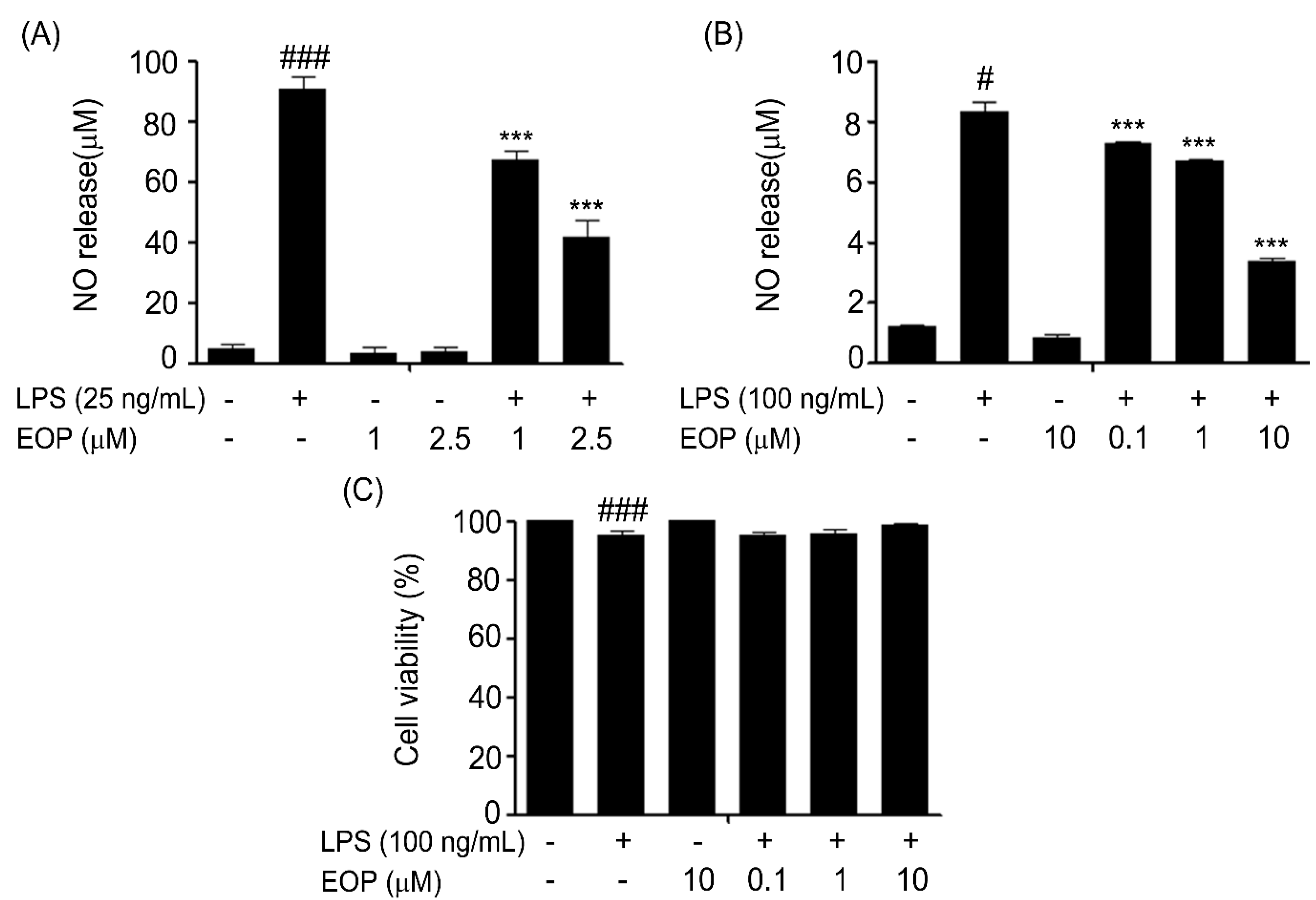
2.2. Effect of EOP on LPS-Induced Production of NO and Cytotoxicity in Rat Primary and Murine BV-2 Microglia

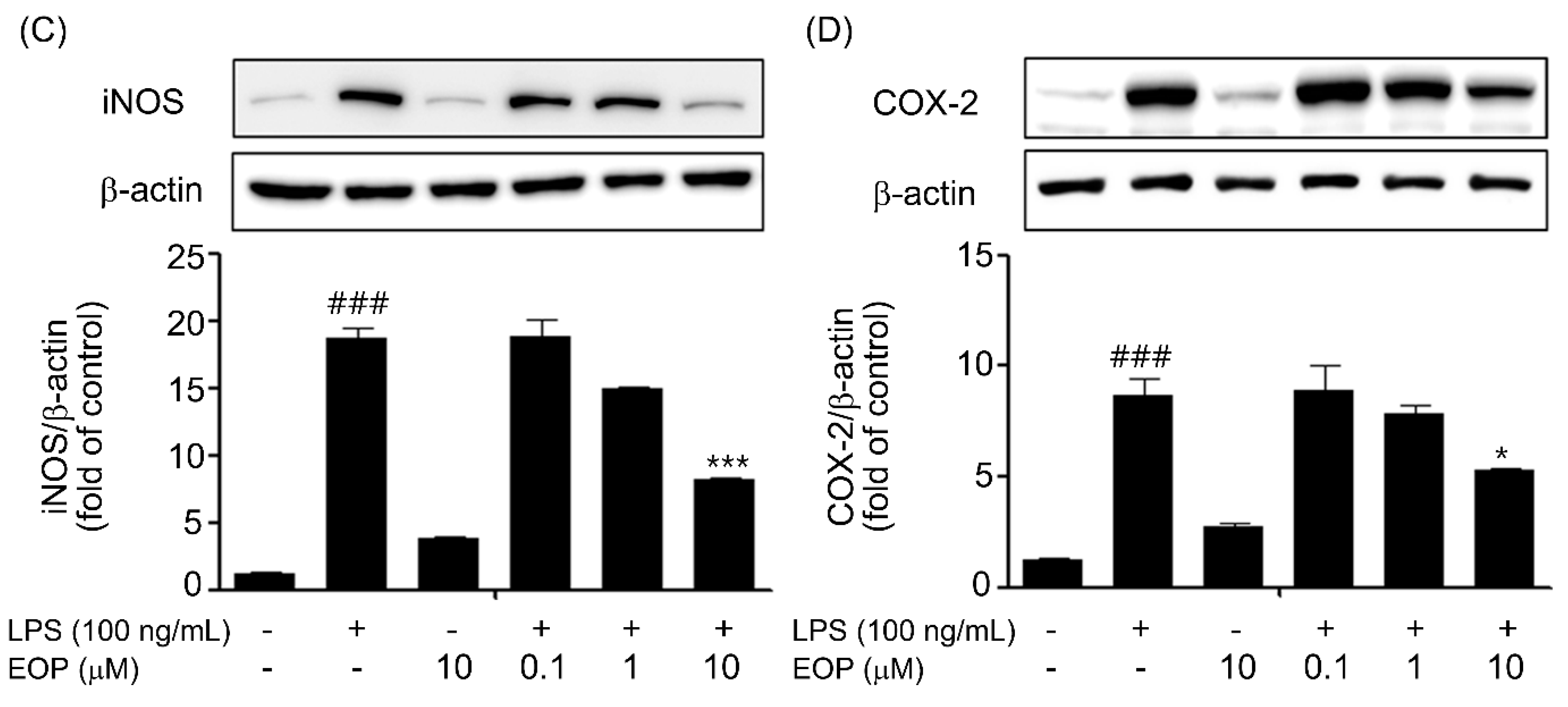
2.3. EOP Attenuates LPS-Mediated Inducible Nitric Oxide Synthase (iNOS) and COX-2 Expression in BV-2 Microglia
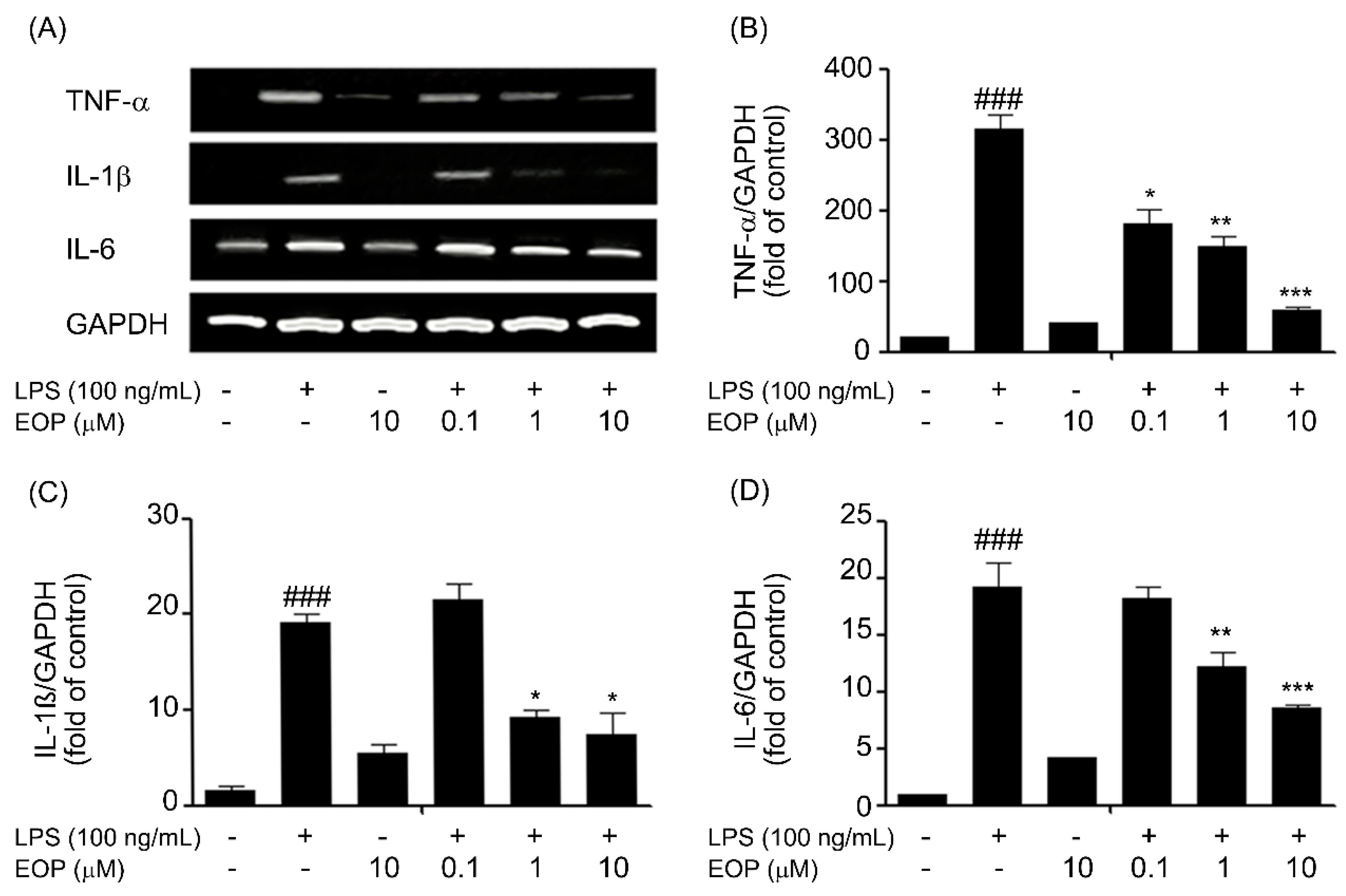
2.4. EOP Mitigates LPS-Induced Expression of Pro-Inflammatory Cytokines in Microglial Cells
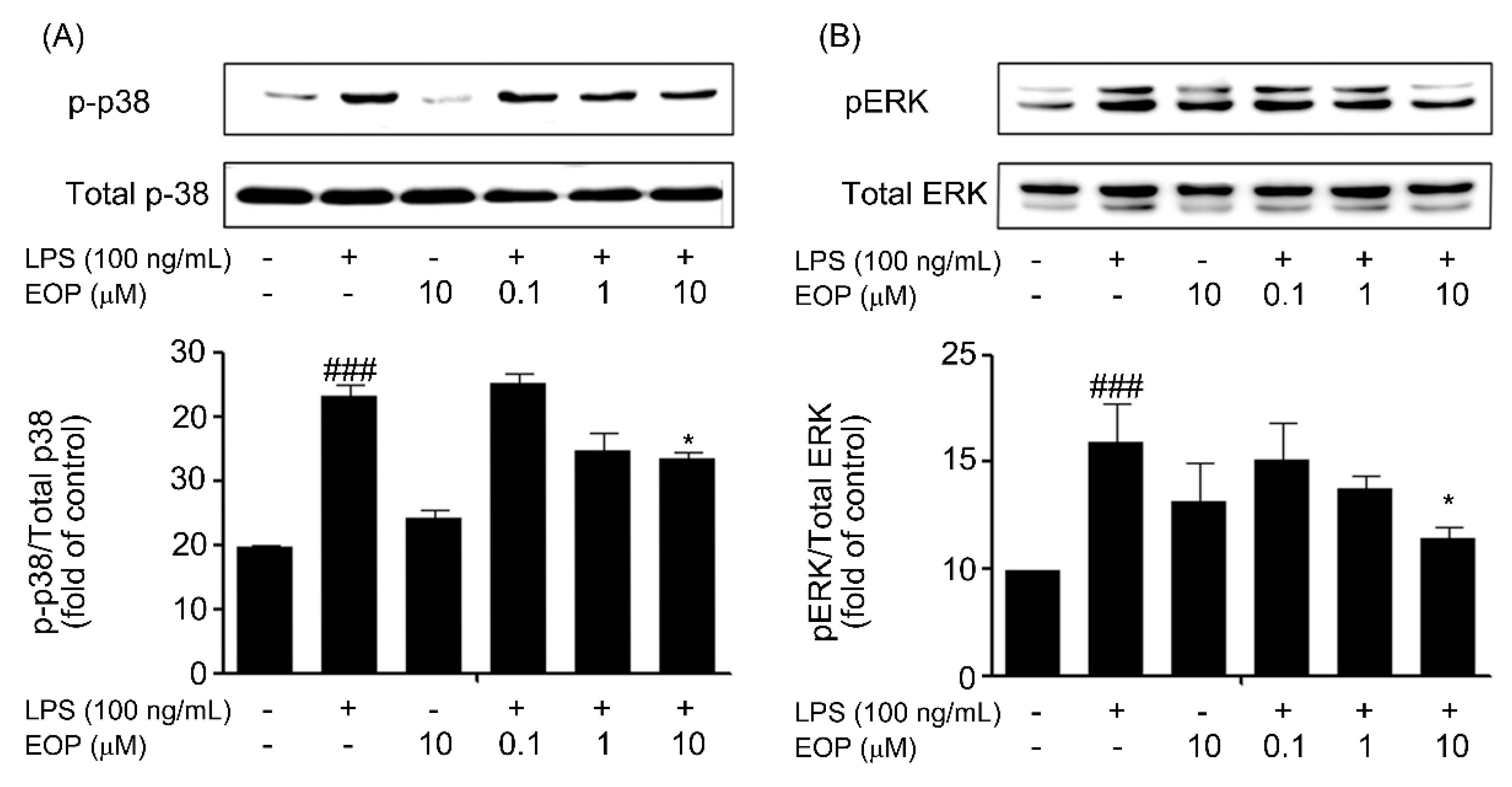
2.5. EOP Inhibits LPS-Induced Phosphorylation of p38 and ERK MAPK in BV-2 Microglial Cells




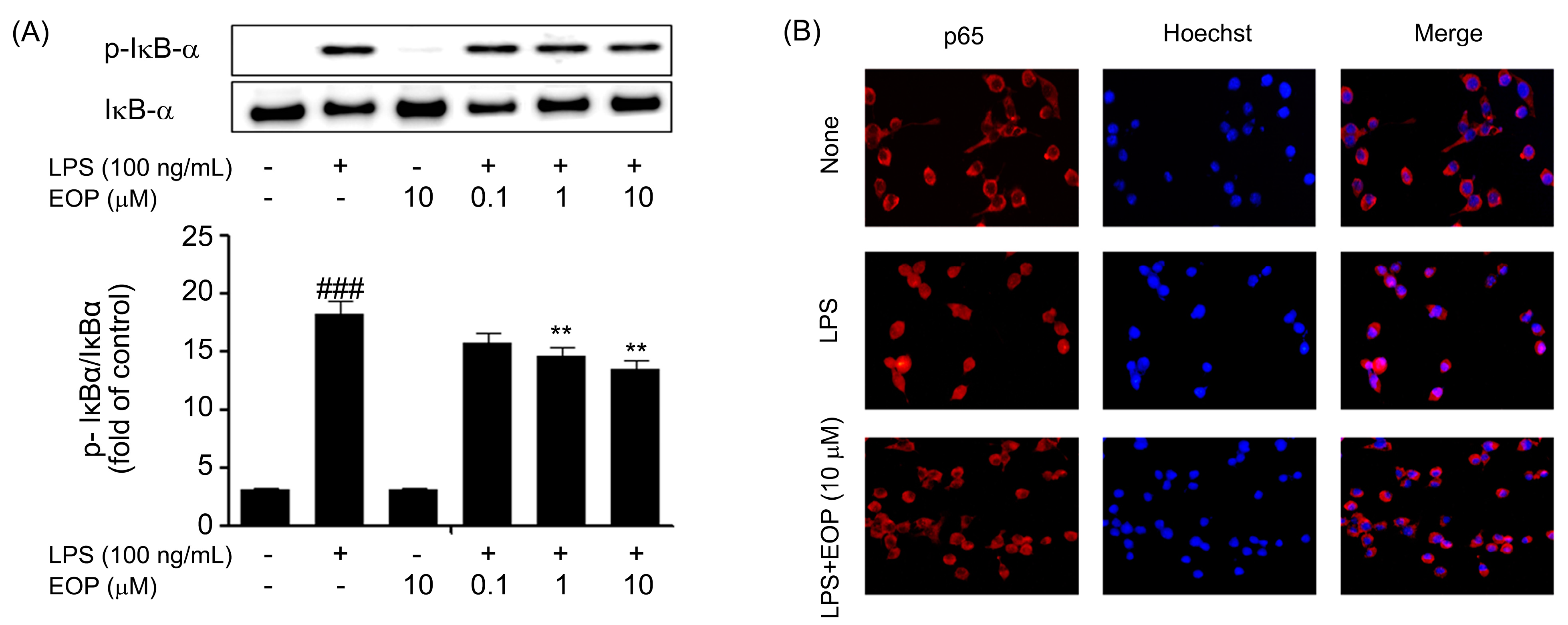
2.6. Inhibitory Effect of EOP on Phosphorylation of IκB-α and Subsequent Activation and Localization of NF-kB in BV-2 Microglial Cells

3. Experimental Section
3.1. Reagents
3.2. Microglial Cell Culture
3.3. NO and Cell Viability Assay
3.4. EOP Synthesis Methodology
3.5. RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
3.6. Western Blot Analysis
3.7. Immunocytochemistry
3.8. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Kim, I.S.; Koppulla, S.; Kim, B.W.; Choi, D.K. Strategic selection of neuroinflammatory models in Parkinson’s disease: Evidence from experimental studies. CNS Neurol. Disord. Drug Targets 2013, 12, 680–697. [Google Scholar]
- Fink, M.P. Ethyl pyruvate. Curr. Opin. Anaesthesiol. 2008, 21, 160–167. [Google Scholar]
- Park, S.Y.; Yi, E.Y.; Jung, M.; Lee, Y.M.; Kim, Y.J. Ethyl pyruvate, an anti-inflammatory agent, inhibits tumor angiogenesis through inhibition of the NF-κB signaling pathway. Cancer Lett. 2011, 303, 150–154. [Google Scholar]
- Dong, N.; Yao, Y.M.; Dong, Y.Q.; Liu, H.; Wei, P.; Yu, Y.; Sheng, Z.Y. Effects of ethyl pyruvate on splenocyte proliferation and apoptosis in burn rats with delayed resuscitation. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 2005, 17, 393–396. [Google Scholar]
- Epperly, M.; Jin, S.; Nie, S.; Cao, S.; Zhang, X.; Franicola, D.; Wang, H.; Fink, M.P.; Greenberger, J.S. Ethyl pyruvate, a potentially effective mitigator of damage after total-body irradiation. Radiat. Res. 2007, 168, 552–559. [Google Scholar]
- Uchiyama, T.; Delude, R.L.; Fink, M.P. Dose-dependent effects of ethyl pyruvate in mice subjected to mesenteric ischemia and reperfusion. Intensive Care Med. 2003, 29, 2050–2058. [Google Scholar]
- Van Zoelen, M.A.; Bakhtiari, K.; Dessing, M.C.; van’t Veer, C.; Spek, C.A.; Tanck, M.; Meijers, J.C.; van der Poll, T. Ethyl pyruvate exerts combined anti-inflammatory and anticoagulant effects on human monocytic cells. Thromb. Haemost. 2006, 96, 789–793. [Google Scholar]
- Venkataraman, R.; Kellum, J.A.; Song, M.; Fink, M.P. Resuscitation with Ringer’s ethyl pyruvate solution prolongs survival and modulates plasma cytokine and nitrite/nitrate concentrations in a rat model of lipopolysaccharide-induced shock. Shock 2002, 18, 507–512. [Google Scholar]
- Yang, R.; Gallo, D.J.; Baust, J.J.; Uchiyama, T.; Watkins, S.K.; Delude, R.L.; Fink, M.P. Ethyl pyruvate modulates inflammatory gene expression in mice subjected to hemorrhagic shock. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G212–G221. [Google Scholar]
- Yang, R.; Uchiyama, T.; Alber, S.M.; Han, X.; Watkins, S.K.; Delude, R.L.; Fink, M.P. Ethyl pyruvate ameliorates distant organ injury in a murine model of acute necrotizing pancreatitis. Crit. Care Med. 2004, 32, 1453–1459. [Google Scholar]
- Han, Y.; Englert, J.A.; Yang, R.; Delude, R.L.; Fink, M.P. Ethyl pyruvate inhibits nuclear factor-κB-dependent signaling by directly targeting p65. J. Pharmacol. Exp. Ther. 2005, 312, 1097–1105. [Google Scholar]
- Johansson, A.S.; Johansson-Haque, K.; Okret, S.; Palmblad, J. Ethyl pyruvate modulates acute inflammatory reactions in human endothelial cells in relation to the NF-κB pathway. Br. J. Pharmacol. 2008, 154, 1318–1326. [Google Scholar]
- Cho, I.H.; Kim, S.W.; Kim, J.B.; Kim, T.K.; Lee, K.W.; Han, P.L.; Lee, J.K. Ethyl pyruvate attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J. Neurosci. Res. 2006, 84, 1505–1511. [Google Scholar]
- Shen, H.; Hu, X.; Liu, C.; Wang, S.; Zhang, W.; Gao, H.; Stetler, R.A.; Gao, Y.; Chen, J. Ethyl pyruvateprotects against hypoxic-ischemic brain injury via anti-cell death and anti-inflammatory mechanisms. Neurobiol. Dis. 2010, 37, 711–722. [Google Scholar]
- Wang, L.Z.; Sun, W.C.; Zhu, X.Z. Ethyl pyruvate protects PC12 cells from dopamine-induced apoptosis. Eur. J. Pharmacol. 2005, 508, 57–68. [Google Scholar]
- Huh, S.H.; Chung, Y.C.; Piao, Y.; Jin, M.Y.; Son, H.J.; Yoon, N.S.; Hong, J.Y.; Pak, Y.K.; Kim, Y.S.; Hong, J.K.; et al. Ethyl pyruvate rescues nigrostriatal dopaminergic neurons by regulating glial activation in a mouse model of Parkinson’s disease. J. Immunol. 2011, 187, 960–969. [Google Scholar]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflammation 2012, 9. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. S1), S210–S212. [Google Scholar]
- Maccioni, R.B.; Rojo, L.E.; Fernandez, J.A.; Kuljis, R.O. The role of neuroimmunomodulation in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2009, 1153, 240–246. [Google Scholar]
- Kao, K.K.; Fink, M.P. The biochemical basis for the anti-inflammatory and cytoprotective actions of ethyl pyruvate and related compounds. Biochem. Pharmacol. 2010, 80, 151–159. [Google Scholar]
- Brand, K. Aerobic glycolysis by proliferating cells: Protection against oxidative stress at the expense of energy yield. J. Bioenerg. Biomembr. 1997, 29, 355–364. [Google Scholar]
- Montgomery, C.M.; Webb, J.L. Metabolic studies on heart mitochondria. II. The inhibitory action of parapyruvate on the tricarboxylic acid cycle. J. Biol. Chem. 1956, 221, 359–368. [Google Scholar]
- Varma, S.D.; Devamanoharan, P.S.; Ali, A.H. Prevention of intracellular oxidative stress to lens by pyruvate and its ester. Free Radic. Res. 1998, 28, 131–135. [Google Scholar]
- Streit, W.J.; Xue, Q.S. Human CNS immune senescence and neurodegeneration. Curr. Opin. Immunol. 2014, 29, 93–96. [Google Scholar]
- Lee, M. Neurotransmitters and microglial-mediated neuroinflammation. Curr. Protein Pept. Sci. 2013, 14, 21–32. [Google Scholar]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-α. FASEB J. 2006, 20, 670–682. [Google Scholar]
- Panaro, M.A.; Cianciulli, A. Current opinions and perspectives on the role of immune system in the pathogenesis of Parkinson’s disease. Curr. Pharm. Des. 2012, 18, 200–208. [Google Scholar]
- Na, K.S.; Jung, H.Y.; Kim, Y.K. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 277–286. [Google Scholar]
- Kim, E.J.; Kwon, K.J.; Park, J.Y.; Lee, S.H.; Moon, C.H.; Baik, E.J. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: Associated with iNOS and COX-2. Brain Res. 2002, 941, 1–10. [Google Scholar]
- Qu, W.S.; Tian, D.S.; Guo, Z.B.; Fang, J.; Zhang, Q.; Yu, Z.Y.; Xie, M.J.; Zhang, H.Q.; Lu, J.G.; Wang, W. Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J. Neuroinflammation 2012, 9. [Google Scholar] [CrossRef]
- Rajan, S.; Ye, J.; Bai, S.; Huang, F.; Guo, Y.L. NF-κB, but not p38 MAP kinase, is required for TNF-alpha-induced expression of cell adhesion molecules in endothelial cells. J. Cell Biochem. 2008, 105, 477–486. [Google Scholar]
- Chung, H.S.; Kim, S.N.; Jeong, J.H.; Bae, H. A novel synthetic compound 4-acetyl-3-methyl-6-(2-bromophenyl)pyrano[3,4-c]pyran-1,8-dione inhibits the production of nitric oxide and proinflammatorycytokines via the NF-κB pathway in lipopolysaccharide-activated microglia cells. Neurochem. Res. 2013, 38, 807–814. [Google Scholar]
- Brown, G.C.; Neher, J.J. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol. Neurobiol. 2010, 41, 242–247. [Google Scholar]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar]
- Yu, D.H.; Noh, D.H.; Song, R.H.; Park, J. Ethyl pyruvate downregulates tumor necrosis factor α and interleukin (IL)-6 and upregulates IL-10 in lipopolysaccharide-stimulated canine peripheral blood mononuclear cells. J. Vet. Med. Sci. 2010, 72, 1379–1381. [Google Scholar]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-κB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar]
- Kim, B.W.; Koppula, S.; Hong, S.S.; Jeon, S.B.; Kwon, J.H.; Hwang, B.Y.; Park, E.J.; Choi, D.K. Regulation of microglia activity by glaucocalyxin-A: Attenuation of lipopolysaccharide-stimulated neuroinflammation through NF-κB and p38 MAPK signaling pathways. PLoS One 2013, 8, e55792. [Google Scholar]
- Bocchini, V.; Mazzolla, R.; Barluzzi, R.; Blasi, E.; Sick, P.; Kettenmann, H. An immortalized cell line expresses properties of activated microglial cells. J. Neurosci. Res. 1992, 31, 616–621. [Google Scholar]
- More, S.V.; Park, J.Y.; Kim, B.W.; Kumar, H.; Lim, H.W.; Kang, S.M.; Koppula, S.; Yoon, S.H.; Choi, D.K. Anti-neuroinflammatory activity of a novel cannabinoid derivative by inhibiting the NF-κB signaling pathway in lipopolysaccharide-induced BV-2 microglial cells. J. Pharmacol. Sci. 2013, 121, 119–130. [Google Scholar]
- Jeon, N.R.; Koppula, S.; Kim, B.W.; Park, S.H.; Lee, H.W.; Choi, D.K. MMHD [(S,E)-2-methyl-1-(2-methylthiazol-4-yl) hexa-1,5-dien-ol], a novel synthetic compound derived from epothilone, suppresses nuclear factor-κB-mediated cytokine expression in lipopolysaccharide-stimulated BV-2 microglia. J. Pharmacol. Sci. 2010, 112, 158–166. [Google Scholar]
- Kim, B.W.; Koppula, S.; Kim, I.S.; Lim, H.W.; Hong, S.M.; Han, S.D.; Hwang, B.Y.; Choi, D.K. Anti-neuroinflammatory activity of Kamebakaurin from Isodon japonicus via inhibition of c-Jun NH(2)-terminal kinase and p38 mitogen-activated protein kinase pathway in activated microglial cells. J. Pharmacol. Sci. 2011, 116, 296–308. [Google Scholar]
- An, H.; Kim, I.S.; Koppula, S.; Kim, B.W.; Park, P.J.; Lim, B.O.; Choi, W.S.; Lee, K.H.; Choi, D.K. Protective effects of Gastrodia elata Blume on MPP+-induced cytotoxicity in human dopaminergic SH-SY5Y cells. J. Ethnopharmacol. 2010, 130, 290–298. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, S.; More, S.V.; Park, J.-Y.; Jeon, S.-B.; Park, S.Y.; Park, E.-J.; Yoon, S.-H.; Choi, D.-K. EOP, a Newly Synthesized Ethyl Pyruvate Derivative, Attenuates the Production of Inflammatory Mediators via p38, ERK and NF-κB Pathways in Lipopolysaccharide-Activated BV-2 Microglial Cells. Molecules 2014, 19, 19361-19375. https://doi.org/10.3390/molecules191219361
Min S, More SV, Park J-Y, Jeon S-B, Park SY, Park E-J, Yoon S-H, Choi D-K. EOP, a Newly Synthesized Ethyl Pyruvate Derivative, Attenuates the Production of Inflammatory Mediators via p38, ERK and NF-κB Pathways in Lipopolysaccharide-Activated BV-2 Microglial Cells. Molecules. 2014; 19(12):19361-19375. https://doi.org/10.3390/molecules191219361
Chicago/Turabian StyleMin, Soon, Sandeep Vasant More, Ju-Young Park, Sae-Bom Jeon, Shin Young Park, Eun-Jung Park, Sung-Hwa Yoon, and Dong-Kug Choi. 2014. "EOP, a Newly Synthesized Ethyl Pyruvate Derivative, Attenuates the Production of Inflammatory Mediators via p38, ERK and NF-κB Pathways in Lipopolysaccharide-Activated BV-2 Microglial Cells" Molecules 19, no. 12: 19361-19375. https://doi.org/10.3390/molecules191219361