
Leishmanicidal Activity of Betulin Derivatives in Leishmania amazonensis; Effect on Plasma and Mitochondrial Membrane Potential, and Macrophage Nitric Oxide and Superoxide Production

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Parasites
2.2. Murine Macrophages
2.3. Compounds
2.4. Promastigote In Vitro Susceptibility
2.5. Cytotoxicity and Selectivity
2.6. Macrophage-Amastigote Model
2.7. Plasma Membrane Potential (ΔΨp)
2.8. Mitochondrial Membrane Potential (ΔΨm)
2.9. Nitric Oxide (NO) Production
2.10. Superoxide (O2−) Production
2.11. Statistics
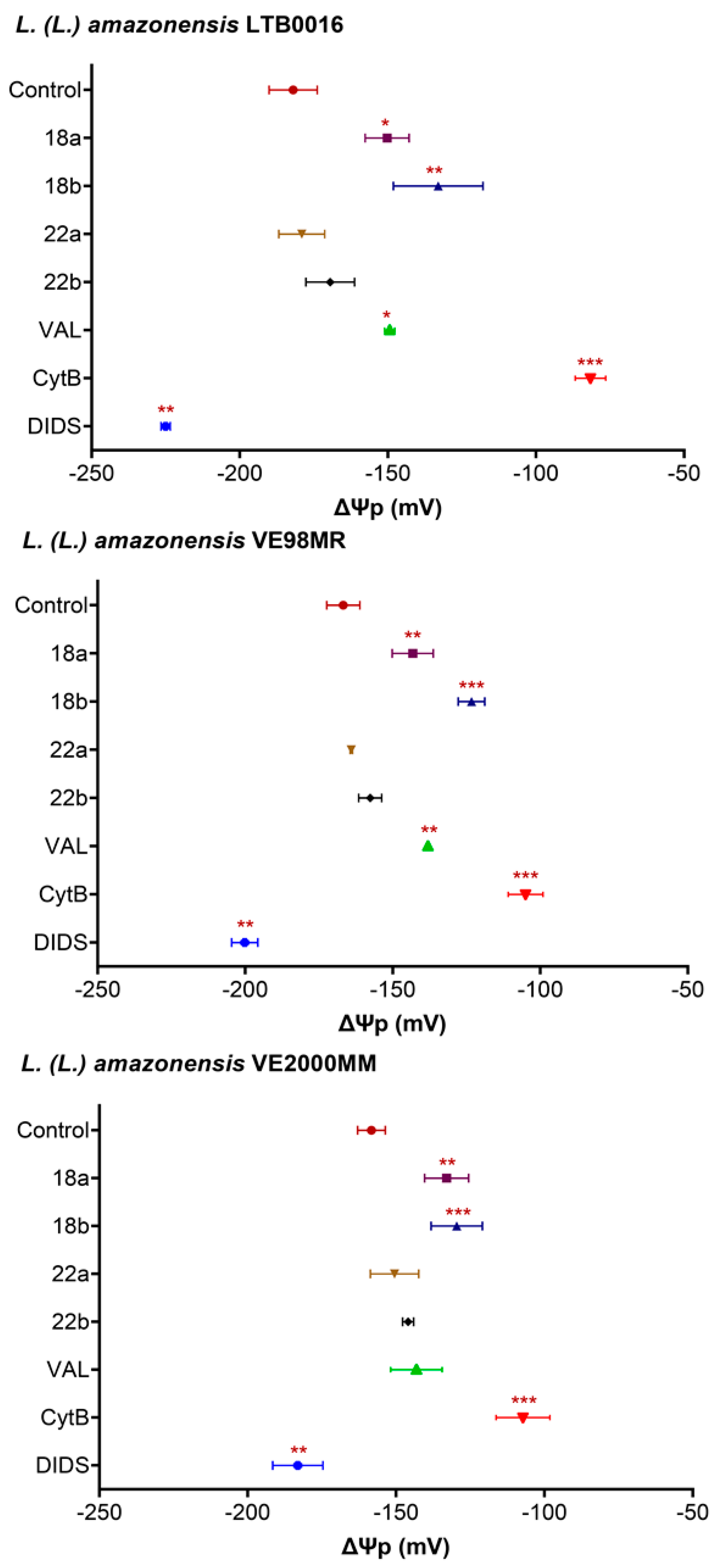
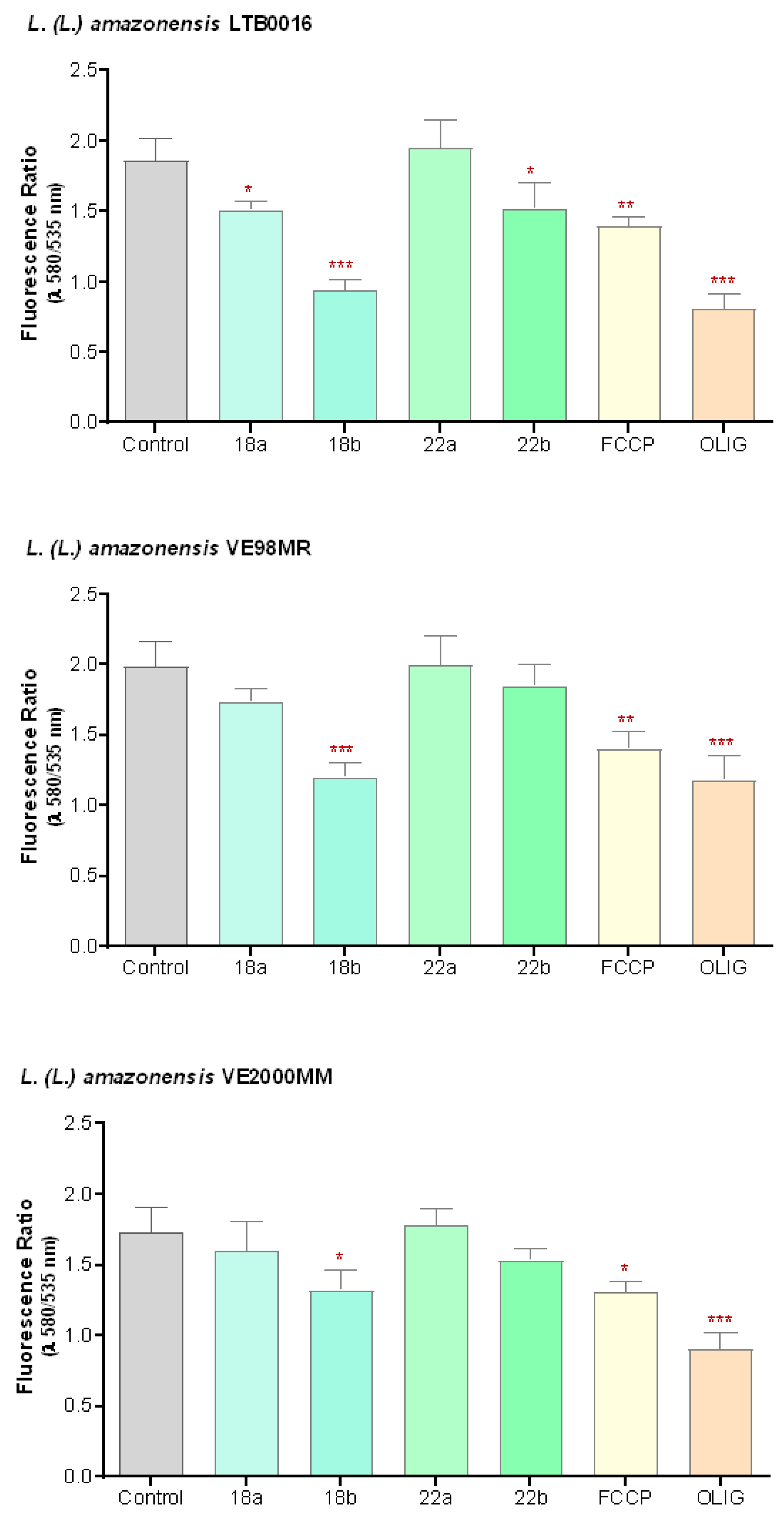
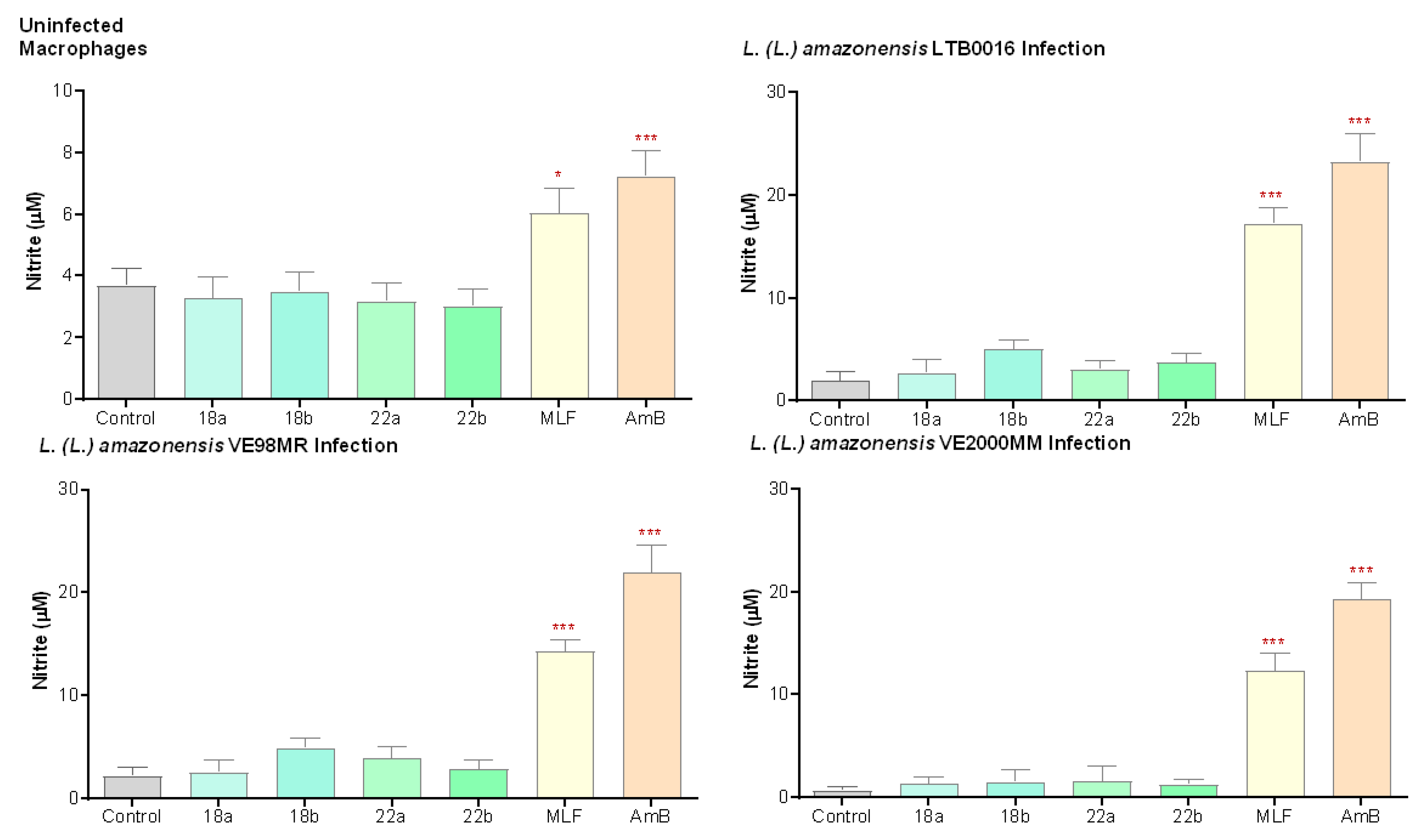
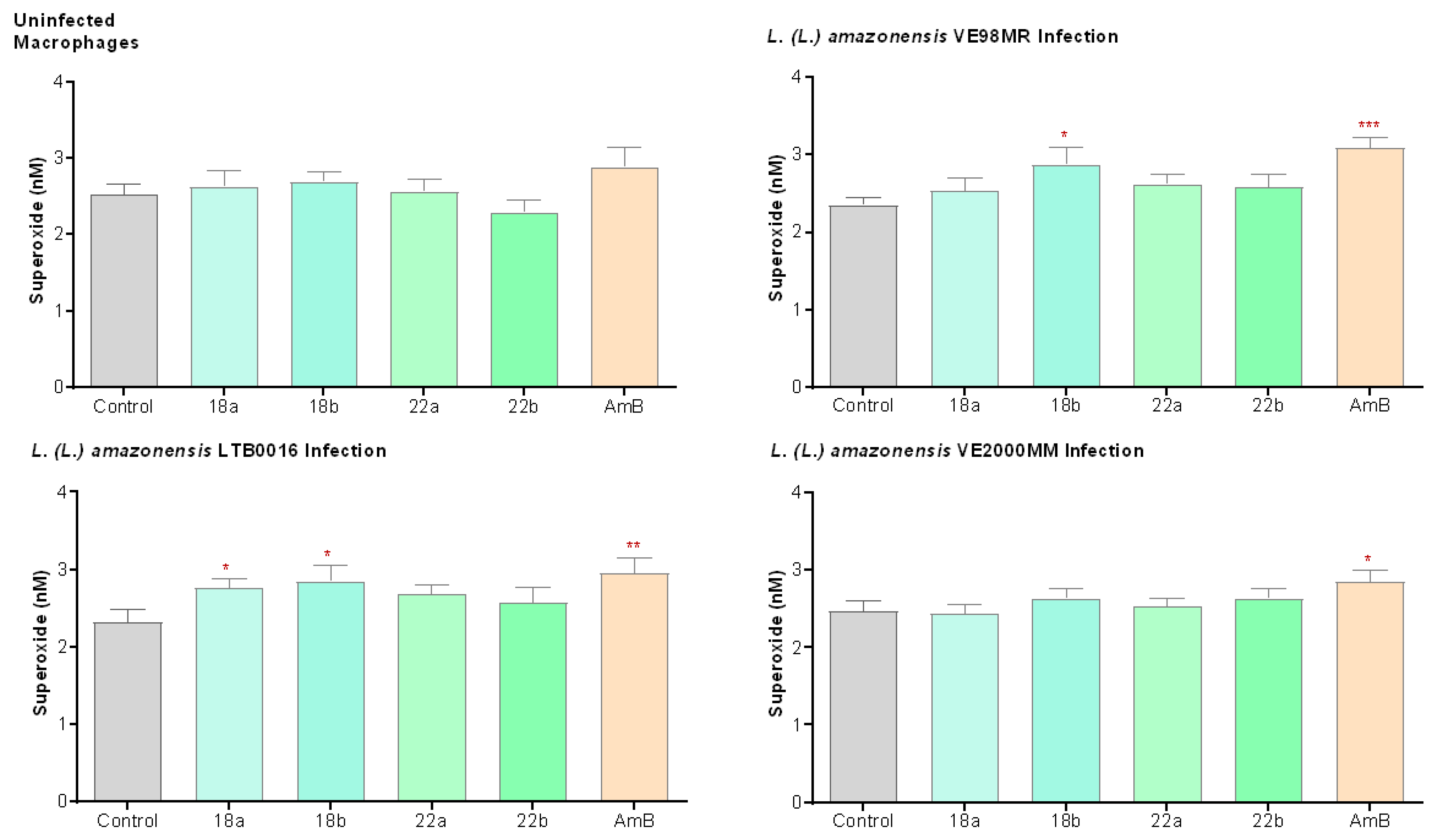
3. Results
3.1. Selection of the Tool Compounds
3.2. Effect of Compounds on Intracellular Amastigotes
3.3. In the Search of the Mechanism of Action of Betulin Derivatives
3.3.1. Plasma Membrane Potential (ΔΨp) in Promastigotes
3.3.2. Mitochondrial Membrane Potential (ΔΨm) in Promastigotes
3.3.3. Nitric Oxide Production (NO) in Macrophages
3.3.4. Superoxide Production (O2−) in Macrophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savoia, D. Recent updates and perspectives on leishmaniasis. J. Infect. Dev. Ctries 2015, 9, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makala, L.H.C.; Baban, B. Novel therapeutic approaches to Leishmania infection. In Leishmaniasis—Trends in Epidemiology, Diagnosis and Treatment; Claborn, D.M., Ed.; InTechOpen: London, UK, 2014; pp. 495–523. Available online: https://www.intechopen.com/books/-novel-therapeutic-approaches-to-leishmania-infection (accessed on 11 October 2020).
- Sundar, S.; Chakravarty, J. Visceral leishmaniasis. In Drug Resistance in Leishmania Parasites: Consequences, Molecular Mechanisms and Possible Treatments, 2nd ed.; Ponte-Sucre, A., Padrón-Nieves, M., Eds.; Springer Nature: Cham, Switzerland, 2018; pp. 159–176. [Google Scholar]
- World Health Organization. Investing to Overcome the Global Impact of Neglected Tropical Diseases; Third Who Report on Neglected Tropical Diseases; WHO: Geneva, Switzerland, 2015; pp. 118–126. [Google Scholar]
- Guevara, J.R. MPPS. Estadísticas de Leishmaniasis Visceral en Venezuela Histórico/Año 1990–2018; Servicio Autónomo Instituto de Biomedicina “Dr. Jacinto Convit”: Caracas, Venezuela, 2018. [Google Scholar]
- Zerpa, O.; Ulrich, M.; Borges, R.; Rodrìguez, V.; Centeno, M.; Negrón, E.; Belizario, D.; Convit, J. Epidemiological aspects of human and canine visceral leishmaniasis in Venezuela. Rev. Panam Salud Publica 2003, 13, 239–245. [Google Scholar] [CrossRef]
- Rodríguez, N.M.; De Guglielmo, Z.; Barrios, M.A.; Barrios, R.M.; Zerpa, O.; Feliciangeli, M.D. Nueva Esparta Genetic homogeneity within Leishmania (L.) infantum isolated from human and dogs: The relationship with the sandfly fauna distribution in endemic areas of Nueva Esparta State, Venezuela. Parasitology 2005, 130, 611–619. [Google Scholar] [CrossRef]
- Hernández, D.; Rojas, E.; Scorza, J.V.; Jorguera, A. Infectividad del perro (Canis familiaris) para Lutzomyia youngi en Trujillo, Venezuela. Biomédica 2006, 26, 242–248. [Google Scholar] [CrossRef]
- Guevara, J.R. MPPS. Estadísticas de Leishmaniasis Cutánea en Venezuela Histórico/Año 1990–2018; Servicio Autónomo Instituto de Biomedicina “Dr. Jacinto Convit”: Caracas, Venezuela, 2018. [Google Scholar]
- Peña, I.; Manzano, P.; Cantizani, J.; Kessler, A.; Alonso-Padilla, J.; Bardera, A.I.; Alvarez, E.; Colmenarejo, G.; Cotillo, I.; Roquero, I. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: An open resource. Sci. Rep. 2015, 5, 1–47. [Google Scholar] [CrossRef] [Green Version]
- De Vries, H.J.C.; Reedijk, S.H.; Schallig, H.D.F.H. Cutaneous leishmaniasis: Recent developments in diagnosis and management. Am. J. Clin. Dermatol. 2015, 16, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Reveiz, L.; Maia-Elkhoury, A.N.S.; Nicholls, R.S.; Sierra Romero, G.A.; Yadon, Z.E. Interventions for American cutaneous and mucocutaneous leishmaniasis: A systematic review update. PLoS ONE 2013, 8, e61843. [Google Scholar] [CrossRef] [Green Version]
- De Menezes, J.P.B.; Guedes, C.E.S.; Petersen, A.L.D.O.A.; Fraga, D.B.M.; Veras, P.S.T. Advances in development of new treatment for leishmaniasis. BioMed Res. Int. 2015, 2015, 815023. [Google Scholar] [CrossRef]
- Joshi, S.; Rawat, K.; Yadav, N.K.; Kumar, V.; Siddiqi, M.I.; Dube, A. Visceral leishmaniasis: Advancements in vaccine development via classical and molecular approaches. Front. Immunol. 2014, 5, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Kumar, M.; Singh, R.K. Leishmaniasis: Current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 2012, 5, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Ponte-Sucre, A.; Diaz, E. Leishmaniasis: The Biology of a Parasite. In Drug Resistance in Leishmania Parasites: Consequences, Molecular Mechanisms and Possible Treatments, 2nd ed.; Ponte-Sucre, A., Padrón-Nieves, M., Eds.; Springer Nature: Cham, Switzerland, 2018; pp. 1–16. [Google Scholar]
- Gupta, S.K.; Dandekar, T. Bioinformatics in Leishmania Drug Design. In Drug Resistance in Leishmania Parasites: Consequences, Molecular Mechanisms and Possible Treatments, 2nd ed.; Ponte-Sucre, A., Padrón-Nieves, M., Eds.; Springer Nature: Cham, Switzerland, 2018; pp. 297–317. [Google Scholar]
- Vermelho, A.B.; Supuran, C.T.; Cardoso, V.; Menezes, D.; De Andrade Silva, J.R.; Pinto Ferreira, J.L.; Fernandez Amaral, A.C.; Rodrigues, I.A. Leishmaniasis: Possible new strategies for treatment. In Leishmaniasis—Trends in Epidemiology, Diagnosis and Treatment; Claborn, D.M., Ed.; InTechOpen: London, UK, 2014; pp. 351–376. Available online: https://www.intechopen.com/books/leishmaniasis-trends-in-epidemiology-diagnosis-and-treatment/leishmaniasis-possible-new-strategies-for-treatment (accessed on 11 October 2020).
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Spinks, D.; Ong, H.B.; Mpamhanga, C.P.; Shanks, E.J.; Robinson, D.A.; Collie, I.T.; Read, K.D.; Fearson, J.A.; Wyatt, P.G.; Brenk, R.; et al. Design, synthesis and biological evaluation of novel inhibitors of Trypanosoma brucei pteridine reductase 1. Chem. Med. Chem. 2011, 6, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Prada, C.F.; Alvarez-Velilla, R.; Balaña-Fouce, R.; Prieto-Sánchez, R.; Calvo-Álvarez, C.; Escudero-Martínez, E.; Requena, J.M.; Ordoñez, J.M.; Desideri, C.; Pérez-Pertejo, A.; et al. Gimatecan and other camptothecin derivatives poison Leishmania DNA-topoisomerase IB leading to a strong leishmanicidal effect. Biochem. Pharmacol. 2013, 85, 1433–1440. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.H.; López, S.E.; Rodríguez, N.M.; Oviedo, H. Anti-leishmanial activity, structure–activity relationship of series of 2-(trifluoromethyl)benzo[b][1,8]naphthyridin-4(1H)-ones. Arch. Pharm. 2018, 351, 1–9. [Google Scholar] [CrossRef]
- Rodríguez, M.; Gutiérrez, J.; Domínguez, J.; Pixoto, P.A.; Fernández, A.; Rodríguez, N.M.; Deffieux, D.; Rojas, L.B.; Quideau, S.; Pouysegu, L.; et al. Synthesis and leishmanicidal evaluation of sulfanyl and sulfonyl-tethered functionalized benzoate derivatives featuring a nitroimidazole moiety. Arch. Pharm. 2020, 353, e2000002. [Google Scholar] [CrossRef]
- Brito, A.M.; Santos, D.; Rodrigues, S.; Brito, R.G.; Xavier-Filho, L. Plants with anti-Leishmania activity: Integrative review from 2000 to 2011. Pharmacogn. Rev. 2013, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, L.A.; Pereira Da Silva, L.H.; Fett-Neto, A.G.; De Azevedo, W.F., Jr.; De Souza Moreira, I.; Palma, M.S.; Calixto, J.B.; Filho, S.A.; Ribero Dos Santos, R.; Pereira Soares, M.B.; et al. The use of biodiversity as source of new chemical entities against defined molecular targets for treatment of malaria, tuberculosis, and T-cell mediated diseases—A review. Mem. Inst. Oswaldo Cruz 2005, 100, 475–506. [Google Scholar] [CrossRef] [Green Version]
- Krasutsky, P. Birch bark research and development. Nat. Prod. Rep. 2006, 23, 919–942. [Google Scholar] [CrossRef]
- Sousa, M.C.; Varandas, R.; Santos, R.C.; Santos-Rosa, M.; Alves, V.; Salvador, J.A.R. Anti-leishmanial activity of semisynthetic lupane triterpenoids betulin and betulinic acid derivatives: Synergistic effects with miltefosine. PLoS ONE 2014, 9, e89939. [Google Scholar] [CrossRef] [Green Version]
- Alcazar, W.; López, A.S.; Alakurtti, S.; Tuononen, M.L.; Yli-Kauhaluoma, J.; Ponte-Sucre, A. Betulin derivatives impair Leishmania braziliensis viability and host–parasite interaction. Bioorg. Med. Chem. 2014, 22, 6220–6226. [Google Scholar] [CrossRef]
- Alakurtti, S.; Bergström, P.; Sacerdoti-Sierra, N.; Jaffe, C.L.; Yli-Kauhaluoma, J. Anti-leishmanial activity of betulin derivatives. J. Antibiot. 2010, 63, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Wert, L.; Alakurtti, S.; Corral, M.J.; Sánchez-Fortún, S.; Yli-Kauhaluoma, J.; Alunda, J.M. Toxicity of betulin derivatives and in vitro effect on promastigotes and amastigotes of Leishmania infantum and L. donovani. J. Antibiot. 2011, 64, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, N.M.; Guzmán, B.; Rodas, A.; Takiff, H.; Bloom, B.R.; Convit, J. Diagnoses of cutaneous leishmaniasis and species discrimination of parasites by PCR and hybridization. J. Clin. Microbiol. 1994, 32, 2246–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, M.; Pharoah, M.; Ashall, F.; Miles, M.A. Human urine stimulates growth of Leishmania in vitro. Trans. R. Soc. Trop. Med. Hyg. 1991, 85, 477–479. [Google Scholar] [CrossRef]
- Allahverdiyev, A.M.; Bagirova, M.; Elcicek, S.; Cakir Koc, R.; Oztel, O.N. Effect of human urine on cell cycle and infectivity of Leismania species promastigotes in vitro. Am. J. Trop. Med. Hyg. 2011, 85, 639–643. [Google Scholar] [CrossRef]
- Ralph, P.; Nakoinz, I. Phagocytosis and cytolysis by a macrophage tumour and its cloned cell line. Nature 1975, 257, 393–394. [Google Scholar] [CrossRef]
- Le Pape, P.; Pagniez, F.; Abdala, H. A new automatized fluorometric assay for anti-Leishmania drug screening. Acta Parasitol. 2002, 47, 79–81. [Google Scholar]
- Tong, F.P. Statistical Methods for Dose-Response Assays; University of California: Berkeley, CA, USA, 2010; Available online: http://escholarship.org/uc/item/9295t422# (accessed on 11 October 2020).
- Borborema, S.E.T.; Schwendener, R.A.; Osso, J.A.; De Andrade, H.F., Jr.; Do Nascimento, N. Uptake and anti-leishmanial activity of meglumine antimoniate-containing liposomes in Leishmania major infected macrophages. Int. J. Antimicrob. Agents 2011, 38, 341–347. [Google Scholar] [CrossRef]
- Nwaka, S.; Hudson, A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discov. 2006, 5, 941–955. [Google Scholar] [CrossRef]
- Neal, R.A.; Croft, S.L. An in-vitro system for determining the activity of compounds against the intracellular amastigote form of Leishmania donovani. J. Antimicrob. Chemother. 1984, 14, 463–475. [Google Scholar] [CrossRef]
- Da Luz, R.I.; Vermeersch, M.; Dujardin, J.C.; Cos, P.; Maes, L. In vitro sensitivity testing of Leishmania clinical field isolates: Preconditioning of promastigotes enhances infectivity for macrophage host cells. Antimicrob. Agents Chemother. 2009, 53, 5197–5203. [Google Scholar] [CrossRef] [Green Version]
- Lira, R.; Sundar, S.; Makharia, A.; Kenney, R.; Gam, A.; Saraiva, E.; Sacks, D. Evidence that the high incidence of treatment failures in Indian kala-azar is due to the emergence of antimony-resistant strains of Leishmania donovani. J. Infect. Dis. 1999, 180, 564–567. [Google Scholar] [CrossRef] [Green Version]
- Vieira, L.; Slotki, I.; Cabantchik, Z.I. Chloride conductive pathways which support electrogenic H+ pumping by Leishmania major promastigotes. J. Biol. Chem. 1995, 270, 5299–5304. [Google Scholar] [CrossRef] [Green Version]
- Padrón-Nieves, M.; Machuca, C.; Díaz, E.; Cotrim, P.; Rodríguez, N.M.; Ponte-Sucre, A. Correlation between glucose uptake and membrane potential in Leishmania parasites isolated from DCL patients with therapeutic failure: A proof of concept. Parasitol. Res. 2014, 113, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Padrón-Nieves, M.; Ponte-Sucre, A. Cellular Markers for the Identification of Chemo-Resistant Isolates in Leishmania. In Methods in Molecular Biology. Trypanosomatids Methods and Protocols; Michels, P., Ginger, M., Zilberstein, D., Eds.; Springer Nature: Cham, Switzerland; Humana Press: New York, NY, USA, 2019; Chapter 46; pp. 755–769. [Google Scholar]
- De Macedo-Silva, S.T.; De Oliveira Silva, T.L.A.; Urbina, J.A.; Souza, W.D.; Rodrigues, J.C.F. Antiproliferative, ultrastructural, and physiological effects of amiodarone on promastigote and amastigote forms of Leishmania amazonensis. Mol. Biol. Int. 2011, 2011, 876021. [Google Scholar] [CrossRef] [Green Version]
- Ponte-Sucre, A.; Mendoza-León, A.; Moll, H. Experimental leishmaniasis: Synergistic effect of ion channel blockers and interferon-gamma on the clearance of Leishmania major by macrophages. Parasitol. Res. 2001, 87, 27–31. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Vicik, R.; Schultheis, M.; Schirmeister, T.; Moll, H. Aziridine-2,3-dicarboxylates, peptidomimetic cysteine protease inhibitors with anti-leishmanial activity. Antimicrob. Agents Chemother. 2006, 50, 2439–2447. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.L. Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol. 1996, 268, 142–151. [Google Scholar]
- Decuypere, S.; Vanaerschot, M.; Brunker, K.; Imamura, H.; Müller, S.; Khanal, B.; Rijal, S.; Dujardin, J.C.; Coombs, G.H. Molecular mechanisms of drug resistance in natural Leishmania populations vary with genetic background. PLoS Negl. Trop. Dis. 2012, 6, e1514. [Google Scholar] [CrossRef] [PubMed]
- De Morais-Teixeira, E.; Damasceno, Q.S.; Galuppo, M.K.; Romanha, A.J.; Rabello, A. The in vitro leishmanicidal activity of hexadecylphosphocholine (miltefosine) against four medically relevant Leishmania species of Brazil. Mem. Inst. Oswaldo Cruz 2011, 106, 475–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, M.; Monzote, L.; Montalvo, A.M.; Scull, R. Screening of medicinal plants against Leishmania amazonensis. Pharm. Biol. 2010, 48, 1053–1058. [Google Scholar] [CrossRef]
- García, M.C.F.; Soares, D.C.; Santana, R.C.; Saraiva, E.M.; Siani, A.C.; Ramos, M.F.S.; Danielli, M.G.M.; Souto-Padron, T.C.; Pinto da Silva, L.H. The in vitro anti-leishmanial activity of essential oil from Aloysia gratissima and guaiol, its major sesquiterpene against Leishmania amazonensis. Parasitology 2018, 145, 1219–1227. [Google Scholar] [CrossRef]
- Al-Sokari, S.S.; Ali, N.A.A.; Monzote, L.; Al-Fatimi, M.A. Evaluation of anti-leishmanial activity of albaha medicinal plants against Leishmania amazonensis. BioMed Res. Int. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Souza-Fagundes, E.M.; Cota, B.B.; Rosa, L.H.; Romanha, A.J.; Corrêa-Oliveira, R.; Zani, C.L.; Texeira-Carvalho, A.; Martins-Filho, O.A. In vitro activity of hypnophilin from Lentinus strigosus: A potential prototype for Chagas disease and leishmaniasis chemotherapy. Braz. J. Med. Biol. Res. 2010, 43, 1054–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alakurtti, S.; Heiska, T.; Kiriazis, A.; Sacerdoti-Sierra, N.; Jaffe, C.L.; Yli-Kauhaluoma, J. Synthesis and anti-leishmanial activity of heterocyclic betulin derivatives. Bioorg. Med. Chem. 2010, 18, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Haldar, A.K.; Sen, P.; Roy, S. Use of antimony in the treatment of leishmaniasis: Current status and future directions. Mol. Biol. Int. 2011, 2011, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzmuller, P.; Sereno, D.; Lemesre, J.L. Lower nitric oxide susceptibility of trivalent antimony-resistant amastigotes of Leishmania infantum. Antimicrob. Agents Chemother. 2005, 49, 4406–4409. [Google Scholar] [CrossRef] [Green Version]
- Vanaerschot, M.; Maes, I.; Ouakad, M.; Adaui, I.; Maes, L.; De Doncker, S. Linking in vitro and in vivo survival of clinical Leishmania donovani strains. PLoS ONE 2010, 5, e12211. [Google Scholar] [CrossRef] [Green Version]
- Prayong, P.; Barusrux, S.; Weerapreeyakul, N. Cytotoxic activity screening of some indigenous Thai plants. Fitoterapia 2008, 79, 598–601. [Google Scholar] [CrossRef]
- Chowdhury, S.; Mukherjee, T.; Sengupta, S.; Chowdhury, S.R.; Mukhopadhyay, S.; Majumder, H. Novel betulin derivatives as anti-leishmanial agents with mode of action targeting type IB DNA topoisomerase. Mol. Pharmacol. 2011, 80, 694–703. [Google Scholar] [CrossRef] [Green Version]
- De Sá, M.S.; Costa, J.F.O.; Krettli, A.U.; Zalis, M.G.; de Azevedo, G.L.M.; Sette, I.V.M.F.; de Amorim, C.C.; Filho, J.M.B.; Giulietti-Harley, A.M.; Dos Santos, R.R.; et al. Antimalarial activity of betulinic acid and derivatives in vitro against Plasmodium falciparum and in vivo in P. berghei-infected mice. Parasitol. Res. 2009, 105, 275–279. [Google Scholar] [CrossRef]
- Takahashi, M.; Fuchino, H.; Sekita, S.; Satake, M. In vitro leishmanicidal activity of some scarce natural products. Phytother. Res. 2004, 18, 573–578. [Google Scholar] [CrossRef]
- Sauvain, M.; Kunesch, N.; Poisson, J.; Gantier, J.C.; Gayral, P.; Dedet, J.P. Isolation of leishmanicidal triterpenes and lignans from the amazonian liana Doliocarpus dentatus (Dilleniaceae). Phytother. Res. 2002, 10, 1–4. [Google Scholar] [CrossRef]
- Chowdhury, S.; Mukherjee, T.; Chowdhury, S.R.; Sengupta, S.; Mukhopadhyay, S.; Parasuraman, J.; Majumder, H.K. Disuccinyl betulin triggers metacaspase-dependent endonuclease G-mediated cell death in unicellular protozoan parasite Leishmania donovani. Antimicrob. Agents Chemother. 2014, 58, 2186–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudagar, P.; Dubey, V.K. Cloning, expression, characterization and inhibition studies on trypanothione synthetase, a drug target enzyme, from Leishmania donovani. Biol. Chem. 2011, 392, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Saudagar, P.; Dubey, V.K. Molecular mechanisms of in vitro betulin-induced apoptosis of Leishmania donovani. Am. J. Trop. Med. Hyg. 2014, 90, 354–360. [Google Scholar] [CrossRef]
- Zhang, Y.; Xhaard, H.; Ghemtio, L. Predictive classification models and targets identification for betulin derivatives as Leishmania donovani inhibitors. J. Cheminform. 2018, 10, 1–16. [Google Scholar] [CrossRef]
- Camargos, H.S.; Moreira, R.A.; Mendanha, S.A.; Fernandes, K.S.; Dorta, M.L.; Alonso, A. Terpenes increase the lipid dynamics in the Leishmania plasma membrane at concentrations similar to their IC50 values. PLoS ONE 2014, 9, e104429. [Google Scholar] [CrossRef]
- Azas, N.; Di Giorgio, C.; Delmas, F.; Gasquet, M.; Timon-David, P. Assessment of amphotericin B susceptibility in Leishmania infantum promastigotes by flow cytometric membrane potential assay. Cytometry 1997, 28, 165–169. [Google Scholar] [CrossRef]
- Zilberstein, D.; Dwyer, D.M. Antidepressants cause lethal disruption of membrane function in the human protozoan parasite Leishmania. Science 1984, 226, 977–979. [Google Scholar] [CrossRef]
- Díaz-Achirica, P.; Ubach, J.; Guinea, A.; Andreu, D.; Rivas, L. The plasma membrane of Leishmania donovani promastigotes is the main target for CA(1-8)M(1-18), a synthetic cecropin A-melittin hybrid peptide. Biochem. J. 1998, 330, 453–460. [Google Scholar] [CrossRef]
- Mazoir, N.; Benharref, A.; Bailén, M.; Reina, M.; González-Coloma, A.; Martínez-Díaz, R.A. Anti-leishmanial and antitrypanosomal activity of triterpene derivatives from latex of two Euphorbia species. Z. Naturforsch. 2011, 66, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Jawed, J.J.; Das, M.C.; Sandhu, P.; De Utpal, C.; Dinda, B.; Akhter, Y.; Bhattacharjee, S. Anti-leishmanial and immunomodulatory activity of lupeol a triterpene compound isolated from Sterculia villosa. J. Antimicrob. Agents 2017, 50, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, S. Evaluation of in vitro and in vivo anti-leishmanial potential of bergenin rich Bergenia ligulata (Wall.) Engl. root extract against visceral leishmaniasis in inbred BALB/c mice through immunomodulation. J. Tradit. Complement. Med. 2018, 8, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Assreuy, J.; Cunha, F.Q.; Epperlein, M.; Noronha-Dutra, A.; O’Donnell, A.; Liew, F.Y.; Moncada, S. Production of nitric oxide and superoxide by activated macrophages and killing of Leishmania major. Eur. J. Immunol. 1994, 24, 672–676. [Google Scholar] [CrossRef]
- Carneiro, P.P.; Conceiçao, J.; Macedo, M.; Magalhães, V.; Carvalho, E.M.; Bacellar, O. The role of nitric oxide and reactive oxygen species in the killing of Leishmania braziliensis by monocytes from patients with cutaneous leishmaniasis. PLoS ONE 2016, 11, e0148084. [Google Scholar] [CrossRef] [Green Version]
- Gantt, K.R.; Goldman, T.L.; McCormick, M.L.; Miller, M.A.; Jeronimo, S.M.B.; Nascimento, E.T.; Britigan, B.E.; Wilson, M.E. Oxidative responses of human and murine macrophages during phagocytosis of Leishmania chagasi. J. Immunol. 2001, 167, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukbel, R.M.; Patten, C., Jr.; Gibson, K.; Ghosh, M.; Petersen, C.; Jones, D.E. Macrophage killing of Leishmania amazonensis amastigotes requires both nitric oxide and superoxide. Am. J. Trop. Med. Hyg. 2007, 76, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Acuña, S.M.; Aoki, J.I.; Laranjeira-Silva, M.F.; Zampieri, R.A.; Fernandes, J.C.R.; Muxel, S.M.; Floeter-Winter, M.L. Arginase expression modulates nitric oxide production in Leishmania (Leishmania) amazonensis. PLoS ONE 2017, 12, e0187186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calegari-Silva, T.C.; Pereira, R.M.S.; Barbosa De-Melo, L.D.; Saraiva, E.M.; Soares, D.C.; Bellio, M.; Ulisses, G.; Lopes, U.G. NF-κB-mediated repression of iNOS expression in Leishmania amazonensis macrophage infection. Immunol. Lett. 2009, 127, 19–26. [Google Scholar] [CrossRef]
- Isnard, A.; Shio, M.T.; Olivier, M. Impact of Leishmania metalloprotease GP63 on macrophage signaling. Front. Cell Infect. Microbiol. 2012, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Betulin Derivatives | Parasite Growth % [50 µM] | Promastigote (GI50) | Macrophages J774.1 (CC50) | SI | ||||
|---|---|---|---|---|---|---|---|---|
| (µM) | IL | SL | (µM) | IL | SL | CC50/GI50 | ||
| 11 | 19.76 | 19.33 | 14.14 | 24.53 | 25.08 | 8.95 | 41.22 | 1.30 |
| 171 | 6.49 | 1.35 | −0.03 | 2.74 | 19.26 | 10.10 | 28.41 | 14.23 |
| 181 | 3.32 | 0.90 | 0.24 | 1.55 | 48.09 | 33.17 | 63.01 | 53.63 |
| 222 | 7.98 | 1.98 | 0.99 | 2.98 | 61.13 | 52.91 | 69.35 | 30.82 |
| 242 | 10.26 | 9.05 | 6.95 | 11.15 | 51.06 | 34.78 | 67.34 | 5.64 |
| 252 | 11.70 | 6.46 | 2.63 | 10.30 | 65.93 | 51.76 | 80.10 | 10.20 |
| 262 | 4.28 | 3.85 | −0.23 | 7.93 | 21.86 | 15.72 | 28.00 | 5.68 |
| 272 | 7.34 | 4.23 | 0.42 | 8.04 | 21.12 | 16.86 | 25.38 | 4.99 |
| SbIII | ND | 5.51 | 4.92 | 6.08 | ND | ND | ND | ND |
| AmB | ND | 0.068 | 0.05 | 0.08 | ND | ND | ND | ND |
| PNT | ND | 0.93 | 0.51 | 1.35 | ND | ND | ND | ND |
| MLF | ND | 4.17 | 3.79 | 4.54 | ND | ND | ND | ND |
| GI50 (µM) | MØ J774.1 | SI (CC50/GI50) | |||||
|---|---|---|---|---|---|---|---|
| LTB0016 | VE98MR | VE2000MM | CC50 1 (µM) | LTB0016 | VE98MR | VE2000MM | |
| 18 | 0.20 | 1.76 | 0.72 | 48.09 | 240.45 | 27.32 | 66.79 |
| 22 | 0.21 | 0.63 | 1.69 | 61.13 | 291.10 | 97.03 | 36.17 |
| AmB | 0.020 | 0.073 | 0.080 | >10 | >500 | >136 | >125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcazar, W.; Alakurtti, S.; Padrón-Nieves, M.; Tuononen, M.L.; Rodríguez, N.; Yli-Kauhaluoma, J.; Ponte-Sucre, A. Leishmanicidal Activity of Betulin Derivatives in Leishmania amazonensis; Effect on Plasma and Mitochondrial Membrane Potential, and Macrophage Nitric Oxide and Superoxide Production. Microorganisms 2021, 9, 320. https://doi.org/10.3390/microorganisms9020320
Alcazar W, Alakurtti S, Padrón-Nieves M, Tuononen ML, Rodríguez N, Yli-Kauhaluoma J, Ponte-Sucre A. Leishmanicidal Activity of Betulin Derivatives in Leishmania amazonensis; Effect on Plasma and Mitochondrial Membrane Potential, and Macrophage Nitric Oxide and Superoxide Production. Microorganisms. 2021; 9(2):320. https://doi.org/10.3390/microorganisms9020320
Chicago/Turabian StyleAlcazar, Wilmer, Sami Alakurtti, Maritza Padrón-Nieves, Maija Liisa Tuononen, Noris Rodríguez, Jari Yli-Kauhaluoma, and Alicia Ponte-Sucre. 2021. "Leishmanicidal Activity of Betulin Derivatives in Leishmania amazonensis; Effect on Plasma and Mitochondrial Membrane Potential, and Macrophage Nitric Oxide and Superoxide Production" Microorganisms 9, no. 2: 320. https://doi.org/10.3390/microorganisms9020320