The Expanding Computational Toolbox for Engineering Microbial Phenotypes at the Genome Scale


1
Department of Bioengineering, University of California, San Diego, San Diego, CA 92093, USA
2
Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800 Lyngby, Denmark
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Microorganisms 2020, 8(12), 2050; https://doi.org/10.3390/microorganisms8122050
Submission received: 16 November 2020
/
Revised: 7 December 2020
/
Accepted: 16 December 2020
/
Published: 21 December 2020
(This article belongs to the Special Issue Genome-Scale Modeling of Microorganisms in the Real World)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Microbial strains are being engineered for an increasingly diverse array of applications, from chemical production to human health. While traditional engineering disciplines are driven by predictive design tools, these tools have been difficult to build for biological design due to the complexity of biological systems and many unknowns of their quantitative behavior. However, due to many recent advances, the gap between design in biology and other engineering fields is closing. In this work, we discuss promising areas of development of computational tools for engineering microbial strains. We define five frontiers of active research: (1) Constraint-based modeling and metabolic network reconstruction, (2) Kinetics and thermodynamic modeling, (3) Protein structure analysis, (4) Genome sequence analysis, and (5) Regulatory network analysis. Experimental and machine learning drivers have enabled these methods to improve by leaps and bounds in both scope and accuracy. Modern strain design projects will require these tools to be comprehensively applied to the entire cell and efficiently integrated within a single workflow. We expect that these frontiers, enabled by the ongoing revolution of big data science, will drive forward more advanced and powerful strain engineering strategies.
1. Introduction
Microbes have been engineered for a broad number of applications. As cell factories, cells have been designed to convert low-value substrates into valuable chemical products, including biofuels [1], commodity chemicals [2], bioactive compounds [3], and foods [4]. To benefit the environment, microbes have been engineered for bioremediation [5] and biosensing [6] of toxic compounds and pollutants. As engineered tools, microbes have been programmed using cell circuits to exhibit elaborate behaviors, from synchronized fluorescence [7] to hunting down tumors to deliver chemotherapeutics [8]. Finally, as cellular products, microbes themselves are increasingly of interest for probiotic and nutritional supplements [9].
The experimental workflow to engineer a new microbial strain has a number of common steps, although the order may vary (Figure 1A) [10]. First, a background organism and strain is chosen for the application of interest. Genes may be knocked out, introduced, knocked down, or overexpressed for a variety of purposes, such as control of transcriptional regulation, redirection of metabolic flux to desired pathways, or removal of unwanted or wasteful processes. Bioprocess conditions can be optimized through control of various factors including media, feed rate, growth rate, pH, and temperature. Specific sequence variants can be introduced through rational design or selected through screens and adaptive laboratory evolution to control expression, alter enzyme activity, or remove regulatory sites from proteins. The typical strain design workflow thus requires a large number of decisions on how to improve strain behavior. Left to a trial and error approach, the complexity of biological systems makes efficient engineering of strains a daunting task.
To aid strain design efforts, computational tools have been integrated from various fields into the strain design workflow [11]. These tools offer the promise of restricting the experimental search space by either identifying modifications that are more likely to improve strain performance or proposing entirely new designs through mathematical modeling of cell behavior. However, many steps in the strain design process are still driven by rational approaches, rules of thumb, and extensive experimental screening and trial and error. Workflows driven purely by predictive tools would have the advantage of efficiency of execution through fewer experimental steps, reduced time, and ultimately improved performance through careful guidance toward an optimal desired phenotype. We describe two approaches that show promise as systematic tools for cell design: genetic circuits and genome-scale modeling.
One strategy for constructing synthetic strains has been to engineer desired behaviors through the use of genetic circuits [12]. The key concept is to carefully characterize and often mathematically model the behavior of a ‘circuit’, typically a small transcriptional regulatory network, to control a cell phenotype. As greater numbers of these small circuits are characterized, they begin to comprise a ‘parts list’ of available phenotypes from which an engineer can choose or can be assembled automatically by an algorithm [13]. Larger and larger circuits can then be constructed of well-characterized smaller circuits to engineer more complex phenotypes. This strategy has been employed for a number of promising applications [14,15].
Another successful paradigm for computational design of cells is genome-scale network modeling [16]. While genetic circuits approaches utilize highly controllable systems of limited scope, genome-scale models seek to predict cell phenotype by comprehensively modeling all known functions of the cell. As part of the Constraint-based Reconstruction and Analysis (COBRA) framework, genome-scale models of metabolism utilize a metabolic network reconstruction to predict metabolic phenotypes and analyze genome-scale datasets [17]. These models deal with the large scope of the system by utilizing the constraint-based modeling framework, which requires few parameters to generate predictions. The challenge of managing these large-scale models is achieved through community enforcement of rigid requirements, testing, and data standards [18,19]. Although these models were originally developed for metabolism, they have recently been extended to include transcription and translation machinery [20,21,22] and even further to whole-cell kinetic simulations [23].
Although computational methods have undoubtedly augmented rational strain design efforts, there are a number of challenges in a strain design workflow that still cannot be effectively addressed by existing computational tools [24] (Figure 1B). For example: (1) Organisms are often chosen for a strain design project due to historical knowledge and convenience, rather than fundamental benefits provided by the organism that could be calculated computationally a priori, (2) Gaps in gene annotation make choosing non-model organisms a risk, (3) The difficulty in accounting for enzyme kinetics makes the understanding of metabolic and allosteric regulation a challenge, (4) A lack of understanding of regulatory networks impedes the understanding and control of gene expression, and (5) Insufficient annotation of the organism genome makes it difficult to interpret the functional implications of sequence variation. Challenges such as these present major barriers to interpreting data and predicting strain phenotype.
There are many methods currently being developed that may directly meet these challenges to enable fully predictive strain design workflows (Figure 1C). For example, advances in metabolic modeling could enable the optimization of bioprocess conditions or the identification of optimal expression levels of pathway genes [25,26]. However, these models are still in development and have not yet been shown to enable accurate predictions at scale. In this perspective, we describe five frontiers consisting of promising developments in computational strain design that may pave the way toward achieving comprehensive and integrated strain design workflows (Figure 2).
2. Frontier 1: Constraint-Based Reconstruction and Modeling
COBRA methods and tools have been used and refined for over 15 years in the field of systems biology [17,41,42,43]. Many current applications of constraint-based models answer biologically meaningful questions such as max growth rate, metabolite production, limiting nutrients, and gene essentiality. COBRA methods are also often used when little parameter and/or metabolite information is available given the size of the networks. These methods have predominantly been limited by annotation gaps in the underlying reconstructed networks [44]. COBRA models have also been expanded to include other cellular processes, such as transcription and translation, in order to increase the predictive capabilities of the models. Previous models did not have the capacity to accurately model suboptimal states, such as thermal or oxidative stress, nor could they properly describe how microbes interact within a community [24]. In the following sections, we describe recent computational advances that have benefitted COBRA models and methods.
2.1. Proteome Allocation Models
One of the main challenges for omics integration with M-models is that they only indirectly relate expression to metabolic fluxes. Metabolism and macromolecular expression (ME) models were developed to be multiscale models that explicitly include multiple cellular processes such as transcription, translation, and post-translational modifications. Early ME-models were able to compute optimal proteome compositions of a growing cell but suffered from significant model sizes and complexity, which has been addressed with the recent development of the COBRAme platform [20]. ME-models have since been produced for a variety of organisms and can now accurately predict overflow metabolism and cofactor/metal usage given media composition [45,46]. Additionally, ME-models have led to increased confidence in the selection of strain designs with robust growth-coupled production, in addition to having better byproduct secretion predictions than M-models [47,48]. Time course ME simulations are also possible and can predict substrate utilization hierarchy on mixed carbon source medium. These simulations compute distinct proteome compositions over time [49]. Prediction capabilities for ME-models have also expanded to include suboptimal states such as stress and mitigation responses. These ‘StressME’ models are able to more accurately model the cell’s unused proteome that acts as a hedging mechanism for preparedness functions under low pH, thermal, and oxidative stress [37,38,39].
Unfortunately, developing a ME-model requires a deep understanding of the organism’s cellular machinery. For organisms that lack the required annotations, less complex alternatives exist. MOMENT, or Metabolic Modeling with Enzyme Kinetics, was first developed to better predict metabolic fluxes and growth rates by using enzyme turnover rates and molecular weights [50]. GEMs can also be integrated with enzyme constraints using kinetic and omics data, which is also known as GECKO. Since GECKO does not require detailed knowledge about every step in protein synthesis its been applied to other organisms such as the eukaryal Saccharomyces cerevisiae [51,52]. Unlike GECKO, MOMENT does not allow for the direct incorporation of measured enzyme concentrations. Short MOMENT or sMOMENT was recently developed to allow for this type of integration while also greatly reducing the size and complexity of the original MOMENT model [53].
2.2. Communities
Modeling communities or co-cultures are important for healthcare and biotech applications since many times strains are paired based on metabolic coupling. For example, pairing phototrophs with heterotrophs is a promising prospect for sustainable biotechnology, since it enables heterotrophs like Escherichia. coli to grow in minimal media devoid of organic carbon sources. Community metabolic models (CM-models) can be constructed for co-cultures to aid in this strain selection. By modeling and simulating various synthetic microbial co-cultures, researchers are able to identify optimal pairs that produce the most active community [54]. In some cases, dynamic community metabolic models can be generated for co-cultures using each organism’s genome-scale metabolic network. These dynamic simulations are able to predict metabolite concentration profiles for the community as well metabolic exchange flux profiles for individual organisms [55]. Along a similar vein, communities are able to partition metabolic functions among community members like in cases of auxotrophy. These specialized pairings are capable of improving product yield and accomplishing more complex tasks as compared to a single strain [56]. The OptAux algorithm was created to aid in designing auxotrophic strains that need a metabolite cross-feeding co-culture [57].
2.3. Pangenomes and Multistrain Models
Reconstructing GEMs for multiple strains across a single species has enabled a systems-level approach to study and characterize the pan-metabolic capabilities of the species. Pangenomic studies have been accomplished for a wide range of species from Staphylococcus aureus to Klebsiella pneumoniae [58,59,60,61,62,63]. Integration of genomics, phenomics, transcriptomics, and genome-scale modeling for seven commonly used E. coli strains linked molecular features to strain-specific phenotypes. The integrated models showed that certain strains are better suited to produce specific compounds or phenotypes, which has implications for strain selection when choosing a platform strain for microbial engineering [27].
2.4. Gap filling, Discovery, and Annotation
In order to more accurately predict an organism’s phenotype, there needs to be a more complete genome annotation to better understand the organism’s capabilities. Even in E. coli, one of the best-studied model organisms, 35% of its genome lacks functional annotations, with many of these genes being experimentally linked to phenotypes [64]. Recently, there has been a multitude of computational tools that have taken strides in elucidating the possible functions of these genes.
Transposable elements (TEs) are of high interest when engineering a strain due to the deleterious effects they can have if uncontrolled. In platform strains, TEs or insertion sequences (ISs) are often removed in order to preserve the intended genomic content. Issues arise if the organism of interest is poorly characterized and the TEs are not known, however, new machine learning algorithms that utilize genome sequence are able to identify the TEs and ISs in both eukaryotic and prokaryotic species [65,66].
Gap-filling has been commonly used for reconstructing genome-scale metabolic models but over the past few years has quickly advanced in coordination with the recent developments in machine learning. Current methods such as DeepEC are able to predict enzyme commission numbers based on genome sequence with high accuracy [67]. Additionally, machine learning algorithms that use genome sequence can predict systems-wide enzyme promiscuity or candidate genes/enzymes for orphan reactions [68,69]. By filling these annotation gaps and identifying possible cases of promiscuity in microbial networks, researchers will be able to achieve more accurate and comprehensive predictions.
3. Frontier 2: Kinetics and Thermodynamics
Kinetic modeling of metabolism is a field with a long history dating back to the original work understanding enzyme kinetics at the beginning of the 20th century. These ODE-based models offer the promise of mechanistically accounting for the saturation and regulatory state of every enzyme, providing a direct representation of the mechanisms underlying cellular homeostasis [35]. Metabolic control analysis, rooted in steady-state analysis of a kinetic model, paved the way for quantitative analysis of metabolic networks in the early days of metabolic engineering [70]. Sharing much of the same underlying theory, constraint-based thermodynamic models calculate the energetic driving forces underlying metabolic fluxes and can be used to determine physiological constraints on reaction reversibility in the metabolic network [29,71,72,73,74]. Multi-scale models accounting for kinetics and thermodynamics have begun to emerge as well [75,76,77], as discussed in the earlier section on Proteome Allocation Models. The development of kinetic and thermodynamic models has been hampered by the difficulty in acquiring the necessary parameters and validating model behavior at large-scale [78]. Furthermore, the complexity of these models, need for accounting for parameter uncertainty, and additional confounding factors such as numerical instability in kinetic models or constraint infeasibilities in thermodynamic models, substantially increase computational requirements for large-scale modeling. However, recent advances in parameterization and simulation of kinetic and thermodynamic models promise to greatly expand the scope and accuracy of these models.
3.1. Parameterization
The critical step in the construction of a kinetic or thermodynamic model is specifying the values of the necessary parameters. This effort is complicated by the lack of required data which leaves these models largely underdetermined. Algorithms must be developed to fit parameters to available data and account for parameter uncertainty.
For parameterizing kinetic models, a number of approaches are now available: (1) Systems-level fitting, where all parameters of the model are fit simultaneously to systems-level data such as metabolomics and fluxomics [79,80], (2) estimation of kinetic parameters directly from in vivo data without the need of a model [81,82], (3) machine learning to estimate kinetic parameters [83], and (4) bottom-up reconstruction of kinetics on an enzyme by enzyme basis [84]. Similarly, methods to account for parameter uncertainty have advanced through powerful algorithms [85]. Thus, the ‘kinetome’, a genome-scale collection of the kinetic properties of metabolic enzymes, may soon be within reach [86].
In thermodynamic models, the majority of work has focused on estimating the standard Gibbs free energy of reaction, dG0r, which can be readily converted to the reaction equilibrium constant Keq. Experimentally, reaction equilibria are directly measured under a variety of biologically relevant experimental conditions, such as pH, T, IS, and magnesium concentration. To estimate the equilibrium properties of reactions lacking experimental data, the most popular approach has been the group-contribution family of methods [87,88], which has led to software such as eQuilibrator [89]. The ability to correct these estimates accurately for pH [90] and temperature [91] have since been added. However, there are a number of inherent flaws in group-contribution as an estimator, including fundamental limitations of the underlying additivity assumptions [92]. Methods for estimating compound Gibbs energies based on direct quantum chemistry predictions are a promising alternative [93].
3.2. Simulation
Kinetic models also present substantial computational challenges in simulation and analysis. Numerous issues including model stiffness due to poor conditioning, dynamic instability, and complex dynamic properties require sophisticated tools to manage dealing with kinetic models effectively. A number of software packages have emerged to meet this challenge [94,95,96]. Additionally, specialized methods for dealing with large-scale kinetic models have emerged by necessity [23,97,98].
Simulation of the thermodynamic properties of a metabolic network faces a distinct set of challenges. Thermodynamic simulation at the genome-scale, through constraint-based methods such as thermodynamic flux balance analysis (tFBA) [99] and network-embedded thermodynamics (NET) [29], must carefully account for uncertainty in thermodynamic parameters and metabolic concentration constraints. Thermodynamic optimization algorithms, such as tFBA, often involve a mix of integer and linear constraints, and MILP, MIQP, and MINLP algorithms may compute slowly at the genome-scale without efficient solving approaches. Integration of thermodynamic constraints with other biophysical constraints presents further complications due to the non-convexity of the resulting space [100].
4. Frontier 3: 3D Structures
Currently, structures are commonly used for analyzing observed sequence variants or identifying functional sites of a protein for targeted engineering. While structure-guided enzyme design remains a promising application, it is incredibly complex and often coupled to large experimental screens [101,102,103]. Software has been developed to aid in enzyme design, such as the Iterative Protein Redesign and Optimization (IPRO) method, but incorporating enzyme design into a strain design workflow remains difficult due to the required expertise and experimental validation required [104]. On the other hand, integrating protein structures into systems biology has shown promise as a more accessible addition to strain design workflows, but remains a challenge due to differences between the fields causing a steep learning curve. Here, we highlight some of the advances that have lowered the learning curve for using and integrating structures data with systems biology approaches.
4.1. 3D Reconstruction
GEMs have now been expanded to include protein structural information, which has enabled comparative structural proteome analysis between strains and organisms. These GEMs with protein structures (GEM-PROs) allow for a direct mapping of gene to protein structure to phenotype [34,105]. Software has been developed to aid with the construction of high-quality GEM-PROs and to visualize/annotate structures. The pipeline and software, ssbio, is available for use on GitHub (http://github.com/SBRG/ssbio) and is implemented in Python [106]. For cases where a protein is poorly characterized and the necessary mapping fails to exist, homology modeling or tools such as I-TASSER can be used to predict 3D structures and structure-based functional annotations [107]. AlphaFold was also recently announced as the best protein folding solution at the Critical Assessment of protein Structure Prediction (CASP)-14 competition [108]. CASP was founded in 1994 with the goal to establish the state of the art in protein structure prediction based, and AlphaFold 2 has recently achieved predictions competitive with results obtained from experimental methods, something that was once thought to be impossible.
4.2. Applications
Structural information is often used for 3D mutational mapping or visualization but has more capabilities when used in an integrated workflow. For example, GEM-PROs for multiple strains of the same species enables the comparison of sequence variants among conserved genes [105]. Additionally, combining machine learning approaches and 3D structural mutational mapping has identified genetic signatures of antimicrobial resistance evolution to multiple antibiotics in M. tuberculosis [109]. Brunk et al. developed a multiscale workflow to better understand the roles and mechanisms of protein post-translational modifications (PTMs) due to the challenges they present in engineering organisms and their interference with drug action [110]. This workflow incorporates genome-scale modeling, genome editing, and molecular enzyme assays to identify specific roles of PTMs and how they regulate cell phenotype [110]. Well-established software incorporating three-dimensional structural information also exists today. Amber is a package suite of computer programs and has been in development for over 40 years. Amber simulates molecular dynamics for proteins and other biomolecules using structural information and molecular mechanical force fields [111,112].
5. Frontier 4: Genome Sequence and Phenotype Prediction
The genome sequence lies at the heart of a strain design workflow. Heterologous genes must either be added via plasmids with established expression behavior or integrated into the chromosome, and the behavior of these genes depends on the sequence of both coding and non-coding regions of the genes. Further, mutations occur in any mutagenesis or adaptive laboratory evolution strain that control phenotype. Strain design projects may choose between different strains of a species, and the sequence variations between these strains may lead to diverse differences in behavior. Quantifying the sum of sequence factors to predict strain phenotype such as gene expression is an active area of research. Two related strain design tasks utilize the genome sequence: (1) analysis of observed sequence variation data and (2) prediction of phenotype based on genome sequence.
5.1. Sequence Interpretation
A combination of natural, selected, or randomized sequence variants of different genes will typically be observed or generated throughout a typical strain design project. Interpreting the effect of these mutations presents a significant challenge. Natural variants occurring across strains of a species have been analyzed to understand phenotypes related to antimicrobial resistance [109,113,114,115]. Machine learning models can be trained on a phenotype of interest, and key variables extracted to understand which genetic features are important for determining the phenotype [116]. A well-established example of this type of analysis is the identification of protein-DNA binding motifs, for example by the MEME suite [117]. Sequence variants occurring in the course of adaptive laboratory evolution have also been collected [118], and analyzed [119]. This type of sequence analysis is possible due to the recent development of data structures for contextualizing multiple data types within the genome [120]. Further follow up is required to understand the mechanistic basis establishing the relationship between these genetic features and the phenotype.
5.2. Phenotype Prediction
In addition to analysis of observed mutations, the genome sequence can also be used to directly predict strain phenotypes. The genome sequence directly affects the function of the protein product, the mRNA transcription rate, and the protein translation rate of a transcript, termed the translation efficiency. Not surprisingly, models capturing these effects are of great interest. While classically, the assignment of function to a new gene has been done primarily through sequence homology, more sophisticated methods based on machine learning are being developed that have the potential to better capture the sequence-function relationship [67]. Similarly, a number of machine learning and mechanistic models of methods predicting mRNA expression [121] and translation efficiency [122] from promoter and transcript sequence, respectively, have been developed. Other methods predict protein expression directly from sequence, integrating both transcription and translation effects [123]. These methods often are trained on large assays based on synthetic libraries of sequence variants. A key challenge is to achieve general applicability of these models in new experimental conditions and strain backgrounds.
6. Frontier 5: Regulatory Networks
Transcriptional regulatory networks (TRNs) for multiple organisms have been studied rigorously and have made significant advancements with the expansion of ChIP-seq data. Even with this deluge of experimental data, many questions still remain, even for E. coli with one of the best characterized TRNs. Computational approaches combining experimental datasets with machine learning methods have shown promise in further elucidating the underlying regulatory network, even from old low-resolution datasets. These new approaches and insights have opened the door for more accurately integrating metabolic modeling with regulation, a long time goal in the field of computational systems biology. The following will discuss these methods and their implications.
6.1. Regulatory Network Machine Learning Models
A few methods have been developed for further elucidating an organism’s TRN using machine learning. Applying independent component analysis (ICA), an unsupervised machine learning algorithm, to diverse transcriptomics compendia reveal statistically independent signals that modulate the expression of genes. In E. coli, 66% of the 92 identified signals represent the effects of transcriptional regulators, whereas 27% represent biological or genetic explanations. ICA decomposition has also proven effective for other organisms, such as B. subtilis, and S. aureus [28,59,124]. Models for these organisms and more information on the method can be found in the iModulon database (iModulonDB.org) [28,125]. Another method, Transcriptional Regulatory Network Analysis, or TReNA, combines TF/target gene correlations and TF DNA binding information to create gene regulatory predictions. The resulting predictions approach mechanistic accuracy when using high-resolution data collection techniques like single-cell RNA-seq and ChIP-seq, but bulk mRNA data and low-resolution techniques still provide a coarse-grained result. TReNA thus enables researchers to work along this spectrum [126]. While these processes utilize transcriptomics to predict regulators, algorithms also exist to predict gene expression as a function of transcription factor expression levels [127]. Probabilistic regulation of metabolism, or PROM, is a method for integrating TRNs and metabolic models. PROM uses conditional probabilities to represent gene states and gene-TF interactions, as well as FBA for modeling the metabolic network. The method was one of the first to enable automated integration of transcriptional and metabolic networks using high-throughput datasets [128].
6.2. Network Inference
Being able to predict network structure and identify the roles of specific vs. global regulators is necessary for a more complete understanding of how gene expression changes under varying sample conditions. Rustad et al. created a transcription factor overexpression (TFOE) library by cloning and overexpressing 206 TFs in M. tuberculosis. The resulting strains were used to identify sets of genes affected by TFOE and assembled into a global transcriptional map. The TFOE regulatory map identifies potential regulators of gene sets and was used by Rustad et al. to predict and validate the phenotype of a regulator affecting the susceptibility of isoniazid, an antibiotic used to treat tuberculosis [129]. Kochanowski et al. developed computational methods to identify global vs. specific transcriptional regulation based on promoter activity and to discern metabolites that serve as potential transcriptional regulators. The simple mathematical models use high-throughput measurements of metabolite concentrations and promoter activity. A model of E. coli central carbon metabolism showed that 90% of expression changes are due to a few regulatory metabolites (F1P, FBP, and cAMP) and global TFs (Crp and Cra) [130].
7. Drivers of Advances in Computational Tools for Strain Design
The improvements in both scope and accuracy of the computational tools discussed here have been driven by a combination of experimental and machine learning advances. The availability of large-scale datasets that enable comprehensive modeling has exploded. To list a few examples: (1) Continued determination of experimentally-determined protein 3D structures has empowered the rapid development of homology modeling of protein structures, (2) Falling sequencing costs and improved sequencing quality have resulted in a rapid increase of the number, assembly quality, and annotation quality of available microbial genomes, with similar increases to other sequence-based omics types, such as RNA-seq and ribo-seq, and (3) Improved quantitative proteomics datasets have enabled the large-scale estimation of in vivo enzyme turnover rates [81], greatly empowering kinetic and proteome-allocation modeling approaches. Alongside this increased availability of data, machine learning methods to effectively utilize this experimental data to parameterize biological models have greatly improved in recent years. In addition to machine learning algorithms, any expert in machine learning knows the importance of data curation and data cleaning to successful model development. For this reason, the growth and maintenance of highly curated organism-specific knowledgebases, such as RegulonDB and EcoCyc for E. coli, has been critical to providing a source of reliable data for many computational tools [131,132]. Thus, as these companion fields continue to evolve, it is expected that the accuracy of the computational tools built upon these data and algorithms will continue to rapidly improve.
8. Outlook for Synthetic Genome Design
Although there has been substantial progress in each of the individual fields discussed above, there are additional challenges with integrating these tools into an effective strain design workflow. Workflow: While we discussed many tools as they relate to individual strain design tasks, these tasks must be synthesized into a coherent end-to-end design workflow. The decisions of the order of operations in the development of a strain could greatly benefit from computational predictions, but much work is yet to be done to identify a strain design workflow that maximizes efficiency and minimizes cost and risk. Expertise: Any workflow that integrates many different computational tools will require domain expertise in each tool to decide details of implementation, from parameters to valid use cases. Thus, strain designers will be required to have broad computational skillsets that exceed what is taught by most current training programs. Software: The practical difficulty of implementing many separate computational tools can become a substantial burden, spanning various details from licensing issues to file formats. However, the number of software packages enabling these workflows continues to increase, and we mention many examples in this work (Figure 3). Thanks to these efforts, finding compatible tools for easily integrated workflows is becoming easier. Validation: Tools must be validated to clearly established accuracy metrics under physiological conditions. Validation of tools on individual datasets, for example on a single wild type strain background, is likely to be insufficient as the strain is engineered further from the wild type. To meet these challenges, it is critical to take a systematic approach that includes dedicated training, effective documentation of tools, and extensive validation of tools in real applications. There will be a significant challenge reaching a standard where strain design researchers can effectively conduct analyses and understand results from multiple tools across a typical workflow.
The field is nearing an important milestone in synthetic biology, that of the comprehensive and computationally-driven strain design workflow. We may soon enter an era of ‘computational genome design’, where rational approaches finally give way to biological design algorithms dominated by computational predictions. Thus, one of the early promises of the field of systems biology may finally be nearing its realization. The practical applications of such a cell design workflow are endless, from the chemical industry to the environment to human health.
Author Contributions
Conceptualization, D.C.Z., B.O.P.; writing—original draft preparation, D.C.Z., A.P., B.O.P.; writing—review and editing, D.C.Z., A.P., B.O.P.; visualization, D.C.Z., A.P.; funding acquisition, B.O.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Novo Nordisk Foundation, grant NNF10CC1016517.
Acknowledgments
We would like to thank Patrick Phaneuf and Tobias Alter for helpful comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liao, J.C.; Mi, L.; Pontrelli, S.; Luo, S. Fuelling the Future: Microbial Engineering for the Production of Sustainable Biofuels. Nat. Rev. Microbiol. 2016, 14, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, H.U.; Chae, T.U.; Cho, J.S.; Kim, J.W.; Shin, J.H.; Kim, D.I.; Ko, Y.-S.; Jang, W.D.; Jang, Y.-S. A Comprehensive Metabolic Map for Production of Bio-Based Chemicals. Nat. Catal. 2019, 2, 18–33. [Google Scholar] [CrossRef]
- Kalia, V.C.; Saini, A.K. (Eds.) Metabolic Engineering for Bioactive Compounds: Strategies and Processes; Springer: Singapore, 2017. [Google Scholar]
- Matassa, S.; Boon, N.; Pikaar, I.; Verstraete, W. Microbial Protein: Future Sustainable Food Supply Route with Low Environmental Footprint. Microb. Biotechnol. 2016, 9, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dash, H.R. 1—Microbial Bioremediation: A Potential Tool for Restoration of Contaminated Areas. In Microbial Biodegradation and Bioremediation; Das, S., Ed.; Elsevier: Oxford, UK, 2014; pp. 1–21. [Google Scholar]
- Bereza-Malcolm, L.T.; Mann, G.; Franks, A.E. Environmental Sensing of Heavy Metals Through Whole Cell Microbial Biosensors: A Synthetic Biology Approach. ACS Synth. Biol. 2015, 4, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Danino, T.; Mondragón-Palomino, O.; Tsimring, L.; Hasty, J. A Synchronized Quorum of Genetic Clocks. Nature 2010, 463, 326–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Din, M.O.; Danino, T.; Prindle, A.; Skalak, M.; Selimkhanov, J.; Allen, K.; Julio, E.; Atolia, E.; Tsimring, L.S.; Bhatia, S.N.; et al. Synchronized Cycles of Bacterial Lysis for in Vivo Delivery. Nature 2016, 536, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.; Singh, P.K.; Shukla, P. Metabolic Engineering for Probiotics and Their Genome-Wide Expression Profiling. Curr. Protein Pept. Sci. 2018, 19, 68–74. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, H.U. Systems Strategies for Developing Industrial Microbial Strains. Nat. Biotechnol. 2015, 33, 1061–1072. [Google Scholar] [CrossRef]
- St. John, P.C.; Bomble, Y.J. Approaches to Computational Strain Design in the Multiomics Era. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Brophy, J.A.N.; Voigt, C.A. Principles of Genetic Circuit Design. Nat. Methods 2014, 11, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, A.A.K.; Der, B.S.; Shin, J.; Vaidyanathan, P.; Paralanov, V.; Strychalski, E.A.; Ross, D.; Densmore, D.; Voigt, C.A. Genetic Circuit Design Automation. Science 2016, 352, aac7341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlmayer, F.; Aubel, D.; Fussenegger, M. Synthetic Gene Circuits for the Detection, Elimination and Prevention of Disease. Nat. Biomed. Eng. 2018, 2, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.S.; Collins, J.J. Synthetic Biology: Applications Come of Age. Nat. Rev. Genet. 2010, 11, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Kim, H.U.; Lee, S.Y. Current State and Applications of Microbial Genome-Scale Metabolic Models. Curr. Opin. Syst. Biol. 2017, 2, 10–18. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and Analysis of Biochemical Constraint-Based Models Using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A Systematic Assessment of Current Genome-Scale Metabolic Reconstruction Tools. Genome Biol. 2019, 20, 158. [Google Scholar] [CrossRef] [Green Version]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for Standardized Genome-Scale Metabolic Model Testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, C.J.; Ebrahim, A.; Yang, L.; King, Z.A.; Catoiu, E.; O’Brien, E.J.; Liu, J.K.; Palsson, B.O. COBRAme: A Computational Framework for Genome-Scale Models of Metabolism and Gene Expression. PLoS Comput. Biol. 2018, 14, e1006302. [Google Scholar] [CrossRef]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using Genome-Scale Models to Predict Biological Capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, E.J.; Lerman, J.A.; Chang, R.L.; Hyduke, D.R.; Palsson, B.Ø. Genome-Scale Models of Metabolism and Gene Expression Extend and Refine Growth Phenotype Prediction. Mol. Syst. Biol. 2013, 9, 693. [Google Scholar] [CrossRef]
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B., Jr.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A Whole-Cell Computational Model Predicts Phenotype from Genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCloskey, D.; Palsson, B.Ø.; Feist, A.M. Basic and Applied Uses of Genome-Scale Metabolic Network Reconstructions of Escherichia Coli. Mol. Syst. Biol. 2013, 9, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richelle, A.; David, B.; Demaegd, D.; Dewerchin, M.; Kinet, R.; Morreale, A.; Portela, R.; Zune, Q.; von Stosch, M. Towards a Widespread Adoption of Metabolic Modeling Tools in Biopharmaceutical Industry: A Process Systems Biology Engineering Perspective. NPJ Syst. Biol. Appl. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, S.; Chakrabarti, A.; Soh, K.C.; Burgard, A.; Yang, T.H.; Van Dien, S.; Miskovic, L.; Hatzimanikatis, V. Identification of Metabolic Engineering Targets for the Enhancement of 1,4-Butanediol Production in Recombinant E. Coli Using Large-Scale Kinetic Models. Metab. Eng. 2016, 35, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, J.M.; Koza, A.; Campodonico, M.A.; Machado, D.; Seoane, J.M.; Palsson, B.O.; Herrgård, M.J.; Feist, A.M. Multi-Omics Quantification of Species Variation of Escherichia Coli Links Molecular Features with Strain Phenotypes. Cell Syst. 2016, 3, 238–251.e12. [Google Scholar] [CrossRef] [Green Version]
- Sastry, A.V.; Gao, Y.; Szubin, R.; Hefner, Y.; Xu, S.; Kim, D.; Choudhary, K.S.; Yang, L.; King, Z.A.; Palsson, B.O. The Escherichia Coli Transcriptome Mostly Consists of Independently Regulated Modules. Nat. Commun. 2019, 10, 5536. [Google Scholar] [CrossRef] [Green Version]
- Kümmel, A.; Panke, S.; Heinemann, M. Putative Regulatory Sites Unraveled by Network-Embedded Thermodynamic Analysis of Metabolome Data. Mol. Syst. Biol. 2006, 2, 2006.0034. [Google Scholar] [CrossRef] [Green Version]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A Bilevel Programming Framework for Identifying Gene Knockout Strategies for Microbial Strain Optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- De Groot, D.H.; Lischke, J.; Muolo, R.; Planqué, R.; Bruggeman, F.J.; Teusink, B. The Common Message of Constraint-Based Optimization Approaches: Overflow Metabolism Is Caused by Two Growth-Limiting Constraints. Cell. Mol. Life Sci. 2020, 77, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Zrimec, J.; Börlin, C.S.; Buric, F.; Muhammad, A.S.; Chen, R.; Siewers, V.; Verendel, V.; Nielsen, J.; Töpel, M.; Zelezniak, A. Deep Learning Suggests That Gene Expression Is Encoded in All Parts of a Co-Evolving Interacting Gene Regulatory Structure. Nat. Commun. 2020, 11, 6141. [Google Scholar] [CrossRef]
- Kotte, O.; Zaugg, J.B.; Heinemann, M. Bacterial Adaptation through Distributed Sensing of Metabolic Fluxes. Mol. Syst. Biol. 2010, 6, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunk, E.; Mih, N.; Monk, J.; Zhang, Z.; O’Brien, E.J.; Bliven, S.E.; Chen, K.; Chang, R.L.; Bourne, P.E.; Palsson, B.O. Systems Biology of the Structural Proteome. BMC Syst. Biol. 2016, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.D.; Rocha, M.; Maia, P. A Review of Dynamic Modeling Approaches and Their Application in Computational Strain Optimization for Metabolic Engineering. Front. Microbiol. 2018, 9, 1690. [Google Scholar] [CrossRef] [PubMed]
- Jabarivelisdeh, B.; Waldherr, S. Optimization of Bioprocess Productivity Based on Metabolic-Genetic Network Models with Bilevel Dynamic Programming. Biotechnol. Bioeng. 2018, 115, 1829–1841. [Google Scholar] [CrossRef]
- Chen, K.; Gao, Y.; Mih, N.; O’Brien, E.J.; Yang, L.; Palsson, B.O. Thermosensitivity of Growth Is Determined by Chaperone-Mediated Proteome Reallocation. Proc. Natl. Acad. Sci. USA 2017, 114, 11548–11553. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Yang, L.; Lloyd, C.J.; Fang, X.; Palsson, B.O. Genome-Scale Model of Metabolism and Gene Expression Provides a Multi-Scale Description of Acid Stress Responses in Escherichia Coli. PLoS Comput. Biol. 2019, 15, e1007525. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Mih, N.; Anand, A.; Park, J.H.; Tan, J.; Yurkovich, J.T.; Monk, J.M.; Lloyd, C.J.; Sandberg, T.E.; Seo, S.W.; et al. Cellular Responses to Reactive Oxygen Species Are Predicted from Molecular Mechanisms. Proc. Natl. Acad. Sci. USA 2019, 116, 14368–14373. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Haringa, C.; Tang, W.; Noorman, H.; Chu, J.; Zhuang, Y.; Zhang, S. Coupled Metabolic-Hydrodynamic Modeling Enabling Rational Scale-up of Industrial Bioprocesses. Biotechnol. Bioeng. 2020, 117, 844–867. [Google Scholar] [CrossRef]
- Monk, J.; Nogales, J.; Palsson, B.O. Optimizing Genome-Scale Network Reconstructions. Nat. Biotechnol. 2014, 32, 447–452. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst. Biol. 2013, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, J.G.R.; Jensen, K.; Lieven, C.; Lærke Hansen, A.S.; Galkina, S.; Beber, M.; Özdemir, E.; Herrgård, M.J.; Redestig, H.; Sonnenschein, N. Cameo: A Python Library for Computer Aided Metabolic Engineering and Optimization of Cell Factories. ACS Synth. Biol. 2018, 7, 1163–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-Based Models Predict Metabolic and Associated Cellular Functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef]
- Lerman, J.A.; Hyduke, D.R.; Latif, H.; Portnoy, V.A.; Lewis, N.E.; Orth, J.D.; Schrimpe-Rutledge, A.C.; Smith, R.D.; Adkins, J.N.; Zengler, K.; et al. In Silico Method for Modelling Metabolism and Gene Product Expression at Genome Scale. Nat. Commun. 2012, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.K.; Lloyd, C.; Al-Bassam, M.M.; Ebrahim, A.; Kim, J.-N.; Olson, C.; Aksenov, A.; Dorrestein, P.; Zengler, K. Predicting Proteome Allocation, Overflow Metabolism, and Metal Requirements in a Model Acetogen. PLoS Comput. Biol. 2019, 15, e1006848. [Google Scholar] [CrossRef] [PubMed]
- Dinh, H.V.; King, Z.A.; Palsson, B.O.; Feist, A.M. Identification of Growth-Coupled Production Strains Considering Protein Costs and Kinetic Variability. Metab. Eng. Commun. 2018, 7, e00080. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; O’Brien, E.J.; Feist, A.M.; Palsson, B.O. Literature Mining Supports a next-Generation Modeling Approach to Predict Cellular Byproduct Secretion. Metab. Eng. 2017, 39, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Ebrahim, A.; Lloyd, C.J.; Saunders, M.A.; Palsson, B.O. DynamicME: Dynamic Simulation and Refinement of Integrated Models of Metabolism and Protein Expression. BMC Syst. Biol. 2019, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Adadi, R.; Volkmer, B.; Milo, R.; Heinemann, M.; Shlomi, T. Prediction of Microbial Growth Rate versus Biomass Yield by a Metabolic Network with Kinetic Parameters. PLoS Comput. Biol. 2012, 8, e1002575. [Google Scholar] [CrossRef] [Green Version]
- Massaiu, I.; Pasotti, L.; Sonnenschein, N.; Rama, E.; Cavaletti, M.; Magni, P.; Calvio, C.; Herrgård, M.J. Integration of Enzymatic Data in Bacillus Subtilis Genome-Scale Metabolic Model Improves Phenotype Predictions and Enables in Silico Design of Poly-γ-Glutamic Acid Production Strains. Microb. Cell Fact. 2019, 18, 3. [Google Scholar] [CrossRef]
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.-J.; Kerkhoven, E.J.; Nielsen, J. Improving the Phenotype Predictions of a Yeast Genome-Scale Metabolic Model by Incorporating Enzymatic Constraints. Mol. Syst. Biol. 2017, 13, 935. [Google Scholar] [CrossRef]
- Bekiaris, P.S.; Klamt, S. Automatic Construction of Metabolic Models with Enzyme Constraints. BMC Bioinformatics 2020, 21, 19. [Google Scholar] [CrossRef] [PubMed]
- Zuñiga, C.; Li, T.; Guarnieri, M.T.; Jenkins, J.P.; Li, C.-T.; Bingol, K.; Kim, Y.-M.; Betenbaugh, M.J.; Zengler, K. Synthetic Microbial Communities of Heterotrophs and Phototrophs Facilitate Sustainable Growth. Nat. Commun. 2020, 11, 3803. [Google Scholar] [CrossRef] [PubMed]
- Özcan, E.; Seven, M.; Şirin, B.; Çakır, T.; Nikerel, E.; Teusink, B.; Toksoy Öner, E. Dynamic Co-Culture Metabolic Models Reveal the Fermentation Dynamics, Metabolic Capacities and Interplays of Cheese Starter Cultures. Biotechnol. Bioeng. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Qiao, K.; Edgar, S.; Stephanopoulos, G. Distributing a Metabolic Pathway among a Microbial Consortium Enhances Production of Natural Products. Nat. Biotechnol. 2015, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.J.; King, Z.; Sandberg, T.; Hefner, Y.; Feist, A. Model-Driven Design and Evolution of Non-Trivial Synthetic Syntrophic Pairs. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Monk, J.M.; Charusanti, P.; Aziz, R.K.; Lerman, J.A.; Premyodhin, N.; Orth, J.D.; Feist, A.M.; Palsson, B.Ø. Genome-Scale Metabolic Reconstructions of Multiple Escherichia Coli Strains Highlight Strain-Specific Adaptations to Nutritional Environments. Proc. Natl. Acad. Sci. USA 2013, 110, 20338–20343. [Google Scholar] [CrossRef] [Green Version]
- Poudel, S.; Tsunemoto, H.; Seif, Y.; Sastry, A.V.; Szubin, R.; Xu, S.; Machado, H.; Olson, C.A.; Anand, A.; Pogliano, J.; et al. Revealing 29 Sets of Independently Modulated Genes in Staphylococcus Aureus, Their Regulators, and Role in Key Physiological Response. Proc. Natl. Acad. Sci. USA 2020, 117, 17228–17239. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Attia, H.; Szubin, R.; Yassin, A.S.; Palsson, B.Ø.; Aziz, R.K.; Monk, J.M. Comparative Genome-Scale Metabolic Modeling of Metallo-Beta-Lactamase-Producing Multidrug-Resistant Klebsiella Pneumoniae Clinical Isolates. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef]
- Seif, Y.; Kavvas, E.; Lachance, J.-C.; Yurkovich, J.T.; Nuccio, S.-P.; Fang, X.; Catoiu, E.; Raffatellu, M.; Palsson, B.O.; Monk, J.M. Genome-Scale Metabolic Reconstructions of Multiple Salmonella Strains Reveal Serovar-Specific Metabolic Traits. Nat. Commun. 2018, 9, 3771. [Google Scholar] [CrossRef] [Green Version]
- Prigent, S.; Nielsen, J.C.; Frisvad, J.C.; Nielsen, J. Reconstruction of 24 Penicillium Genome-Scale Metabolic Models Shows Diversity Based on Their Secondary Metabolism. Biotechnol. Bioeng. 2018, 115, 2604–2612. [Google Scholar] [CrossRef]
- Fouts, D.E.; Matthias, M.A.; Adhikarla, H.; Adler, B.; Amorim-Santos, L.; Berg, D.E.; Bulach, D.; Buschiazzo, A.; Chang, Y.-F.; Galloway, R.L.; et al. What Makes a Bacterial Species Pathogenic?: Comparative Genomic Analysis of the Genus Leptospira. PLoS Negl. Trop. Dis. 2016, 10, e0004403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatak, S.; King, Z.A.; Sastry, A.; Palsson, B.O. The Y-Ome Defines the 35% of Escherichia Coli Genes That Lack Experimental Evidence of Function. Nucleic Acids Res. 2019, 47, 2446–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Tang, H. ISEScan: Automated Identification of Insertion Sequence Elements in Prokaryotic Genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep Learning Enables High-Quality and High-Throughput Prediction of Enzyme Commission Numbers. Proc. Natl. Acad. Sci. USA 2019, 116, 13996–14001. [Google Scholar] [CrossRef] [Green Version]
- Hadadi, N.; MohammadiPeyhani, H.; Miskovic, L.; Seijo, M.; Hatzimanikatis, V. Enzyme Annotation for Orphan and Novel Reactions Using Knowledge of Substrate Reactive Sites. Proc. Natl. Acad. Sci. USA 2019, 116, 7298–7307. [Google Scholar] [CrossRef] [Green Version]
- Oberhardt, M.A.; Zarecki, R.; Reshef, L.; Xia, F.; Duran-Frigola, M.; Schreiber, R.; Henry, C.S.; Ben-Tal, N.; Dwyer, D.J.; Gophna, U.; et al. Systems-Wide Prediction of Enzyme Promiscuity Reveals a New Underground Alternative Route for Pyridoxal 5’-Phosphate Production in E. Coli. PLoS Comput. Biol. 2016, 12, e1004705. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Saavedra, E.; Rodríguez-Enríquez, S.; Olín-Sandoval, V. Metabolic Control Analysis: A Tool for Designing Strategies to Manipulate Metabolic Pathways. J. Biomed. Biotechnol. 2008, 2008. [Google Scholar] [CrossRef] [Green Version]
- Noor, E.; Flamholz, A.; Liebermeister, W.; Bar-Even, A.; Milo, R. A Note on the Kinetics of Enzyme Action: A Decomposition That Highlights Thermodynamic Effects. FEBS Lett. 2013, 587, 2772–2777. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Thermodynamics-Based Metabolic Flux Analysis. Biophys. J. 2007, 92, 1792–1805. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.J.; Dwivedi, V.; Reed, J.L. Quantitative Assessment of Thermodynamic Constraints on the Solution Space of Genome-Scale Metabolic Models. Biophys. J. 2013, 105, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, B.; Zielinski, D.C.; Monk, J.M.; Palsson, B.O. Thermodynamic Favorability and Pathway Yield as Evolutionary Tradeoffs in Biosynthetic Pathway Choice. Proc. Natl. Acad. Sci. USA 2018, 115, 11339–11344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvy, P.; Hatzimanikatis, V. ETFL: A Formulation for Flux Balance Models Accounting for Expression, Thermodynamics, and Resource Allocation Constraints. bioRxiv 2019. [Google Scholar] [CrossRef]
- Salvy, P.; Hatzimanikatis, V. Emergence of Diauxie as an Optimal Growth Strategy under Resource Allocation Constraints in Cellular Metabolism. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pandey, V.; Hadadi, N.; Hatzimanikatis, V. Enhanced Flux Prediction by Integrating Relative Expression and Relative Metabolite Abundance into Thermodynamically Consistent Metabolic Models. PLoS Comput. Biol. 2019, 15, e1007036. [Google Scholar] [CrossRef] [Green Version]
- Saa, P.A.; Nielsen, L.K. Formulation, Construction and Analysis of Kinetic Models of Metabolism: A Review of Modelling Frameworks. Biotechnol. Adv. 2017, 35, 981–1003. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, S.; Dash, S.; Maranas, C. K-FIT: An Accelerated Kinetic Parameterization Algorithm Using Steady-State Fluxomic Data. Metab. Eng. 2020, 61, 197–205. [Google Scholar] [CrossRef]
- Saa, P.A.; Nielsen, L.K. Construction of Feasible and Accurate Kinetic Models of Metabolism: A Bayesian Approach. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Davidi, D.; Noor, E.; Liebermeister, W.; Bar-Even, A.; Flamholz, A.; Tummler, K.; Barenholz, U.; Goldenfeld, M.; Shlomi, T.; Milo, R. Global Characterization of in Vivo Enzyme Catalytic Rates and Their Correspondence to in Vitro Kcat Measurements. Proc. Natl. Acad. Sci. USA 2016, 113, 3401–3406. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, D.; Campeau, A.; Lloyd, C.J.; Phaneuf, P.V.; Hefner, Y.; Carrillo-Terrazas, M.; Feist, A.M.; Gonzalez, D.J.; Palsson, B.O. Kinetic Profiling of Metabolic Specialists Demonstrates Stability and Consistency of in Vivo Enzyme Turnover Numbers. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- Heckmann, D.; Lloyd, C.J.; Mih, N.; Ha, Y.; Zielinski, D.C.; Haiman, Z.B.; Desouki, A.A.; Lercher, M.J.; Palsson, B.O. Machine Learning Applied to Enzyme Turnover Numbers Reveals Protein Structural Correlates and Improves Metabolic Models. Nat. Commun. 2018, 9, 5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, D.A.; Vinnakota, K.C.; Wu, F. Detailed Enzyme Kinetics in Terms of Biochemical Species: Study of Citrate Synthase. PLoS ONE 2008, 3, e1825. [Google Scholar] [CrossRef] [Green Version]
- Andreozzi, S.; Miskovic, L.; Hatzimanikatis, V. iSCHRUNK—In Silico Approach to Characterization and Reduction of Uncertainty in the Kinetic Models of Genome-Scale Metabolic Networks. Metab. Eng. 2016, 33, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, A.; Nielsen, J.; Palsson, B.O. Metabolic Models of Protein Allocation Call for the Kinetome. Cell Syst. 2017, 5, 538–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankowski, M.D.; Henry, C.S.; Broadbelt, L.J.; Hatzimanikatis, V. Group Contribution Method for Thermodynamic Analysis of Complex Metabolic Networks. Biophys. J. 2008, 95, 1487–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noor, E.; Haraldsdóttir, H.S.; Milo, R.; Fleming, R.M.T. Consistent Estimation of Gibbs Energy Using Component Contributions. PLoS Comput. Biol. 2013, 9, e1003098. [Google Scholar] [CrossRef] [Green Version]
- Flamholz, A.; Noor, E.; Bar-Even, A.; Milo, R. eQuilibrator—the Biochemical Thermodynamics Calculator. Nucleic Acids Res. 2012, 40, D770–D775. [Google Scholar] [CrossRef] [Green Version]
- Noor, E.; Bar-Even, A.; Flamholz, A.; Lubling, Y.; Davidi, D.; Milo, R. An Integrated Open Framework for Thermodynamics of Reactions That Combines Accuracy and Coverage. Bioinformatics 2012, 28, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Zhang, Z.; Grubner, S.; Yurkovich, J.T.; Palsson, B.O.; Zielinski, D.C. Temperature-Dependent Estimation of Gibbs Energies Using an Updated Group-Contribution Method. Biophys. J. 2018, 114, 2691–2702. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Zielinski, D.C.; Palsson, B.O. Estimating Metabolic Equilibrium Constants: Progress and Future Challenges. Trends Biochem. Sci. 2018, 43, 960–969. [Google Scholar] [CrossRef]
- Jinich, A.; Rappoport, D.; Dunn, I.; Sanchez-Lengeling, B.; Olivares-Amaya, R.; Noor, E.; Even, A.B.; Aspuru-Guzik, A. Quantum Chemical Approach to Estimating the Thermodynamics of Metabolic Reactions. Sci. Rep. 2014, 4, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiman, Z.B.; Zielinski, D.C.; Koike, Y.; Yurkovich, J.T.; Palsson, B.O. MASSpy: Building, Simulating, and Visualizing Dynamic Biological Models in Python Using Mass Action Kinetics. bioRxiv 2020. [Google Scholar] [CrossRef]
- Salvy, P.; Fengos, G.; Ataman, M.; Pathier, T.; Soh, K.C.; Hatzimanikatis, V. pyTFA and matTFA: A Python Package and a Matlab Toolbox for Thermodynamics-Based Flux Analysis. Bioinformatics 2019, 35, 167–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [Green Version]
- Khodayari, A.; Zomorrodi, A.R.; Liao, J.C.; Maranas, C.D. A Kinetic Model of Escherichia Coli Core Metabolism Satisfying Multiple Sets of Mutant Flux Data. Metab. Eng. 2014, 25, 50–62. [Google Scholar] [CrossRef]
- Tokic, M.; Hatzimanikatis, V.; Miskovic, L. Large-Scale Kinetic Metabolic Models of Pseudomonas Putida KT2440 for Consistent Design of Metabolic Engineering Strategies. Biotechnol. Biofuels 2020, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Soh, K.C.; Hatzimanikatis, V. Constraining the Flux Space Using Thermodynamics and Integration of Metabolomics Data. Methods Mol. Biol. 2014, 1191, 49–63. [Google Scholar]
- Akbari, A.; Palsson, B.O. Scalable Computation of Intracellular Metabolite Concentrations. arXiv 2020. [Google Scholar] [CrossRef]
- Chowdhury, R.; Ren, T.; Shankla, M.; Decker, K.; Grisewood, M.; Prabhakar, J.; Baker, C.; Golbeck, J.H.; Aksimentiev, A.; Kumar, M.; et al. PoreDesigner for Tuning Solute Selectivity in a Robust and Highly Permeable Outer Membrane Pore. Nat. Commun. 2018, 9, 3661. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.-S.; Boyken, S.E.; Baker, D. The Coming of Age of de Novo Protein Design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed. Engl. 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantazes, R.J.; Grisewood, M.J.; Li, T.; Gifford, N.P.; Maranas, C.D. The Iterative Protein Redesign and Optimization (IPRO) Suite of Programs. J. Comput. Chem. 2015, 36, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Monk, J.M.; Lloyd, C.J.; Brunk, E.; Mih, N.; Sastry, A.; King, Z.; Takeuchi, R.; Nomura, W.; Zhang, Z.; Mori, H.; et al. iML1515, a Knowledgebase That Computes Escherichia Coli Traits. Nat. Biotechnol. 2017, 35, 904–908. [Google Scholar] [CrossRef]
- Mih, N.; Brunk, E.; Chen, K.; Catoiu, E.; Sastry, A.; Kavvas, E.; Monk, J.M.; Zhang, Z.; Palsson, B.O. Ssbio: A Python Framework for Structural Systems Biology. Bioinformatics 2018, 34, 2155–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Kavvas, E.S.; Catoiu, E.; Mih, N.; Yurkovich, J.T.; Seif, Y.; Dillon, N.; Heckmann, D.; Anand, A.; Yang, L.; Nizet, V.; et al. Machine Learning and Structural Analysis of Mycobacterium Tuberculosis Pan-Genome Identifies Genetic Signatures of Antibiotic Resistance. Nat. Commun. 2018, 9, 4306. [Google Scholar] [CrossRef] [Green Version]
- Brunk, E.; Chang, R.L.; Xia, J.; Hefzi, H.; Yurkovich, J.T.; Kim, D.; Buckmiller, E.; Wang, H.H.; Cho, B.-K.; Yang, C.; et al. Characterizing Posttranslational Modifications in Prokaryotic Metabolism Using a Multiscale Workflow. Proc. Natl. Acad. Sci. USA 2018, 115, 11096–11101. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Kavvas, E.S.; Yang, L.; Monk, J.M.; Heckmann, D.; Palsson, B.O. A Biochemically-Interpretable Machine Learning Classifier for Microbial GWAS. Nat. Commun. 2020, 11, 2580. [Google Scholar] [CrossRef]
- Davis, J.J.; Boisvert, S.; Brettin, T.; Kenyon, R.W.; Mao, C.; Olson, R.; Overbeek, R.; Santerre, J.; Shukla, M.; Wattam, A.R.; et al. Antimicrobial Resistance Prediction in PATRIC and RAST. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A Deep Learning Approach for Predicting Antibiotic Resistance Genes from Metagenomic Data. Microbiome 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugen, S.P.; Ross, W.; Gourse, R.L. Advances in Bacterial Promoter Recognition and Its Control by Factors That Do Not Bind DNA. Nat. Rev. Microbiol. 2008, 6, 507–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Phaneuf, P.V.; Gosting, D.; Palsson, B.O.; Feist, A.M. ALEdb 1.0: A Database of Mutations from Adaptive Laboratory Evolution Experimentation. Nucleic Acids Res. 2019, 47, D1164–D1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phaneuf, P.V.; Yurkovich, J.T.; Heckmann, D.; Wu, M.; Sandberg, T.E.; King, Z.A.; Tan, J.; Palsson, B.O.; Feist, A.M. Causal Mutations from Adaptive Laboratory Evolution Are Outlined by Multiple Scales of Genome Annotations and Condition-Specificity. BMC Genom. 2020, 21, 514. [Google Scholar] [CrossRef]
- Lamoureux, C.R.; Choudhary, K.S.; King, Z.A.; Sandberg, T.E.; Gao, Y.; Sastry, A.V.; Phaneuf, P.V.; Choe, D.; Cho, B.-K.; Palsson, B.O. The Bitome: Digitized Genomic Features Reveal Fundamental Genome Organization. Nucleic Acids Res. 2020, 48, 10157–10163. [Google Scholar] [CrossRef]
- Einav, T.; Phillips, R. How the Avidity of Polymerase Binding to the –35/–10 Promoter Sites Affects Gene Expression. Proc. Natl. Acad. Sci. USA 2019, 116, 13340–13345. [Google Scholar] [CrossRef] [Green Version]
- Tuller, T.; Waldman, Y.Y.; Kupiec, M.; Ruppin, E. Translation Efficiency Is Determined by Both Codon Bias and Folding Energy. Proc. Natl. Acad. Sci. USA 2010, 107, 3645–3650. [Google Scholar] [CrossRef] [Green Version]
- Bonde, M.T.; Pedersen, M.; Klausen, M.S.; Jensen, S.I.; Wulff, T.; Harrison, S.; Nielsen, A.T.; Herrgård, M.J.; Sommer, M.O.A. Predictable Tuning of Protein Expression in Bacteria. Nat. Methods 2016, 13, 233–236. [Google Scholar] [CrossRef]
- Rychel, K.; Sastry, A.V.; Palsson, B.O. Machine Learning Uncovers Independently Regulated Modules in the Bacillus Subtilis Transcriptome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rychel, K.; Decker, K.; Sastry, A.V.; Phaneuf, P.V.; Poudel, S.; Palsson, B.O. iModulonDB: A Knowledgebase of Microbial Transcriptional Regulation Derived from Machine Learning. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ament, S.; Shannon, P.; Richards, M. TReNa: Fit. Transcriptional Regulatory Networks Using Gene Expression, Priors, Machine Learning; Bioconductor: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Fang, X.; Sastry, A.; Mih, N.; Kim, D.; Tan, J.; Yurkovich, J.T.; Lloyd, C.J.; Gao, Y.; Yang, L.; Palsson, B.O. Global Transcriptional Regulatory Network for Escherichia Coli Robustly Connects Gene Expression to Transcription Factor Activities. Proc. Natl. Acad. Sci. USA 2017, 114, 10286–10291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, S.; Price, N.D. Probabilistic Integrative Modeling of Genome-Scale Metabolic and Regulatory Networks in Escherichia Coli and Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef] [Green Version]
- Rustad, T.R.; Minch, K.J.; Ma, S.; Winkler, J.K.; Hobbs, S.; Hickey, M.; Brabant, W.; Turkarslan, S.; Price, N.D.; Baliga, N.S.; et al. Mapping and Manipulating the Mycobacterium Tuberculosis Transcriptome Using a Transcription Factor Overexpression-Derived Regulatory Network. Genom. Biol. 2014, 15, 502. [Google Scholar] [CrossRef]
- Kochanowski, K.; Gerosa, L.; Brunner, S.F.; Christodoulou, D.; Nikolaev, Y.V.; Sauer, U. Few Regulatory Metabolites Coordinate Expression of Central Metabolic Genes in Escherichia Coli. Mol. Syst. Biol. 2017, 13, 903. [Google Scholar] [CrossRef]
- Santos-Zavaleta, A.; Salgado, H.; Gama-Castro, S.; Sánchez-Pérez, M.; Gómez-Romero, L.; Ledezma-Tejeida, D.; García-Sotelo, J.S.; Alquicira-Hernández, K.; Muñiz-Rascado, L.J.; Peña-Loredo, P.; et al. RegulonDB v 10.5: Tackling Challenges to Unify Classic and High Throughput Knowledge of Gene Regulation in E. Coli K-12. Nucleic Acids Res. 2019, 47, D212–D220. [Google Scholar] [CrossRef] [Green Version]
- Keseler, I.M.; Mackie, A.; Santos-Zavaleta, A.; Billington, R.; Bonavides-Martínez, C.; Caspi, R.; Fulcher, C.; Gama-Castro, S.; Kothari, A.; Krummenacker, M.; et al. The EcoCyc Database: Reflecting New Knowledge about Escherichia Coli K-12. Nucleic Acids Res. 2017, 45, D543–D550. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Challenges and computational solutions for a typical strain design workflow. (A) Typical experimental steps in the development of a new strain design. (B) Common challenges encountered at each strain design step. (C) Computational tools that may be used to meet the strain design challenges. Note that the design steps, challenges, and computational tools highlighted here are intended to be exemplative rather than comprehensive. 1, Modeling organism capabilities [27]; 2, Network reconstruction [18]; 3, Top-down data-driven regulons [28]; 4, Kinetic and COBRA modeling [26]; 5, Kinetic and thermodynamic models [29]; 6, COBRA modeling of gene knockouts [30]; 7, Overflow models [31]; 8, Expression tuning ML models [32]; 9, Kinetic models including regulation [33]; 10, Protein structural analysis [34]; 11, Models using enzyme kinetics [35]; 12, Bioprocess models [36]; 13, StressME models [37,38,39]; 14, Analysis of bioreactor omics data [40].
Figure 1.
Challenges and computational solutions for a typical strain design workflow. (A) Typical experimental steps in the development of a new strain design. (B) Common challenges encountered at each strain design step. (C) Computational tools that may be used to meet the strain design challenges. Note that the design steps, challenges, and computational tools highlighted here are intended to be exemplative rather than comprehensive. 1, Modeling organism capabilities [27]; 2, Network reconstruction [18]; 3, Top-down data-driven regulons [28]; 4, Kinetic and COBRA modeling [26]; 5, Kinetic and thermodynamic models [29]; 6, COBRA modeling of gene knockouts [30]; 7, Overflow models [31]; 8, Expression tuning ML models [32]; 9, Kinetic models including regulation [33]; 10, Protein structural analysis [34]; 11, Models using enzyme kinetics [35]; 12, Bioprocess models [36]; 13, StressME models [37,38,39]; 14, Analysis of bioreactor omics data [40].

Figure 2.
Overview of frontiers in the computational design of synthetic organisms. Frontier 1: Constraint-based Reconstruction and Modeling, consisting of tools for analyzing pan-genomes, microbial communities, gap-filling metabolic networks, and modeling proteome allocation. Frontier 2: Kinetics and Thermodynamics, consisting of tools for parameterizing and simulating kinetic and thermodynamic models. Parameterization can utilize the Michaelis-Menten equation where [A] is the substrate concentration, whereas simulation uses dynamic mass balance equations where S is the stoichiometric matrix. Frontier 3: 3D Structures, consisting of methods for the reconstruction of 3D metabolic networks with protein structural information and subsequent applications of these 3D reconstructions. Frontier 4: Genome Sequence and Phenotype Prediction, consisting of workflows for analyzing strain variations in genome sequence as well as building machine learning models based on genome sequence to predict strain phenotype. Frontier 5: Regulatory Networks, consisting of methods for the determination of transcriptional regulatory networks and subsequence models of gene expression and strain phenotype utilizing regulatory network information.
Figure 2.
Overview of frontiers in the computational design of synthetic organisms. Frontier 1: Constraint-based Reconstruction and Modeling, consisting of tools for analyzing pan-genomes, microbial communities, gap-filling metabolic networks, and modeling proteome allocation. Frontier 2: Kinetics and Thermodynamics, consisting of tools for parameterizing and simulating kinetic and thermodynamic models. Parameterization can utilize the Michaelis-Menten equation where [A] is the substrate concentration, whereas simulation uses dynamic mass balance equations where S is the stoichiometric matrix. Frontier 3: 3D Structures, consisting of methods for the reconstruction of 3D metabolic networks with protein structural information and subsequent applications of these 3D reconstructions. Frontier 4: Genome Sequence and Phenotype Prediction, consisting of workflows for analyzing strain variations in genome sequence as well as building machine learning models based on genome sequence to predict strain phenotype. Frontier 5: Regulatory Networks, consisting of methods for the determination of transcriptional regulatory networks and subsequence models of gene expression and strain phenotype utilizing regulatory network information.

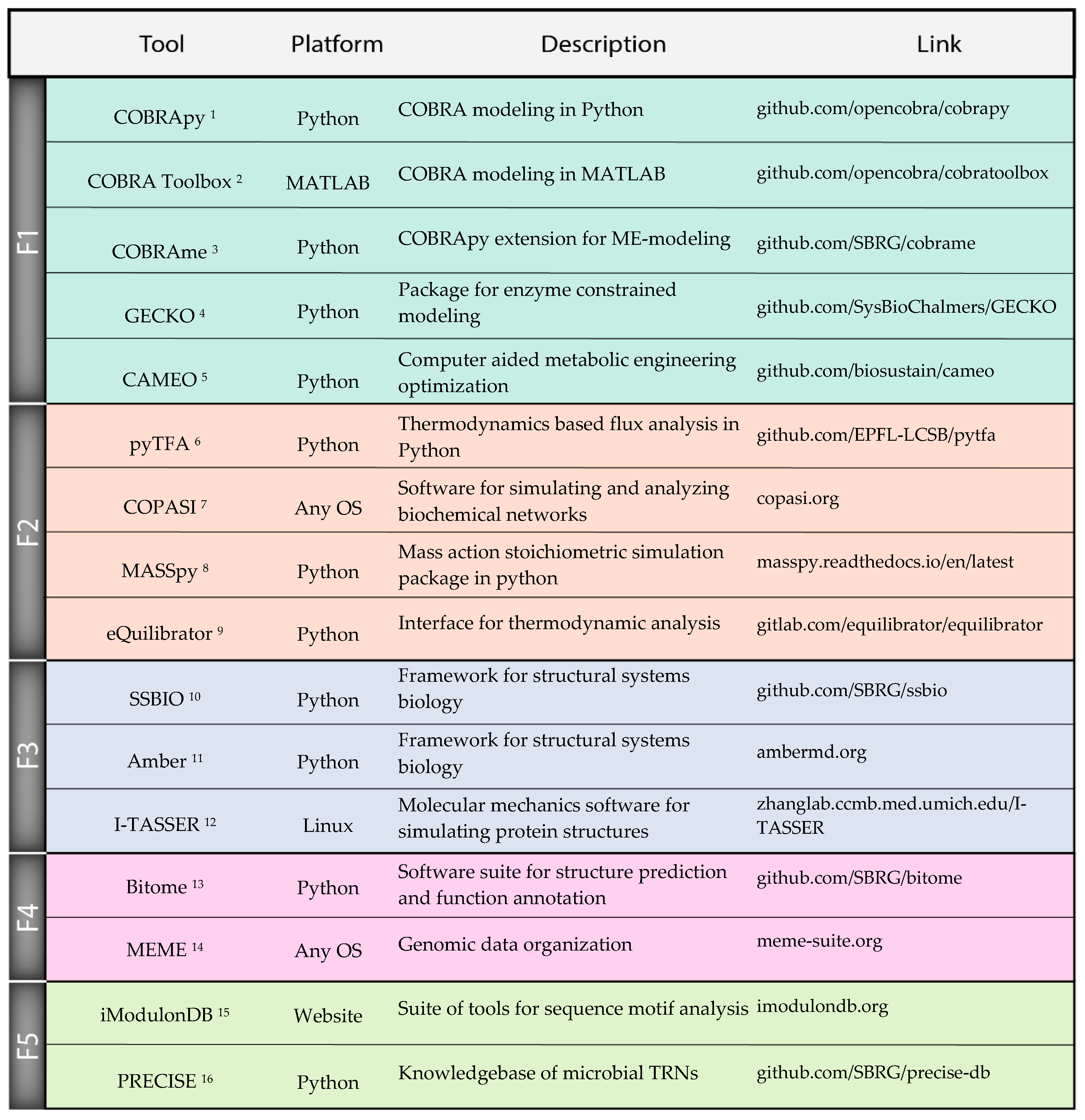
Figure 3.
A selection of actively maintained software for computational design and analysis of microbial phenotypes. We focus on Python tools due to the popularity of the language as well as potential for integration in a single strain design workflow, but also include important packages in other languages and standalone applications. Frontier 1: Packages for constraint-based reconstruction and modeling, proteome allocation modeling, and strain design optimization. Frontier 2: Kinetics and thermodynamics packages for model parameterization, simulation, and thermodynamics constrained modeling. Frontier 3: Software for annotating and visualizing structures as well as integrating 3D structural information with systems biology approaches. Frontier 4: Python package for storing, organizing, and analyzing genome sequences. Frontier 5: Online knowledgebase and software for determining transcriptional regulatory networks using ICA decomposition methods. 1 COBRApy [42]; 2 COBRA Toolbox [17]; 3 COBRAme [20]; 4 GECKO [52]; 5 CAMEO [43]; 6 pyTFA [95]; 7 COPASI [96]; 8 MASSpy [94]; 9 eQuilibrator [89]; 10 SSBIO [106]; 11 Amber [111]; 12 I-TASSER [107]; 13 Bitome [120]; 14 MEME [117]; 15 iModulonDB [125]; 16 PRECISE [28].
Figure 3.
A selection of actively maintained software for computational design and analysis of microbial phenotypes. We focus on Python tools due to the popularity of the language as well as potential for integration in a single strain design workflow, but also include important packages in other languages and standalone applications. Frontier 1: Packages for constraint-based reconstruction and modeling, proteome allocation modeling, and strain design optimization. Frontier 2: Kinetics and thermodynamics packages for model parameterization, simulation, and thermodynamics constrained modeling. Frontier 3: Software for annotating and visualizing structures as well as integrating 3D structural information with systems biology approaches. Frontier 4: Python package for storing, organizing, and analyzing genome sequences. Frontier 5: Online knowledgebase and software for determining transcriptional regulatory networks using ICA decomposition methods. 1 COBRApy [42]; 2 COBRA Toolbox [17]; 3 COBRAme [20]; 4 GECKO [52]; 5 CAMEO [43]; 6 pyTFA [95]; 7 COPASI [96]; 8 MASSpy [94]; 9 eQuilibrator [89]; 10 SSBIO [106]; 11 Amber [111]; 12 I-TASSER [107]; 13 Bitome [120]; 14 MEME [117]; 15 iModulonDB [125]; 16 PRECISE [28].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zielinski, D.C.; Patel, A.; Palsson, B.O. The Expanding Computational Toolbox for Engineering Microbial Phenotypes at the Genome Scale. Microorganisms 2020, 8, 2050. https://doi.org/10.3390/microorganisms8122050
AMA Style
Zielinski DC, Patel A, Palsson BO. The Expanding Computational Toolbox for Engineering Microbial Phenotypes at the Genome Scale. Microorganisms. 2020; 8(12):2050. https://doi.org/10.3390/microorganisms8122050
Chicago/Turabian StyleZielinski, Daniel Craig, Arjun Patel, and Bernhard O. Palsson. 2020. "The Expanding Computational Toolbox for Engineering Microbial Phenotypes at the Genome Scale" Microorganisms 8, no. 12: 2050. https://doi.org/10.3390/microorganisms8122050
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.