The Domestic Environment and the Lung Mycobiome

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Obtention of Broncho-Alveolar Lavage Samples by Bronchoscopy
2.3. Environmental Sampling
2.4. Cultures
2.5. DNA Isolation
2.6. Illumina High-Throughput ITS2 Region Sequencing and Bioinformatic Analyses
3. Results
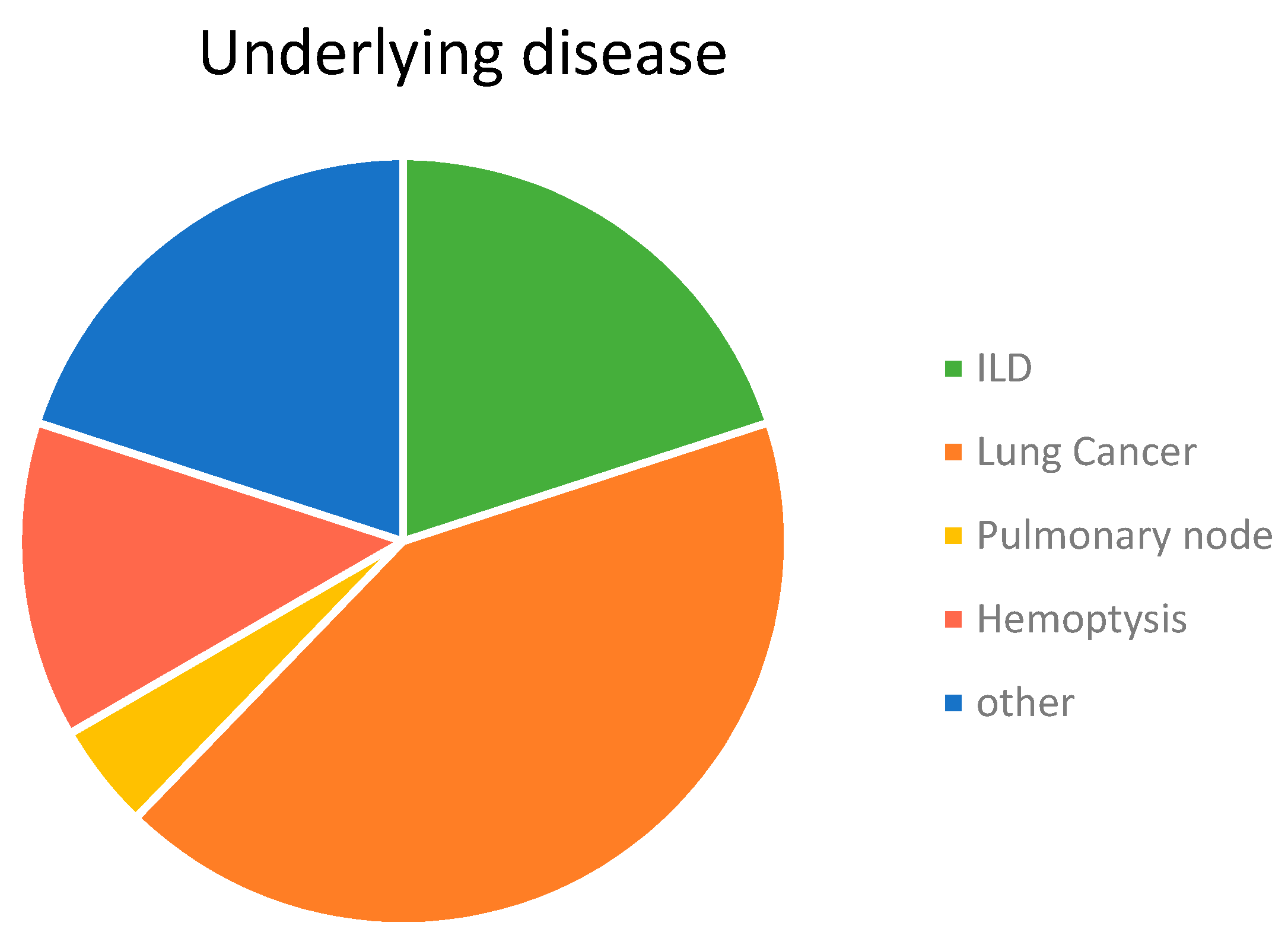
3.1. Characteristics of Participants
3.2. HTS-Based Mycobiome Characterization
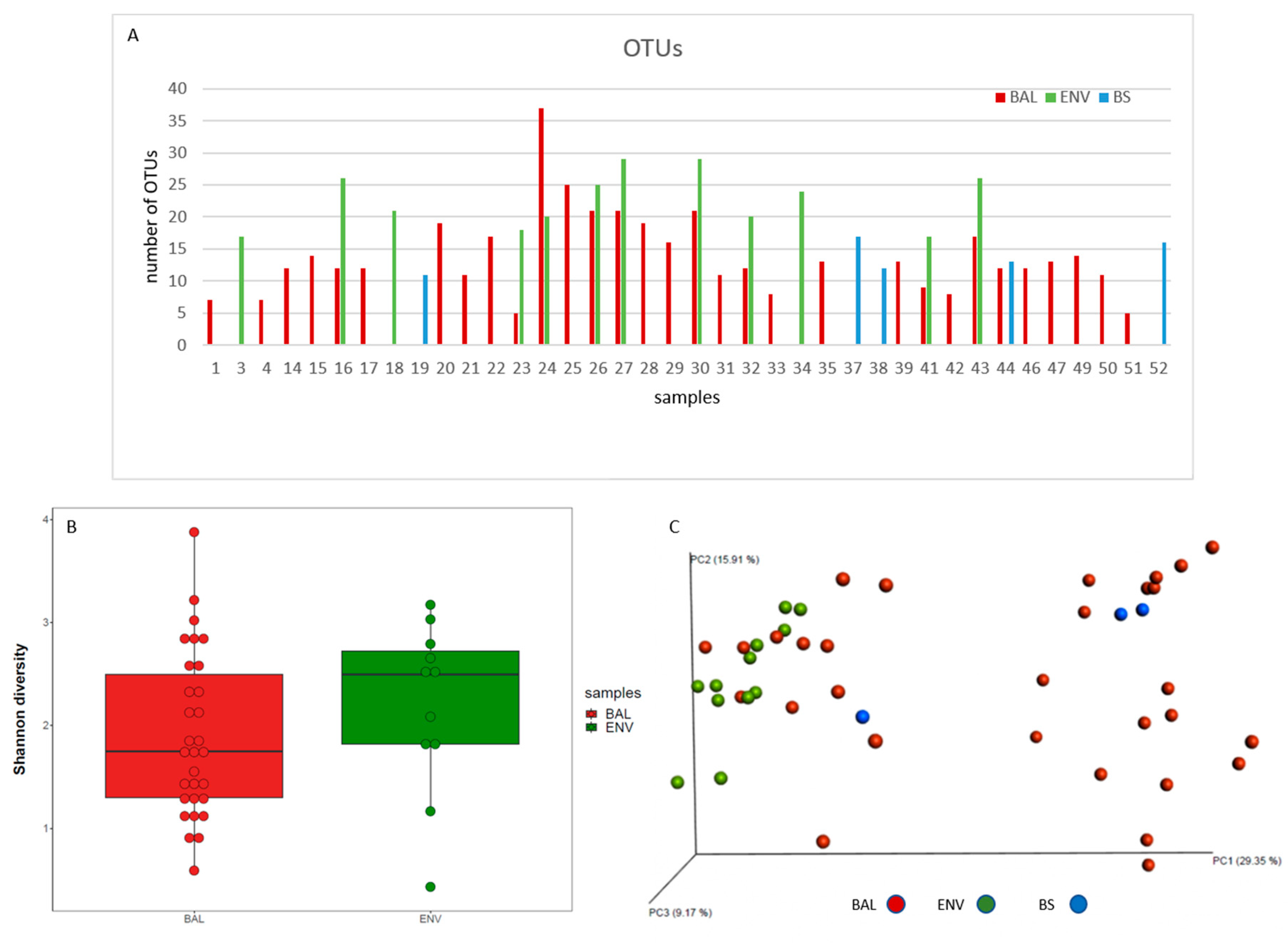
3.2.1. Richness and Diversity
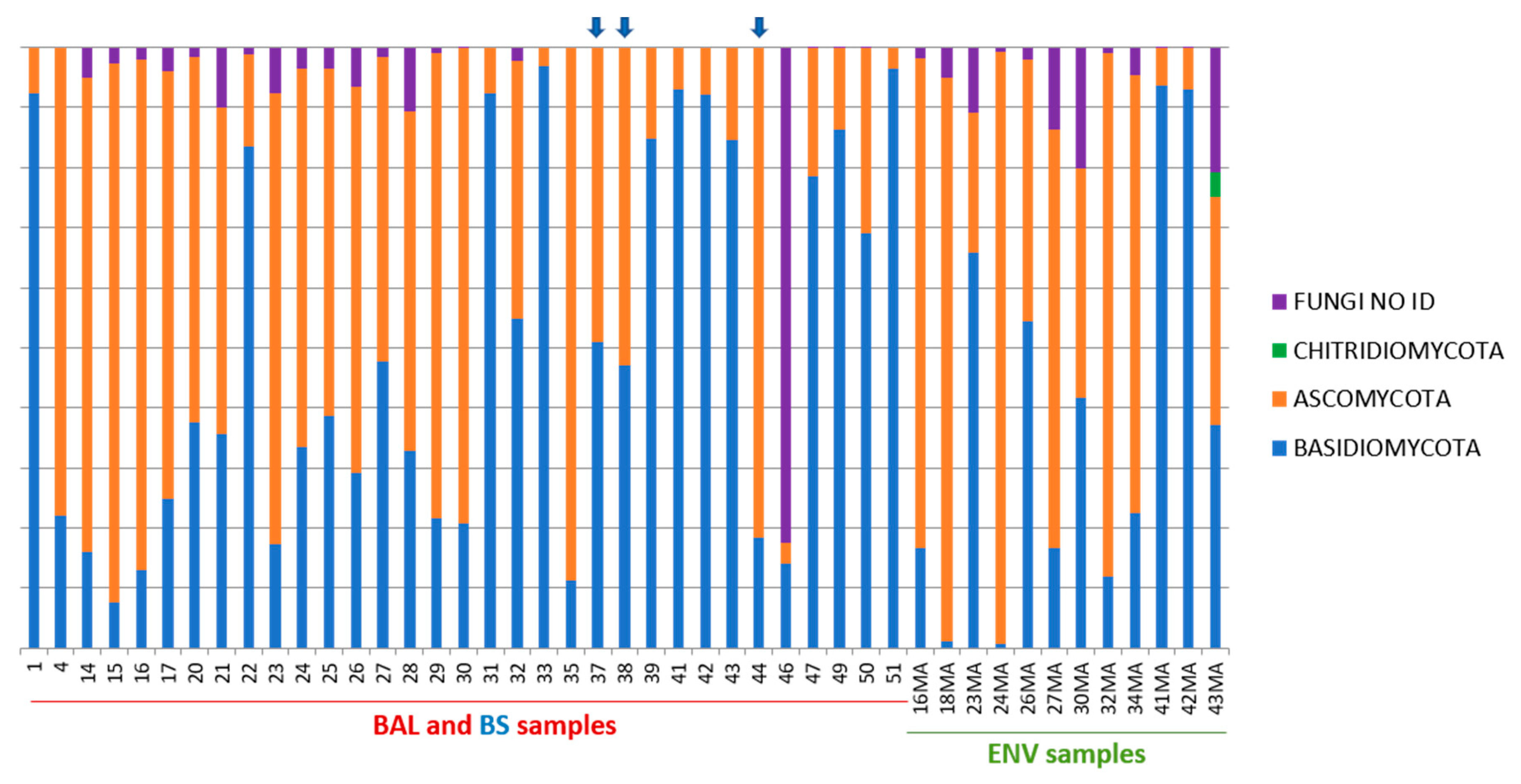
3.2.2. Taxonomy
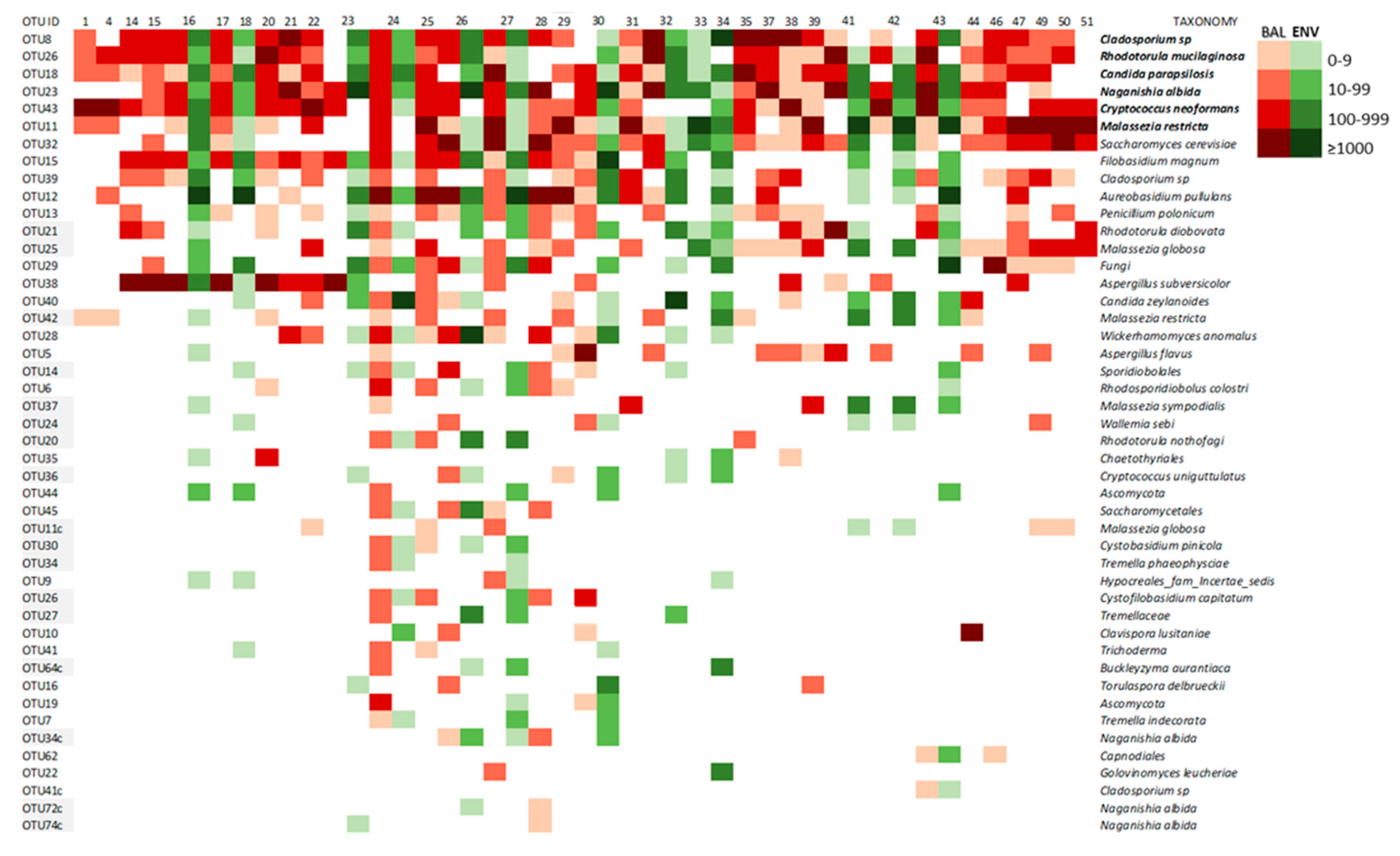
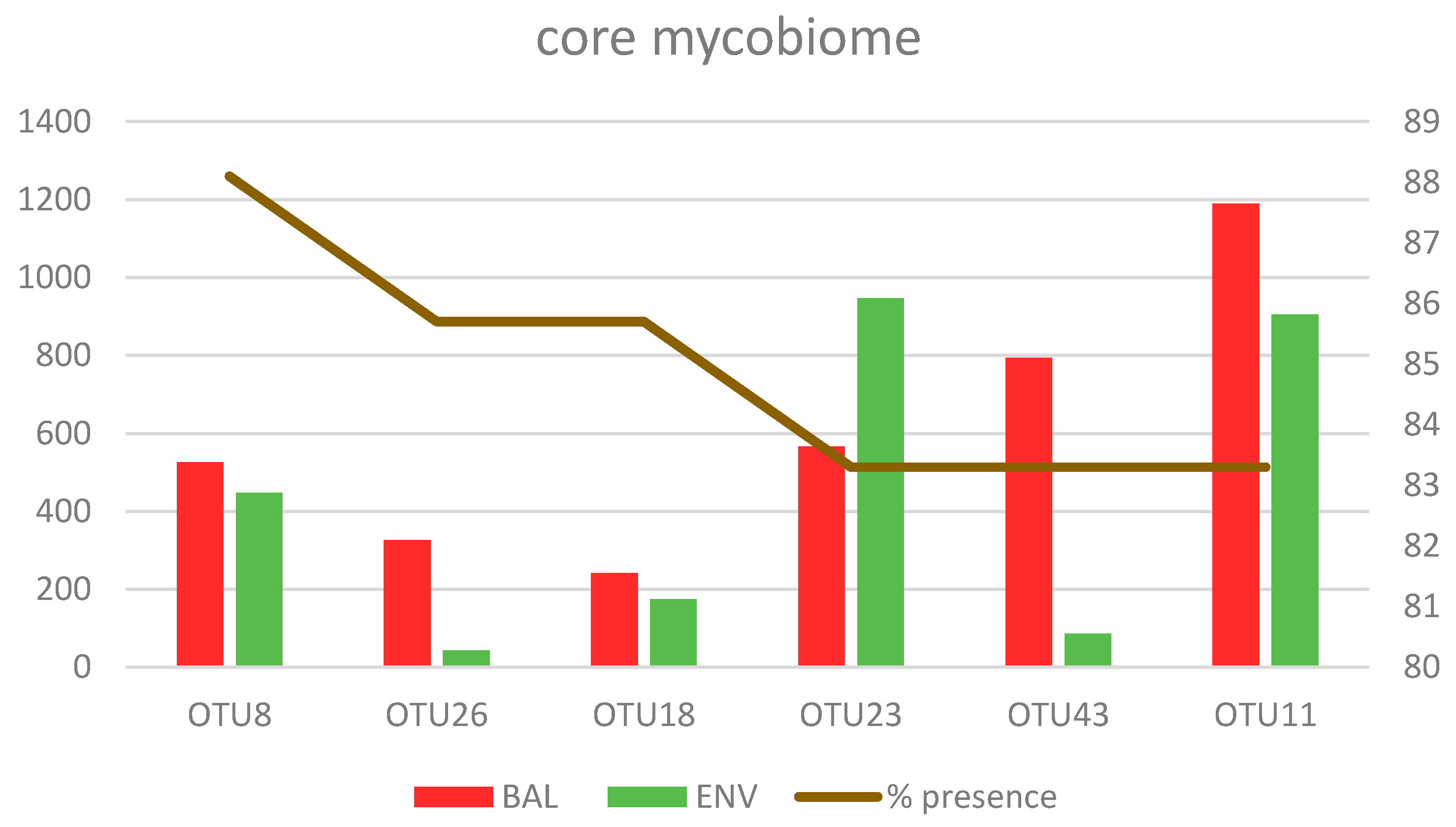
3.2.3. Core Mycobiome
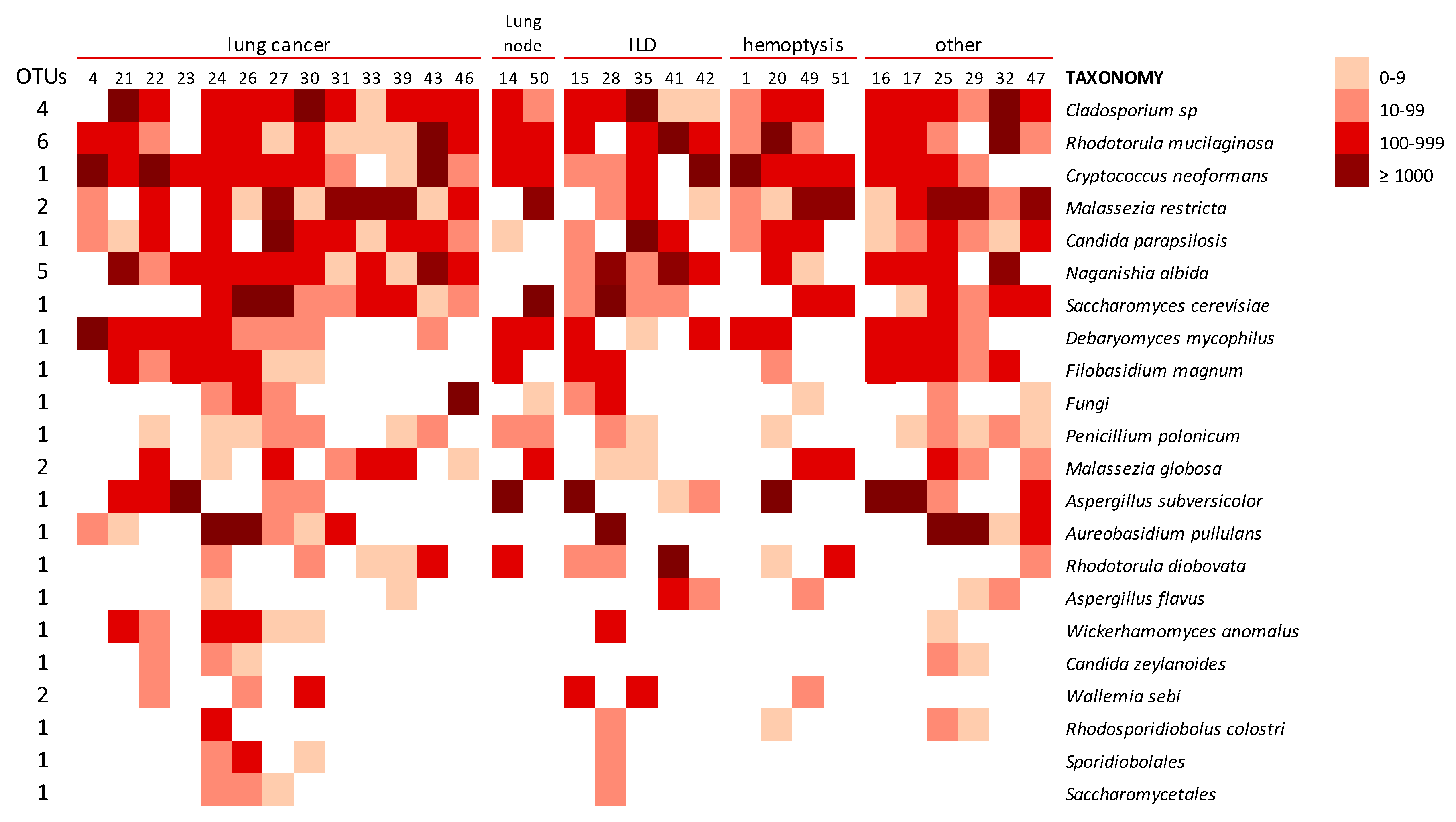
3.2.4. Lung Mycobiome
3.2.5. The Environmental Mycobiome
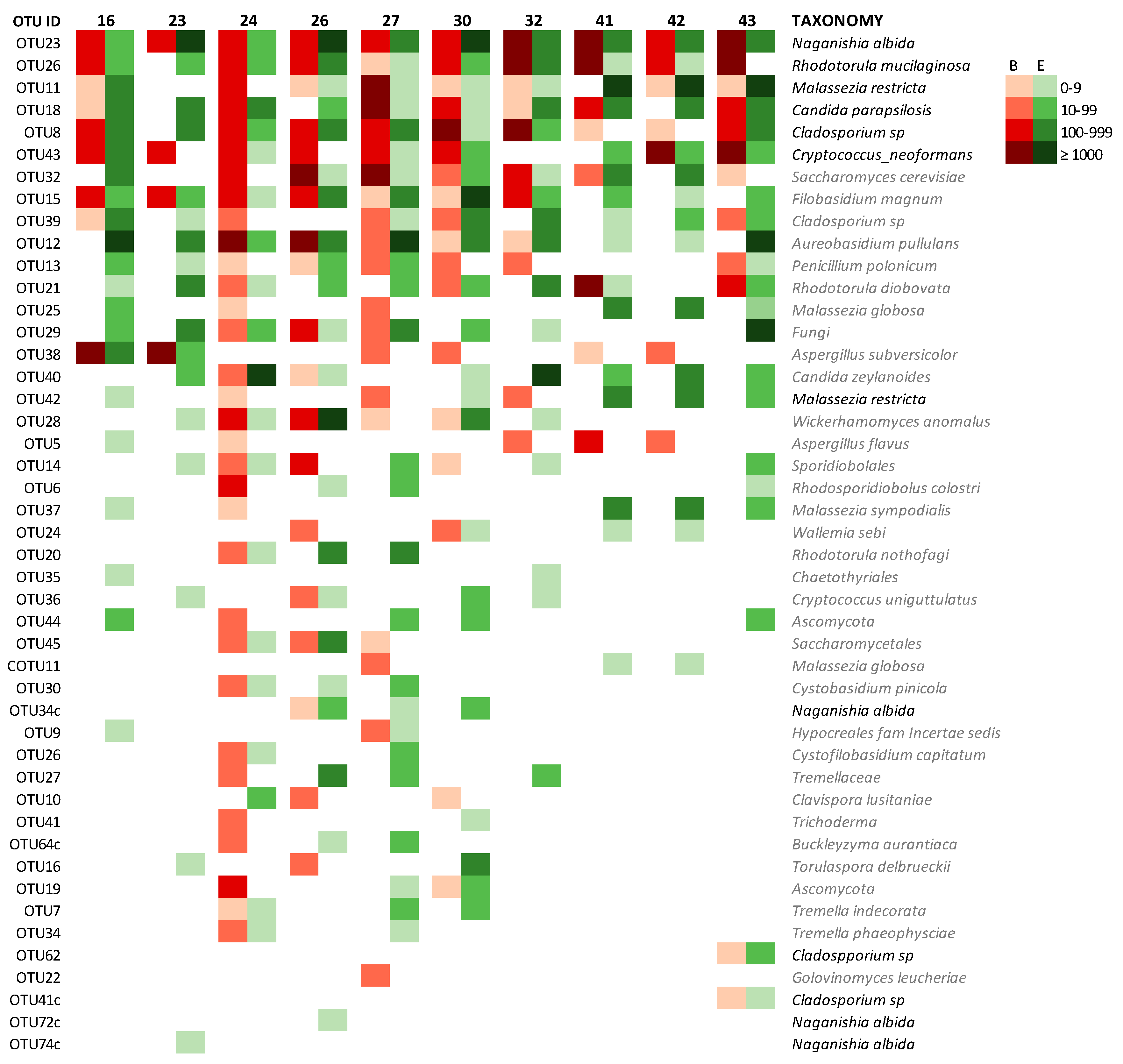
3.2.6. Mycobiome of Lungs and Environments
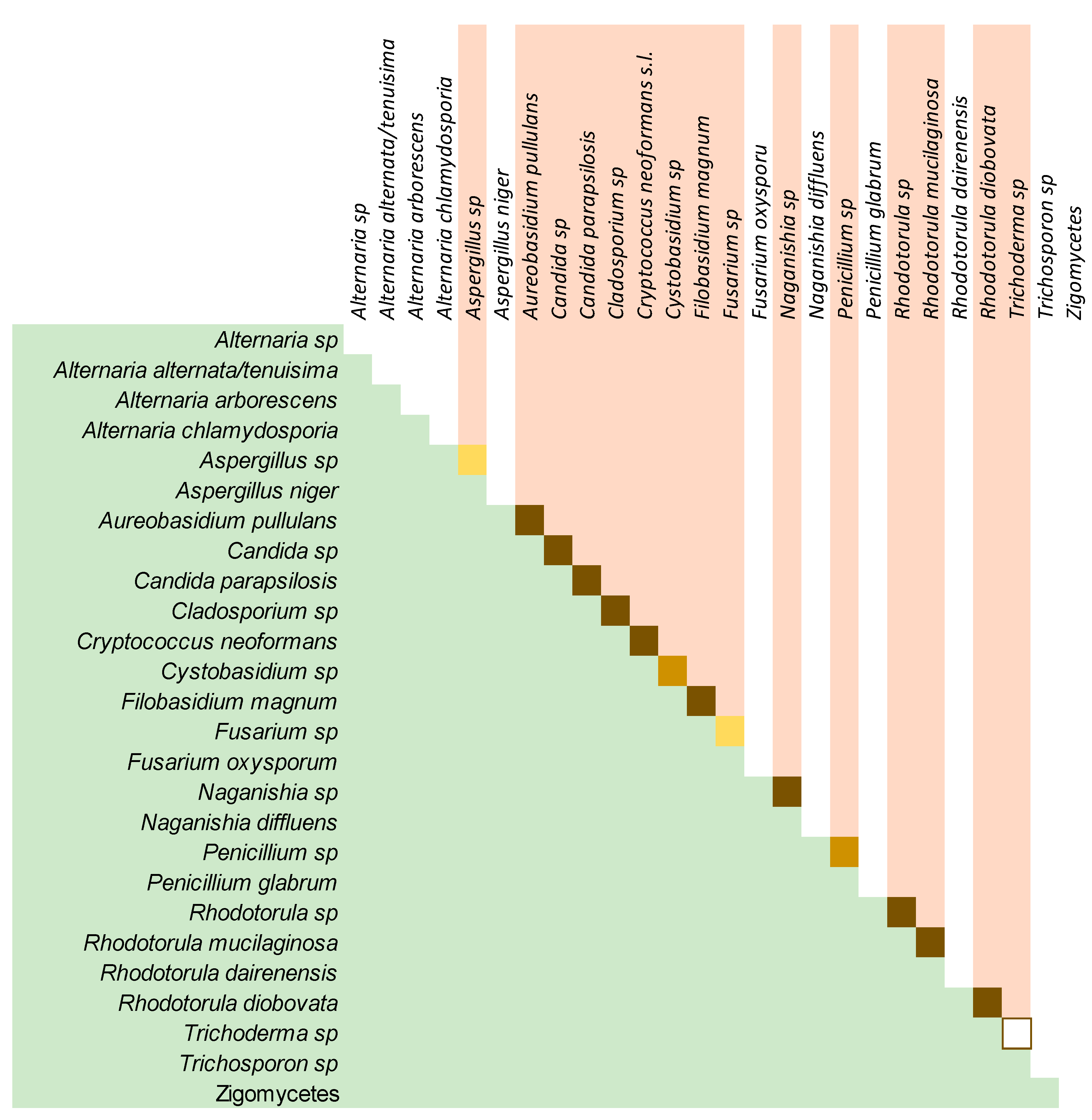
3.3. The Culturable Mycobiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lai, G.C.; Tan, T.G.; Pavelka, N. The mammalian mycobiome: A complex system in a dynamic relationship with the host. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1438. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Izard, J.; Walsh, E.; Batich, K.; Chongsathidkiet, P.; Clarke, G.; Sela, D.A.; Muller, A.J.; Mullin, J.M.; Albert, K.; et al. The Host Microbiome Regulates and Maintains Human Health: A Primer and Perspective for Non-Microbiologists. Cancer Res. 2017, 77, 1783–1812. [Google Scholar] [CrossRef] [Green Version]
- Enaud, R.; Vandenborght, L.E.; Coron, N.; Bazin, T.; Prevel, R.; Schaeverbeke, T.; Berger, P.; Fayon, M.; Lamireau, T.; Delhaes, L. The Mycobiome: A Neglected Component in the M Microbiota-Gut-Brain Axis. Microorganisms 2018, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Huffnagle, G.B.; Noverr, M.C. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limon, J.J.; Skalski, J.H.; Underhill, D.M. Commensal Fungi in Health and Disease. Cell Host Microbe 2017, 22, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Seed, P.C. The Human Mycobiome. Cold Spring Harb. Perspect Med. 2015, 5, a019810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliouat-Denis, C.M.; Chabéa, M.; Delhaes, L.; Dei-Cas, E. Aerially transmitted human fungal pathogens: What can we learn from metagenomics and comparative genomics? Rev. Iberoam. Micol. 2014, 31, 54–61. [Google Scholar] [CrossRef]
- Richardson, M.; Bowyer, P.; Sabino, R. The human lung and Aspergillus: You are what you breathe in? Med. Mycol. 2019, 57, S145–S154. [Google Scholar] [CrossRef] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 7695. [Google Scholar] [CrossRef]
- Beck, J.M.; Young, V.B.; Huffnagle, G.B. The Microbiome of the Lung. Transl. Res. 2012, 160, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Soret, P.; Vandenborght, L.E.; Francis, F.; Coron, N.; Enaud, R.; Avalos, M.; Schaeverbeke, T.; Berger, P.; Fayon, M.; Thiebaut, R.; et al. Respiratory mycobiome and suggestion of inter-kingdom network during acute pulmonary exacerbation in cystic fibrosis. Sci. Rep. 2020, 10, 3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, T.L.; Dominguez-Bello, M.G.; Heisel, T.; Al-Ghalith, G.; Knights, D.; Gale, C.A. Development of the human mycobiome over the first month of life and across body sites. mSystems 2018, 3, e00140-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittinger, K.; Charlson, E.S.; Loy, E.; Shirley, D.J.; Haas, A.R.; Laughlin, A.; Yi, Y.; Wu, G.D.; Lewis, J.D.; Frank, I.; et al. Improved characterization of medically relevant fungi in the human respiratory tract using next-generation sequencing. Genome Biol. 2014, 15, 487. [Google Scholar] [CrossRef]
- Cui, L.; Morris, A.; Huang, L.; Beck, J.M.; Twigg, H.L.; von Mutius, E.; Ghedin, E. The Microbiome and the Lung. Ann. Am. Thorac. Soc. 2014, 11, S227–S232. [Google Scholar] [CrossRef] [Green Version]
- Hyytiäinen, H.K.; Jayaprakash, B.; Kirjavainen, P.V.; Saari, S.E.; Holopainen, R.; Keskinen, J.; Hämeri, K.; Hyvärinen, A.; Boor, B.E.; Täubel, M. Crawling-induced floor dust resuspension affects the microbiota of the infant breathing zone. Microbiome 2018, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Respaldiza, N.; Medrano, F.J.; Medrano, A.C.; Varela, J.M.; de la Horra, C.; Montes-Cano, M.; Ferrer, S.; Wichmann, I.; Gargallo-Viola, D.; Calderon, E.J. High seroprevalence of Pneumocystis infection in Spanish children. Clin. Microbiol. Infect 2004, 10, 1029–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, P.; Friaza, V.; García, E.; de la Horra, C.; Vargas, S.; Calderón, E.; Pavón, A. Early Acquisition of Pneumocystis jirovecii Colonization and Potential Association with Respiratory Distress Syndrome in Preterm Newborn Infants. Clin. Infect. Dis. 2017, 65, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.; Norris, K.A. Colonization by Pneumocystis jirovecii and its role in disease. Clin. Microbiol. Rev. 2012, 25, 297–317. [Google Scholar] [CrossRef] [Green Version]
- Iliev, I.D.; Underhill, D.M. Striking a Balance: Fungal Commensalism versus Pathogenesis. Curr. Opin. Microbiol. 2013, 16, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Dannemiller, K.C.; Gent, J.F.; Leaderer, B.P.; Peccia, J. Indoor microbial communities: Influence on asthma severity in atopic and non-atopic children. J. Allergy Clin. Immunol. 2016, 138, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Pakarinen, J.; Hyvarinen, A.; Salkinoja-Salonen, M.; Laitinen, S.; Nevalainen, A.; Makela, M.J.; Haahtela, T.; von Hertzen, L. Predominance of Gram positive bacteria in house dust in the low-allergy risk Russian Karelia. Environ. Microbiol. 2008, 10, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Von Hertzen, L.; Hyvarinen, A.; Laatikainen, T.; Makela, M.J.; Nevalainen, A.; Vartiainen, E.; Haahtela, T. Risk of atopy associated with microbial components in house dust. Ann. Allergy Asthma. Immunol. 2010, 104, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Green, B.J.; Lemonsa, A.R.; Parkb, Y.; Cox-Ganserb, J.M.; Parkb, J.H. Assessment of fungal diversity in a water-damaged office building. J. Occup. Environ. Hyg. 2017, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Green, B.J. Emerging Insights into the Occupational Mycobiome. Curr. Allergy Asthma. Rep. 2018, 18, 62. [Google Scholar] [CrossRef]
- Yashiro, E.; Savova-Bianchi, D.; Niculita-Hirzel, H. Major Differences in the Diversity of Mycobiomes Associated with Wheat Processing and Domestic Environments: Significant Findings from High-Throughput Sequencing of Fungal Barcode ITS1. Int. J. Environ. Res. Public Health 2019, 16, 2335. [Google Scholar] [CrossRef] [Green Version]
- Krause, R.; Moissl-Eichinger, C.; Halwachs, B.; Gorkiewicz, G.; Berg, G.; Valentin, T.; Prattes, J.; Högenauer, C.; Zollner-Schwetz, I. Mycobiome in the lower respiratory tract—A clinical perspective. Front. Microbiol. 2017, 7, 2169. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Lucht, L.; Tipton, L.; Rogers, M.B.; Fitch, A.; Kessinger, C.; Camp, D.; Kingsley, L.; Leo, N.; Greenblatt, R.M.; et al. Topographic Diversity of the Respiratory Tract Mycobiome and Alteration in HIV and Lung Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 932–942. [Google Scholar] [CrossRef]
- Du Rand, I.A.; Blaikley, J.; Booton, R.; Chaudhuri, N.; Gupta, V.; Khalid, S.; Mandal, S.; Martin, J.; Mills, J.; Navani, N.; et al. British Thoracic Society Guideline for Diagnostic Flexible Bronchoscopy in Adults. Thorax 2013, 68, i1–i44. [Google Scholar] [CrossRef] [Green Version]
- Google Maps. Available online: https://www.google.es/maps (accessed on 19 November 2018).
- Colom, M.F.; Hagen, F.; Gonzalez, A.; Mellado, A.; Morera, N.; Linares, C.; García, D.F.; Peñataro, J.S.; Boekhout, T.; Sánchez, M. Ceratonia siliqua (carob) trees as natural habitat and source of infection by Cryptococcus gattii in the Mediterranean environment. Med. Mycol. 2012, 50, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Pemán, J.; Martín Mazuelos, E.; Rubio, M.C. Guia Práctica de identificación y Diagnóstico en Micología Clínica, 2nd ed.; Revista Iberoamericana de Micologia-Elsevier: Bilbao, Spain, 2007. [Google Scholar]
- De Hoog, G.S.; Guarro, J.; Gené, J.M.; Figueras, M.J. Atlas of clinical fungi, 2nd ed.; ©Centraalbureau voor Schimmelcultures: Utrech, The Netherlands; Universitat Rovira I Virgili: Reus, Spain, 2000; p. 1126. [Google Scholar]
- White, T.J.; Bruns, T.; Lee, S.; Tailor, S. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innins, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Ferrer, C.; Colom, M.F.; Frasés, S.; Mulet, E.; Alió, J. Detection and Identification of fungal pathogens by PCR and by ITS2 5.8S rDNA Typing in ocular infections. J. Clin. Microbiol. 2001, 39, 2873–2879. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Aho, V.T.E.; Pereira, P.A.B.; Haahtela, T.; Pawankar, R.; Auvinen, P.; Koskinen, K. The microbiome of the human lower airways: A next generation sequencing perspective. World Allergy Organ. J. 2015, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.J.; Xu, Z.; Amir, A.; Peddada, S.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vazquez-Baeza, Y.; Birmingham, A.; et al. Effects of library size variance, sparsity, and compositionality on the analysis of microbiome data. PeerJ PrePrints 2015, 3, e1157v1. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Hanson, B.; Zhoua, Y.; Bautista, E.J.; Urch, B.; Speck, M.; Silverman, F.; Muilenberg, M.; Phipatanakul, W.; Weinstock, G.; Sodergren, E.; et al. Characterization of the bacterial and fungal microbiome in indoor dust and outdoor air samples: A pilot study. Environ. Sci. Process Impacts 2016, 18, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.I.; Miletto, M.; Taylor, J.W.; Bruns, T.D. Dispersal in microbes: Fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 2013, 7, 1262–1273. [Google Scholar] [CrossRef] [Green Version]
- Summerbell, R.C.; Saib, F.; Dales, R.; Nolard, N.; Kane, J.; Zwanenburg, H.; Burnett, R.; Krajden, S.; Fung, D.; Leong, D. Ecology of fungi in human dwellings. J. Med. Vet. Mycol. 1992, 30, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Pitkaranta, M.; Meklin, T.; Hyvarinen, A.; Paulin, L.; Auvinen, P.; Nevalainen, A.; Rintala, H. Analysis of Fungal Flora in Indoor Dust by Ribosomal DNA Sequence Analysis, Quantitative PCR, and Culture. Appl. Environ. Microbiol. 2008, 74, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Tipton, L.; Müller, C.L.; Kurtz, Z.D.; Huang, L.; Kleerup, E.; Morris, A.; Bonneau, R.; Ghedin, E. Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome 2018, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mac Aogáin, M.; Chandrasekaran, R.; Lim, A.Y.H.; Low, T.B.; Tan, G.L.; Hassan, T.; Ong, T.H.; Ng, A.H.Q.; Bertrand, D.; Koh, J.Y.; et al. Immunological corollary of the pulmonary mycobiome in bronchiectasis: The CAMEB study. Eur. Respir. J. 2018, 52, 1800766. [Google Scholar] [CrossRef] [PubMed]
- Van Woerden, H.C.; Gregory, C.; Brown, R.; Marchesi, J.R.; Hoogendoorn, B.; Matthews, I.P. Differences in fungi present in induced sputum samples from asthma patients and non-atopic controls: A community-based case control study. BMC Infect. Dis. 2013, 13, 69. [Google Scholar] [CrossRef] [Green Version]
- Willger, S.D.; Grim, S.L.; Dolben, E.L.; Shipunova, A.; Hampton, T.H.; Morrison, H.G.; Filkins, L.M.; O’Toole, G.A.; Moulton, L.A.; Ashare, A.; et al. Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome 2014, 2, 40. [Google Scholar] [CrossRef] [Green Version]
- Zinter, M.S.; Dvorak, C.C.; Mayday, M.Y.; Iwanaga, K.; Ly, N.P.; McGarry, M.E.; Church, G.D.; Faricy, L.E.; Rowan, C.M.; Hume, J.R.; et al. Pulmonary Metagenomic Sequencing Suggests Missed Infections in Immunocompromised Children. Clin. Infect. Dis. 2019, 68, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Delhaes, L.; Monchy, S.; Frealle, E.; Hubans, C.; Salleron, J.; Leroy, S.; Prevotat, A.; Wallet, F.; Wallaert, B.; Dei-Cas, E.; et al. The Airway Microbiota in Cystic Fibrosis: A Complex Fungal and Bacterial Community—Implications for Therapeutic Management. PLoS ONE 2012, 7, e36313. [Google Scholar] [CrossRef] [PubMed]
- Ghannoum, M.A.; Jurevic, R.J.; Mukherjee, P.K.; Cui, F.; Sikaroodi, M.; Naqvi, A.; Gillevet, P.M. Characterization of the Oral Fungal Microbiome (Mycobiome) in Healthy Individuals. PLoS Pathog. 2010, 6, e1000713. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Graeber, S.Y.; Weitnauer, M.; Panitz, J.; Stahl, M.; Clausznitzer, D.; Kaderali, L.; Einarsson, G.; Tunney, M.M.; Elborn, J.S.; et al. Comparison of Microbiomes from Different Niches of Upper and Lower Airways in Children and Adolescents with Cystic Fibrosis. PLoS ONE 2015, 10, e0116029. [Google Scholar] [CrossRef] [Green Version]
- Chambers, D.C.; Gellatly, S.L.; Hugenholtz, P.; Hansbro, P.M. JTD special edition ‘Hot Topics in COPD’—The microbiome in COPD. J. Thorac. Dis. 2014, 6, 1525–1531. [Google Scholar] [CrossRef]
- Krause, R.; Halwachs, B.; Thallinger, G.G.; Klymiuk, I.; Gorkiewicz, G.; Hoenigl, M.; Prattes, J.; Valentin, T.; Heidrich, K.; Buzina, W.; et al. Characterisation of Candida within the Mycobiome/Microbiome of the Lower Respiratory Tract of ICU Patients. PLoS ONE 2016, 11, e0155033. [Google Scholar] [CrossRef]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar]
- Liu, X.Z.; Wang, Q.M.; Göker, M.; Groenewald, M.; Kachalkin, A.V.; Lumbsch, H.T.; Millanes, A.M.; Wedin, M.; Yurkov, A.M.; Boekhout, T.; et al. Towards an integrated phylogenetic classification of the Tremellomycetes. Stud. Mycol. 2015, 81, 85–147. [Google Scholar] [CrossRef] [Green Version]
- Choe, Y.Z.; Blatt, D.B.; Yalcindag, A.; Geffert, S.F.; Bobenchik, A.M.; Michelow, I.C. Cryptococcus albidus Fungemia in an Immunosuppressed Child: Case Report and Systematic Literature Review. J. Pediatric Infect. Dis. Soc. 2020, 9, 100–105. [Google Scholar] [CrossRef]
- Kramer, R.; Sauer-Heilborn, A.; Welte, T.; Guzman, C.A.; Abraham, W.-R.; Höfle, M.G. Cohort study of airway mycobiome in adult cystic fibrosis patients: Differences in community structure between fungi and bacteria reveal predominance of transient fungal elements. J. Clin. Microbiol. 2015, 53, 2900–2907. [Google Scholar] [CrossRef] [Green Version]
- Hamm, P.S.; Taylor, J.W.; Cook, J.A.; Natvig, D.O. Decades-old studies of fungi associated with mammalian lungs and modern DNA sequencing approaches help define the nature of the lung mycobiome. PLoS Pathog. 2020, 16, e1008684. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; Ladaub, J.; Leffa, J.W.; Pollard, K.S.; Menningerd, H.L.; Dunnd, R.R.; Fierera, N. Continental-scale distributions of dust-associated bacteria and fungi. Proc. Natl. Acad. Sci. USA 2015, 112, 5756–5761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, T.; Makimura, K.; Abe, M.; Shiota, R.; Nakamura, Y.; Kano, R.; Hasegawa, A.; Sugita, T.; Shibuya, S.; Watanabe, S.; et al. Revised Culture-Based System for Identification of Malassezia Species. J. Clin. Microbiol. 2007, 45, 3737–3742. [Google Scholar] [CrossRef] [Green Version]
- Guintoli, D.; Stringer, S.L.; Stringer, J.R. Extraordinarily low number of ribosomal RNA genes in P. carinii. J. Eukaryot. Microbiol. 1994, 41, 88S. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AGE RANGE | PROFESSION | ||
|---|---|---|---|
| 40–49 | 2 | Shoes industry | 9 |
| 50–59 | 15 | Building | 7 |
| 60–69 | 13 | Administrative | 4 |
| 70–79 | 10 | House wife | 3 |
| 80–89 | 5 | Grocery | 4 |
| SEX | 25 MEN (55.6%) | Other (less than 2 each) | 20 |
| Sample | Total Before Cleaning | Total Clean | Mean/Sample |
|---|---|---|---|
| BAL | 5,046,424 | 2,826,988 | 62,822 |
| ENV | 1,861,275 | 1,110,227 | 58,433 |
| BS | 108,178 | 75,328 | 15,066 |
| TOTAL | 7,015,877 | 4,012,543 |
| Fungal Class | BAL | ENV | BS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Mean | % | Total | Mean | % | Total | Mean | % | |
| ASCOMYCETES | 85,242 | 2750 | 45.85 | 39,348 | 3279 | 54.65 | 5256 | 2628 | 43.8 |
| BASIDIOMYCETES | 91,948 | 2966 | 49.44 | 27,521 | 2293 | 38.22 | 6744 | 3372 | 56.2 |
| CHYTRIDIOMYCETES | 0 | 0 | 0 | 247 | 20.6 | 0.33 | 0 | 0 | 0 |
| FUNGI UNIDENTIFIED | 8774 | 283 | 4.7 | 4884 | 407 | 6.8 | 0 | 0 | 0 |
| TOTAL | 185,964 | 99.99 | 72,000 | 100 | 12,000 | 100 | |||
| alpha diversity | 1.901600765 | 2.144806055 | 1.693532242 | ||||||
| OTU ID | Taxonomy UNITE | LBA | ENV | ||||
|---|---|---|---|---|---|---|---|
| Samples | % | SEQ | Samples | % | SEQ | ||
| OTU29 | Fungi | 10 | 32.3 | 5470 | 10 | 83.3 | 2588 |
| OTU19 | Fungi; Ascomycota | 2 | 6.4 | 301 | 2 | 16.7 | 15 |
| OTU44 | Fungi; Ascomycota | 1 | 3.2 | 34 | 5 | 41.7 | 228 |
| OTU35 | Fungi; Ascomycota; Eurotiomycetes; Chaetothyriales | 2 | 6.4 | 130 | 3 | 25 | 76 |
| OTU45 | Fungi; Ascomycota; Saccharomycetes; Saccharomycetales | 4 | 12.9 | 131 | 2 | 16.7 | 351 |
| OTU14 | Fungi; Basidiomycota; Microbotryomycetes; Sporidiobolales | 4 | 12.9 | 257 | 6 | 50 | 60 |
| OTU27 | Fungi; Basidiomycota; Tremellomycetes; Tremellales; Tremellaceae | 1 | 3.2 | 64 | 3 | 25 | 283 |
| OTU9 | Fungi; Ascomycota; Sordariomycetes; Hypocreales; Hypocreales fam Incertae sedis | 1 | 3.2 | 75 | 4 | 33.3 | 9 |
| OTU N° | PRESENCE IN SAMPLES (%) | N° OF SEQUENCES (Mean Value) | Taxonomy | ||||
|---|---|---|---|---|---|---|---|
| BAL | MA | TOTAL | BAL | MA | TOTAL | ||
| OTU8 | 90.9 | 83.3 | 88.10% | 526.3 | 447 | 563 | Cladosporium spp. |
| OTU26 | 86.7 | 83.3 | 85.70% | 326.3 | 43.3 | 244 | Rhodotorula mucilaginosa |
| OTU43 | 86.7 | 75 | 83.30% | 794.5 | 85.6 | 611 | Cryptococcus neoformans |
| OTU18 | 80 | 100 | 85.70% | 241.6 | 174.4 | 224 | Candida parapsilosis |
| OTU23 | 76.7 | 100 | 83.30% | 566.8 | 947.4 | 650 | Naganishia albida |
| OTU11 | 83.3 | 83.3 | 83.30% | 1189.1 | 905 | 1023 | Malassezia restricta |
| PATIENT ID | 16 | 23 | 24 | 26 | 27 | 30 | 32 | 41 | 42 | 43 |
|---|---|---|---|---|---|---|---|---|---|---|
| coincident OTUs (present + absent) | 61 | 63 | 58 | 61 | 55 | 61 | 63 | 61 | 60 | 56 |
| % coincidence (present + absent OTUs) | 79.2% | 81.8% | 75.3% | 79.2% | 71.4% | 79.2% | 81.8% | 79.2% | 77.9% | 72.7% |
| OTUs present | 27 | 19 | 38 | 31 | 36 | 33 | 23 | 21 | 21 | 32 |
| Total sequences (BAL + ENV) | 141,606 | 183,668 | 761,132 | 214,953 | 85,412 | 234,499 | 140,075 | 34,002 | 17,158 | 83,962 |
| coincident OTUs (present) | 11 | 4 | 19 | 15 | 14 | 17 | 9 | 5 | 4 | 11 |
| % coincidence (present OTUs) | 40.7% | 21.1% | 46.2% | 48.4% | 38.9% | 51.5% | 39.1% | 23.8% | 19% | 34.4% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio-Portillo, E.; Orts, D.; Llorca, E.; Fernández, C.; Antón, J.; Ferrer, C.; Gálvez, B.; Esteban, V.; Revelles, E.; Pérez-Martín, C.; et al. The Domestic Environment and the Lung Mycobiome. Microorganisms 2020, 8, 1717. https://doi.org/10.3390/microorganisms8111717
Rubio-Portillo E, Orts D, Llorca E, Fernández C, Antón J, Ferrer C, Gálvez B, Esteban V, Revelles E, Pérez-Martín C, et al. The Domestic Environment and the Lung Mycobiome. Microorganisms. 2020; 8(11):1717. https://doi.org/10.3390/microorganisms8111717
Chicago/Turabian StyleRubio-Portillo, Esther, David Orts, Eleuterio Llorca, Cleofé Fernández, Josefa Antón, Consuelo Ferrer, Beatriz Gálvez, Violeta Esteban, Elena Revelles, Carlos Pérez-Martín, and et al. 2020. "The Domestic Environment and the Lung Mycobiome" Microorganisms 8, no. 11: 1717. https://doi.org/10.3390/microorganisms8111717