Shorter Antibacterial Peptide Having High Selectivity for E. coli Membranes and Low Potential for Inducing Resistance

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. 35409-Derived Peptide Synthesis
2.2. 35409-Derived Peptides’ Purification and Characterisation
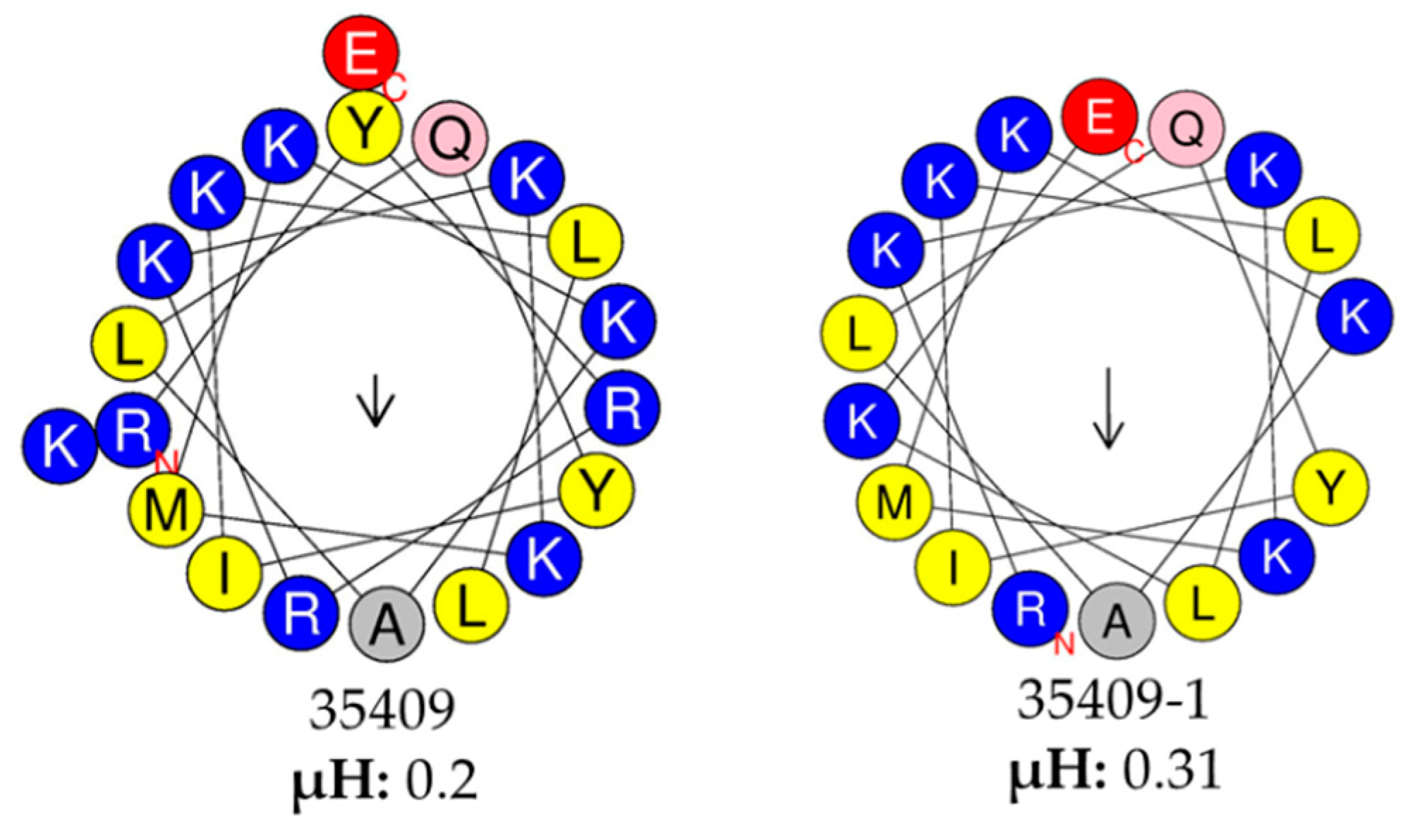
2.3. Bioinformatics Analysis of the Peptide Sequences
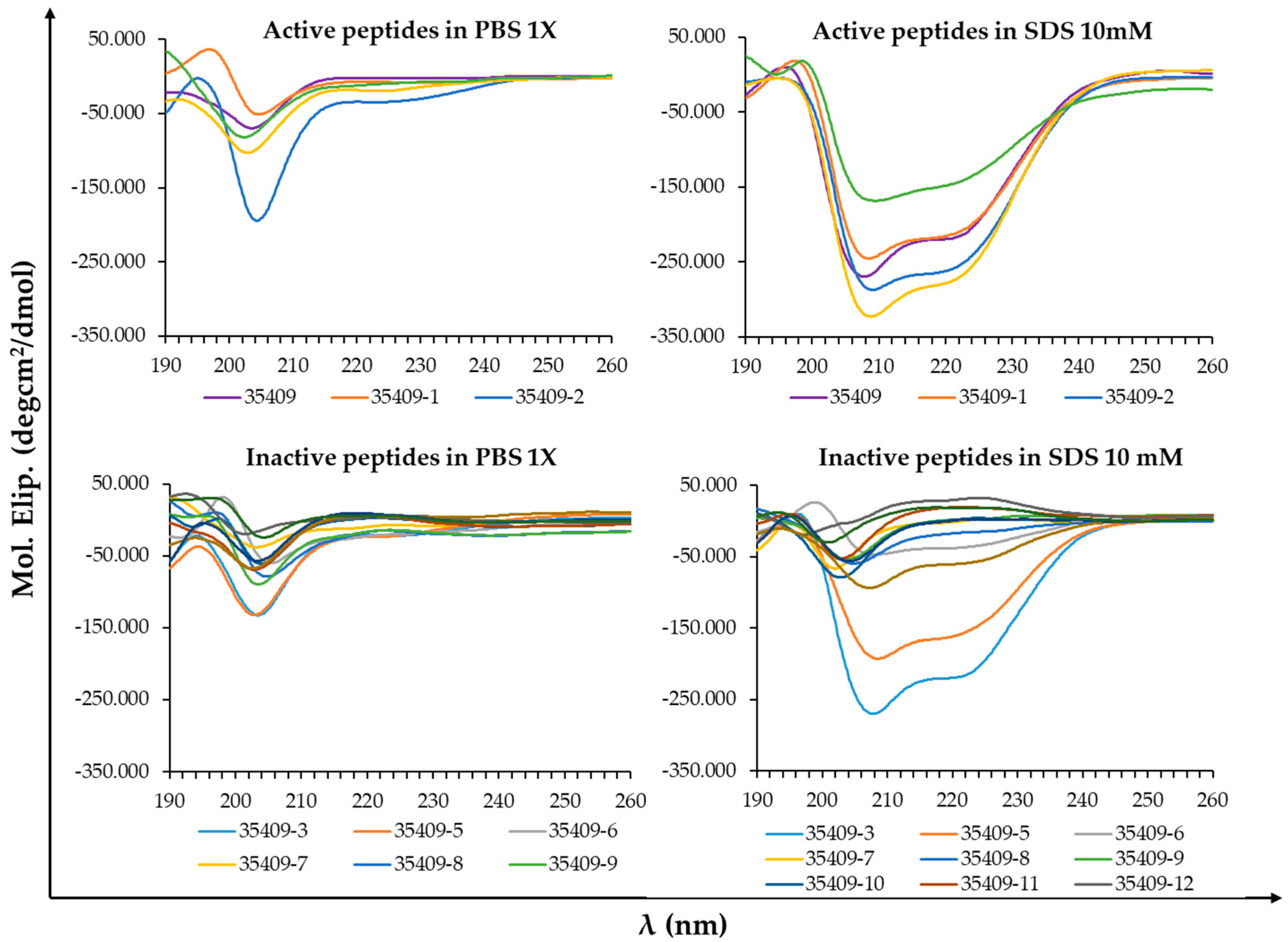
2.4. Circular Dichroism (CD)
2.5. Determining MIC by Broth Microdilution
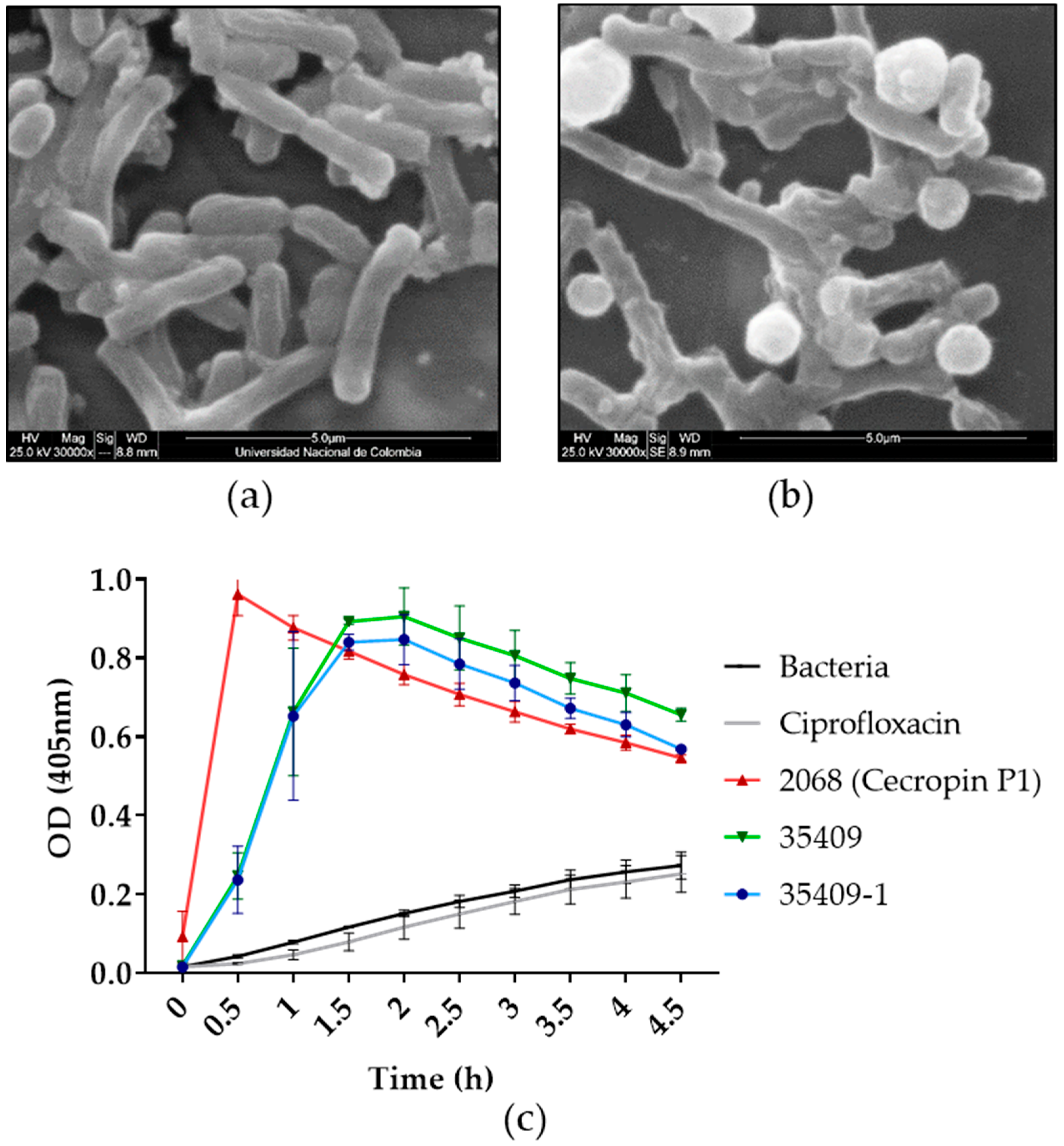
2.6. Bactericidal Activity
2.7. Antibacterial Activity in the Presence of Human Sera
2.8. Antibiotic Synergy
2.9. Haemolytic Activity
2.10. Scanning Electron Microscopy (SEM)
2.11. Permeabilising E. coli ML35 Internal Membrane
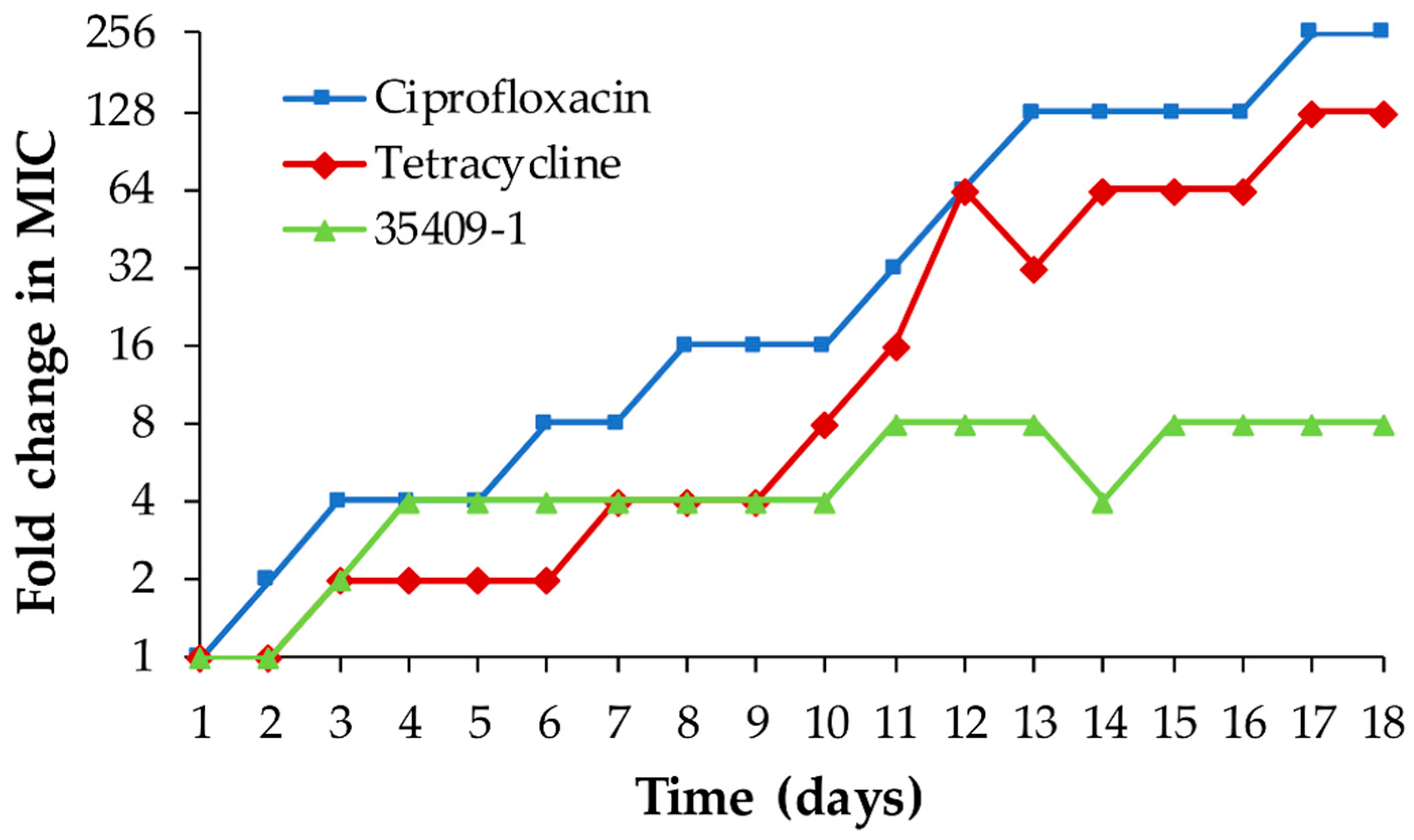
2.12. Evaluating the Potential for Inducing Bacterial Resistance in E. coli ATCC 25922
2.13. Ethics Statement
3. Results
3.1. 35409-Derived Sequences
3.2. Antibacterial and Haemolytic Activity
3.3. The Peptide Sequences’ Physicochemical Properties
3.4. Antibiotic Synergy
3.5. Spectrum of Activity against E. coli
- Isolate 4 (from urine): extended spectrum beta-lactamase (ESBL) (-), resistant to ampicillin, quinolones and co-trimoxazole.
- Isolate 40 (from blood): ESBL (-), resistant to ampicillin, 1st-generation cephalosporins and co-trimoxazole.
- Isolate 44 (from blood): ESBL (+), resistant to aminoglycosides, quinolones, β-lactams (including 1st-, 3rd- and 4th-generation cephalosporins).
3.6. Activity in the Presence of Human Sera
3.7. Mechanism of Action
3.8. Developing Resistance in E. coli ATCC 25922 Bacteria
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
| E. coli Clinical Isolates | Isolate ID | 4 | 40 | 44 | ||
| Sample Origin | Urine | Blood | Blood | |||
| Type of antibiotic | Antibiotic | MIC (µg/mL) | MIC (µg/mL) | MIC (µg/mL) | ||
| Resistance profile | Aminoglycosides | Amikacin | ≤2 (S) | ≤2 (S) | 32 (I) | |
| Gentamicin | ≤1 (S) | ≤1 (S) | ≥16 (R) | |||
| β-lactamases | Ampicillin | ≥32 (R) | ≥32 (R) | ≥32 (R) | ||
| β-lactam +β-lactamase inhibitor | Ampicillin/sulbactam | 8 (S) | 16 (I) | ≥32 (R) | ||
| Piperacillin/Tazobactam | ≤4 (S) | ≤4 (S) | ≥128 (R) | |||
| β-lactamases (cephalosporins) +β-lactamase resistant | 1st-generation | Cephalothin | 4 (S) | 16 (I) | ≥64 (R) | |
| 2nd-generation | Cefoxitin | ≤4 (S) | ≤4 (S) | ≤4 (S) | ||
| 3rd-generation | Cefotaxime | ≤1 (S) | ≤1 (S) | ≥64 (R) | ||
| Ceftazidime | ≤1 (S) | ≤1 (S) | ≥64 (R) | |||
| 4th-generation | Cefepime | ≤1 (S) | ≤1 (S) | ≥64 (R) | ||
| β-lactamases | Carbapenems | Imipenem | ≤1 (S) | ≤1 (S) | ≤1 (S) | |
| Meropenem | ≤0.25 (S) | ≤0.25 (S) | ≤0.25 (S) | |||
| ESBL | - | - | + | |||
| Quinolones | Ciprofloxacin | ≥4 (R) | ≤0.25 (S) | ≥4 (R) | ||
| Nalidixic acid | ≥32 (R) | ≤2 (S) | ≥32 (R) | |||
| Nitrofuran | Nitrofurantoin | ≤16 (S) | ≤16 (S) | ≤16 (S) | ||
| Co-trimoxazole | Trimethoprim/sulfamethoxazole | ≥320 (R) | ≤20 (S) | ≤20 (S) | ||
References
- WHO. Global Action Plan on Antimicrobial Resistance; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- O’Neill, J. Tackling drug-Resistant Infections Globally: Final Report and Recommendations; Review on Antimicrobial Resistance: London, UK, 2016; pp. 1–84. [Google Scholar]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Soblosky, L.; Ramamoorthy, A.; Chen, Z. Membrane interaction of antimicrobial peptides using E. coli lipid extract as model bacterial cell membranes and SFG spectroscopy. Chem Phys. Lipids 2015, 187, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, C.F.; Fang, C.M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Chou, H.T.; Wen, H.W.; Kuo, T.Y.; Lin, C.C.; Chen, W.J. Interaction of cationic antimicrobial peptides with phospholipid vesicles and their antibacterial activity. Peptides 2010, 31, 1811–1820. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, A.; Thennarasu, S.; Lee, D.K.; Tan, A.; Maloy, L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 2006, 91, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Henzler-Wildman, K.A.; Martinez, G.V.; Brown, M.F.; Ramamoorthy, A. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry 2004, 43, 8459–8469. [Google Scholar] [CrossRef]
- Bobone, S.; Stella, L. Selectivity of Antimicrobial Peptides: A Complex Interplay of Multiple Equilibria. Adv. Exp. Med. Biol. 2019, 1117, 175–214. [Google Scholar] [CrossRef]
- Alfred, R.L.; Palombo, E.A.; Panozzo, J.F.; Bhave, M. The antimicrobial domains of wheat puroindolines are cell-penetrating peptides with possible intracellular mechanisms of action. PLoS ONE 2013, 8, e75488. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.K.; Song, J.W.; Gong, F.; Li, S.B.; Chang, H.Y.; Xie, H.M.; Gao, H.W.; Tan, Y.X.; Ji, S.P. Design of an alpha-helical antimicrobial peptide with improved cell-selective and potent anti-biofilm activity. Sci. Rep. 2016, 6, 27394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol 2013, 31, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Santamaria, A.; Patarroyo, M.E.; Curtidor, H. Designing and optimizing new antimicrobial peptides: All targets are not the same. Crit. Rev. Clin. Lab. Sci. 2019, 56, 351–373. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.C.L.; Miranda, A.; da Silva, P.I., Jr. Human Antimicrobial Peptide Isolated from Triatoma infestans Haemolymph, Trypanosoma cruzi-Transmitting Vector. Front. Cell Infect. Microbiol. 2018, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- Tanhaiean, A.; Azghandi, M.; Razmyar, J.; Mohammadi, E.; Sekhavati, M.H. Recombinant production of a chimeric antimicrobial peptide in E. coli and assessment of its activity against some avian clinically isolated pathogens. Microb. Pathog. 2018, 122, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Said, D.G.; Nubile, M.; Mastropasqua, L.; Dua, H.S. Cathelicidin-Derived Synthetic Peptide Improves Therapeutic Potential of Vancomycin against Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 2190. [Google Scholar] [CrossRef] [Green Version]
- Munzker, L.; Oddo, A.; Hansen, P.R. Chemical Synthesis of Antimicrobial Peptides. Methods Mol. Biol. 2017, 1548, 35–49. [Google Scholar] [CrossRef]
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides under Clinical Trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef]
- Giles, F.J.; Rodriguez, R.; Weisdorf, D.; Wingard, J.R.; Martin, P.J.; Fleming, T.R.; Goldberg, S.L.; Anaissie, E.J.; Bolwell, B.J.; Chao, N.J.; et al. A phase III, randomized, double-blind, placebo-controlled, study of iseganan for the reduction of stomatitis in patients receiving stomatotoxic chemotherapy. Leuk. Res. 2004, 28, 559–565. [Google Scholar] [CrossRef]
- Flamm, R.K.; Rhomberg, P.R.; Farrell, D.J.; Jones, R.N. In vitro spectrum of pexiganan activity; bactericidal action and resistance selection tested against pathogens with elevated MIC values to topical agents. Diagn. Microbiol. Infect. Dis. 2016, 86, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Flamm, R.K.; Rhomberg, P.R.; Simpson, K.M.; Farrell, D.J.; Sader, H.S.; Jones, R.N. In vitro spectrum of pexiganan activity when tested against pathogens from diabetic foot infections and with selected resistance mechanisms. Antimicrob. Agents Chemother. 2015, 59, 1751–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottler, L.M.; Ramamoorthy, A. Structure, membrane orientation, mechanism, and function of pexiganan—A highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta 2009, 1788, 1680–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics (Basel) 2020, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, C.; Zhu, Y.; Lai, Z.; Tan, P.; Shan, A. Antimicrobial peptides with protease stability: Progress and perspective. Future Med. Chem. 2019, 11, 2047–2050. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Zaalishvili, G.; Karapetian, M.; Nasrashvili, T.; Kuljanishvili, N.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Grigolava, M.; et al. De novo Design and in Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals (Basel) 2019, 12, 82. [Google Scholar] [CrossRef] [Green Version]
- Suárez, D. Evaluación y Determinación de la CMI del péptido 35409 sobre Bacterias Gram-Negativas y Gram-Positivas. Master’s Thesis, Pontificia Universidad Javeriana, Bogotá, Colombia, 2012. [Google Scholar]
- Barreto-Santamaria, A.; Curtidor, H.; Arevalo-Pinzon, G.; Herrera, C.; Suarez, D.; Perez, W.H.; Patarroyo, M.E. A New Synthetic Peptide Having Two Target of Antibacterial Action in E. coli ML35. Front. Microbiol. 2016, 7, 2006. [Google Scholar] [CrossRef] [Green Version]
- Merrifield, B. Solid phase synthesis. Science 1986, 232, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Vergel Galeano, C.F.; Rivera Monroy, Z.J.; Rosas Pérez, J.E.; García Castañeda, J.E. Efficient synthesis of peptides with 4-methylpiperidine as Fmoc removal reagent by solid phase synthesis. J. Mex. Chem. Soc. 2014, 58, 386–392. [Google Scholar]
- Huang, H.; Rabenstein, D.L. A cleavage cocktail for methionine-containing peptides. J. Pept. Res. 1999, 53, 548–553. [Google Scholar] [CrossRef]
- Insuasty Cepeda, D.S.; Pineda Castaneda, H.M.; Rodriguez Mayor, A.V.; Garcia Castaneda, J.E.; Maldonado Villamil, M.; Fierro Medina, R.; Rivera Monroy, Z.J. Synthetic Peptide Purification via Solid-Phase Extraction with Gradient Elution: A Simple, Economical, Fast, and Efficient Methodology. Molecules 2019, 24, 1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G. Improved methods for classification, prediction, and design of antimicrobial peptides. Methods Mol. Biol. 2015, 1268, 43–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef] [PubMed]
- Daleke, D.L. Regulation of phospholipid asymmetry in the erythrocyte membrane. Curr. Opin. Hematol. 2008, 15, 191–195. [Google Scholar] [CrossRef]
- Ebenhan, T.; Gheysens, O.; Kruger, H.G.; Zeevaart, J.R.; Sathekge, M.M. Antimicrobial peptides: Their role as infection-selective tracers for molecular imaging. Biomed. Res. Int. 2014, 2014, 867381. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Park, S.C.; Jung, M.; Shin, M.K.; Kang, H.L.; Baik, S.C.; Cheong, G.W.; Jang, M.K.; Lee, W.K. Cell-selectivity of tryptophan and tyrosine in amphiphilic alpha-helical antimicrobial peptides against drug-resistant bacteria. Biochem. Biophys. Res. Commun. 2018, 505, 478–484. [Google Scholar] [CrossRef]
- Sitaram, N.; Chandy, M.; Pillai, V.N.; Nagaraj, R. Change of glutamic acid to lysine in a 13-residue antibacterial and hemolytic peptide results in enhanced antibacterial activity without increase in hemolytic activity. Antimicrob. Agents Chemother. 1992, 36, 2468–2472. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012; Volume 32. [Google Scholar]
- Shurko, J.F.; Galega, R.S.; Li, C.; Lee, G.C. Evaluation of LL-37 antimicrobial peptide derivatives alone and in combination with vancomycin against S. aureus. J. Antibiot. (Tokyo) 2018, 71, 971–974. [Google Scholar] [CrossRef] [Green Version]
- Bonapace, C.R.; Bosso, J.A.; Friedrich, L.V.; White, R.L. Comparison of methods of interpretation of checkerboard synergy testing. Diagn. Microbiol. Infect. Dis. 2002, 44, 363–366. [Google Scholar] [CrossRef]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Bisht, G.S.; Rawat, D.S.; Kumar, A.; Kumar, R.; Maiti, S.; Pasha, S. Interaction studies of novel cell selective antimicrobial peptides with model membranes and E. coli ATCC 11775. Biochim. Biophys. Acta 2010, 1798, 1864–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.; Berditsch, M.; Hawecker, J.; Ardakani, M.F.; Gerthsen, D.; Ulrich, A.S. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob. Agents Chemother. 2010, 54, 3132–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, P.; Gao, W.; Chen, H.; Li, D.; Yang, N.; Zhu, J.; Feng, X.; Liu, C.; Li, Z. The Central Hinge Link Truncation of the Antimicrobial Peptide Fowlicidin-3 Enhances Its Cell Selectivity without Antibacterial Activity Loss. Antimicrob. Agents Chemother. 2016, 60, 2798–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.; Wang, J.; Shang, L.; Akhtar, M.U.; Wang, Z.; Shi, B.; Feng, X.; Shan, A. Short, symmetric-helical peptides have narrow-spectrum activity with low resistance potential and high selectivity. Biomater. Sci. 2019, 7, 2394–2409. [Google Scholar] [CrossRef]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-derived short antimicrobial peptide KR-12-a5 and its d-amino acid substituted analogs with cell selectivity, anti-biofilm activity, synergistic effect with conventional antibiotics, and anti-inflammatory activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef]
- Jin, L.; Bai, X.; Luan, N.; Yao, H.; Zhang, Z.; Liu, W.; Chen, Y.; Yan, X.; Rong, M.; Lai, R.; et al. A Designed Tryptophan- and Lysine/Arginine-Rich Antimicrobial Peptide with Therapeutic Potential for Clinical Antibiotic-Resistant Candida albicans Vaginitis. J. Med. Chem. 2016, 59, 1791–1799. [Google Scholar] [CrossRef]
- Juban, M.M.; Barkley, M.D. Circular Dichroism Studies of Secondary Structure of Peptides. In Antibacterial Peptide Protocols. Methods In Molecular Biology; Shafer, W.M., Ed.; Humana Press: Totowa, NJ, USA, 1997; Volume 78. [Google Scholar]
- De la Fuente-Salcido, N.M.; Villarreal-Prieto, J.M.; Díaz León, M.Á.; García Pérez, A.P. Evaluación de la actividad de los agentes antimicrobianos ante el desafío de la resistencia bacteriana. Rev. Mex. Cienc. Farm. 2015, 46, 7–16. [Google Scholar]
- Umerska, A.; Cassisa, V.; Bastiat, G.; Matougui, N.; Nehme, H.; Manero, F.; Eveillard, M.; Saulnier, P. Synergistic interactions between antimicrobial peptides derived from plectasin and lipid nanocapsules containing monolaurin as a cosurfactant against Staphylococcus aureus. Int. J. Nanomed. 2017, 12, 5687–5699. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, S.; Soares, J.W.; Meehan, A.M.; Marek, P.; Kirby, R. Membrane permeability and antimicrobial kinetics of cecropin P1 against Escherichia coli. J. Pept. Sci. 2009, 15, 398–403. [Google Scholar] [CrossRef] [PubMed]
- FDA. Drug Approval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/019537s49_19847s27_19857s31_20780s13TOC.cfm (accessed on 28 May 2020).
- Ageitos, J.M.; Sanchez-Perez, A.; Calo-Mata, P.; Villa, T.G. Antimicrobial peptides (AMPs): Ancient compounds that represent novel weapons in the fight against bacteria. Biochem. Pharm. 2017, 133, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Spohn, R.; Daruka, L.; Lazar, V.; Martins, A.; Vidovics, F.; Grezal, G.; Mehi, O.; Kintses, B.; Szamel, M.; Jangir, P.K.; et al. Integrated evolutionary analysis reveals antimicrobial peptides with limited resistance. Nat. Commun. 2019, 10, 4538. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta 2007, 1768, 2500–2509. [Google Scholar] [CrossRef] [Green Version]
- Zawadzka, K.; Bernat, P.; Felczak, A.; Rozalska, S.; Lisowska, K. Antibacterial activity of high concentrations of carvedilol against Gram-positive and Gram-negative bacteria. Int. J. Antimicrob. Agents 2018, 51, 458–467. [Google Scholar] [CrossRef]
- Park, Y.; Park, S.C.; Park, H.K.; Shin, S.Y.; Kim, Y.; Hahm, K.S. Structure-activity relationship of HP (2-20) analog peptide: Enhanced antimicrobial activity by N-terminal random coil region deletion. Biopolymers 2007, 88, 199–207. [Google Scholar] [CrossRef]
- Subbalakshmi, C.; Nagaraj, R.; Sitaram, N. Biological activities of C-terminal 15-residue synthetic fragment of melittin: Design of an analog with improved antibacterial activity. FEBS Lett. 1999, 448, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Ueno, S.; Minaba, M.; Nishiuchi, Y.; Taichi, M.; Tamada, Y.; Yamazaki, T.; Kato, Y. Generation of novel cationic antimicrobial peptides from natural non-antimicrobial sequences by acid-amide substitution. Ann. Clin. Microbiol. Antimicrob. 2011, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Mulder, K.C.; Lima, L.A.; Miranda, V.J.; Dias, S.C.; Franco, O.L. Current scenario of peptide-based drugs: The key roles of cationic antitumor and antiviral peptides. Front. Microbiol 2013, 4, 321. [Google Scholar] [CrossRef] [Green Version]
- Agadi, N.; Vasudevan, S.; Kumar, A. Structural insight into the mechanism of action of antimicrobial peptide BMAP-28 (1–18) and its analogue mutBMAP18. J. Struct. Biol. 2018, 204, 435–448. [Google Scholar] [CrossRef]
- Jordan, J.; Easton, P.; Hinton, J. Effects of phenylalanine substitutions in gramicidin A on the kinetics of channel formation in vesicles and channel structure in SDS micelles. Biophys. J. 2005, 88, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.N.; Sorensen, K.K.; Johansen, N.T.; Martel, A.; Kirkensgaard, J.J.; Jensen, K.J.; Arleth, L.; Midtgaard, S.R. Dimeric peptides with three different linkers self-assemble with phospholipids to form peptide nanodiscs that stabilize membrane proteins. Soft Matter 2016, 12, 5937–5949. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Jeong, J.H.; Kim, Y. Design, characterization, and antimicrobial activity of a novel antimicrobial peptide derived from bovine lactophoricin. J. Microbiol. Biotechnol. 2017, 27, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides—Using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Llobet, E.; Tomas, J.M.; Bengoechea, J.A. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 2008, 154, 3877–3886. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J. Anaesthesiol. Clin. Pharm. 2017, 33, 300–305. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharm. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, S.; Govender, T.; Kruger, H.G.; de la Torre, B.G.; Albericio, F. Short AntiMicrobial Peptides (SAMPs) as a class of extraordinary promising therapeutic agents. J. Pept. Sci. 2016, 22, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Bottger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Knappe, D.; Henklein, P.; Hoffmann, R.; Hilpert, K. Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob. Agents Chemother. 2010, 54, 4003–4005. [Google Scholar] [CrossRef] [Green Version]
- Starr, C.G.; Wimley, W.C. Antimicrobial peptides are degraded by the cytosolic proteases of human erythrocytes. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2319–2326. [Google Scholar] [CrossRef]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef]
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.Y. Antimicrobial peptides: New hope in the war against multidrug resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [CrossRef]
- Sun, D.; Peyear, T.A.; Bennett, W.D.; Andersen, O.S.; Lightstone, F.C.; Ingólfsson, H.I. Molecular mechanism for gramicidin dimerization and dissociation in bilayers of different thickness. Biophys. J. 2019, 117, 1831–1844. [Google Scholar] [CrossRef] [Green Version]
- Thennarasu, S.; Lee, D.K.; Tan, A.; Prasad Kari, U.; Ramamoorthy, A. Antimicrobial activity and membrane selective interactions of a synthetic lipopeptide MSI-843. Biochim. Biophys. Acta 2005, 1711, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Thennarasu, S.; Huang, R.; Lee, D.K.; Yang, P.; Maloy, L.; Chen, Z.; Ramamoorthy, A. Limiting an antimicrobial peptide to the lipid-water interface enhances its bacterial membrane selectivity: A case study of MSI-367. Biochemistry 2010, 49, 10595–10605. [Google Scholar] [CrossRef] [Green Version]
- Kamysz, E.; Sikorska, E.; Jaskiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Baranska-Rybak, W.; Kamysz, W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12-Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int. J. Mol. Sci. 2020, 21, 887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.; Soblosky, L.; Nguyen, K.; Geng, J.; Yu, X.; Ramamoorthy, A.; Chen, Z. Physiologically-relevant modes of membrane interactions by the human antimicrobial peptide, LL-37, revealed by SFG experiments. Sci. Rep. 2013, 3, 1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallock, K.J.; Lee, D.K.; Ramamoorthy, A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 2003, 84, 3052–3060. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Donhofer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef]
- Lohner, K. Membrane-active Antimicrobial Peptides as Template Structures for Novel Antibiotic Agents. Curr. Top. Med. Chem. 2017, 17, 508–519. [Google Scholar] [CrossRef]
- Rodríguez-Rojas, A.; Moreno-Morales, J.; Mason, A.J.; Rolff, J. Cationic antimicrobial peptides do not change recombination frequency in Escherichia coli. Biol. Lett. 2018, 14, 20180006. [Google Scholar] [CrossRef] [Green Version]
- FDA. The Drug Development Process. Available online: https://www.fda.gov/patients/learn-about-drugand-device-approvals/drug-development-process (accessed on 30 April 2020).
- Muller, P.Y.; Milton, M.N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 2012, 11, 751–761. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Aoki, M.; Yano, Y.; Matsuzaki, K. Interaction of antimicrobial peptide magainin 2 with gangliosides as a target for human cell binding. Biochemistry 2012, 51, 10229–10235. [Google Scholar] [CrossRef]
- Epand, R.F.; Maloy, W.L.; Ramamoorthy, A.; Epand, R.M. Probing the “charge cluster mechanism” in amphipathic helical cationic antimicrobial peptides. Biochemistry 2010, 49, 4076–4084. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.F.; Maloy, L.; Ramamoorthy, A.; Epand, R.M. Amphipathic helical cationic antimicrobial peptides promote rapid formation of crystalline states in the presence of phosphatidylglycerol: Lipid clustering in anionic membranes. Biophys. J. 2010, 98, 2564–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Physicochemical Characteristics | MIC/MBC 1 | MHC 2 | |||
|---|---|---|---|---|---|---|---|
| L | Net Charge | % H aa | E. coli 25922 | P. aeruginosa 27853 | |||
| 35409 | RYRRKKKMKKALQYIKLLKE | 20 | +10 | 30 | 25/50 | >100 | 1.56 |
| 35409-1 | RKKKMKKALQYIKLLKE | 17 | +8 | 35 | 25/25 | 100/100 | >200 |
| 35409-2 | KKKMKKALQYIKLLKE | 16 | +7 | 37 | 25/25 | >100 | >200 |
| 35409-3 | KKMKKALQYIKLLKE | 15 | +6 | 40 | >100 | >100 | >200 |
| 35409-4 | KMKKALQYIKLLKE | 14 | +5 | 42 | 100/100 | >100 | >200 |
| 35409-5 | MKKALQYIKLLKE | 13 | +4 | 46 | >100 | >100 | >200 |
| 35409-6 | ALQYIKLLKE | 10 | +2 | 50 | >100 | >100 | >200 |
| 35409-7 | YIKLLKE | 7 | +2 | 42 | >100 | >100 | >200 |
| 35409-8 | RYRRKKKMKKALQYIKL | 17 | +10 | 29 | >100 | >100 | >200 |
| 35409-9 | RYRRKKKMKKALQY | 14 | +9 | 21 | >100 | >100 | >200 |
| 35409-10 | RYRRKKKMKKA | 11 | +9 | 18 | >100 | >100 | >200 |
| 35409-11 | RYRRKKKMKK | 10 | +9 | 10 | >100 | >100 | >200 |
| 35409-12 | RYRRKKK | 7 | +7 | 0 | >100 | >100 | >200 |
| 35409-13 | KKMKKALQYIKLLK | 14 | +7 | 42 | 50/50 | >100 | >200 |
| 35409-14 | KKMKKALQYIKL | 12 | +6 | 41 | >100 | >100 | 100 |
| 35409-15 | RKKKMKKALQY | 11 | +7 | 27 | >100 | >100 | 200 |
| 35409-16 | KMKKALQY | 8 | +4 | 37 | >100 | >100 | >200 |
| C+ | ciprofloxacin MIC (µg/mL) | - | - | - | 0.008 | 0.256 | - |
| Antibiotic (A) | Peptide (P) | x MIC Individual/Combination | FIC P + A | FIC Index | Interpretation | |
|---|---|---|---|---|---|---|
| Peptide | Antibiotic | |||||
| Ciprofloxacin | 35409-1 | 1/1 | 1/2 | 1 + 2 | 3 | Indifference |
| 35409-2 | 1/1 | 1/2 | 1 + 2 | 3 | Indifference | |
| 35409-4 | 1/1 | 1/2 | 1 + 2 | 3 | Indifference | |
| 35409-13 | 1/1 | 1/2 | 1 + 2 | 3 | Indifference | |
| Gentamicin | 35409-1 | 1/0.25 | 1/0.5 | 0.25 + 0.5 | 0.75 | Indifference |
| 35409-2 | 1/0.25 | 1/0.5 | 0.25 + 0.5 | 0.75 | Indifference | |
| 35409-4 | 1/0.25 | 1/0.5 | 0.25 + 0.5 | 0.75 | Indifference | |
| 35409-13 | 1/0.25 | 1/0.5 | 0.25 + 0.5 | 0.75 | Indifference | |
| Peptide | MIC/MBC (ATCC Strains) | MIC/MBC (Clinical Isolates) | ||||
|---|---|---|---|---|---|---|
| E. coli ML35 43827 | E. coli 35218 | E. coli 11775 | E. coli N°4 | E. coli N°40 | E. coli N°44 | |
| 35409 | 25/25 | 6/6 | >100 | - | - | - |
| 35409-1 | 3/3 | 6/6 | >100 | >100 | 25 | 12.5/100 |
| 35409-2 | 6/12.5 | 12.5/25 | >100 | >100 | 50 | 50/>100 |
| 35409-4 | 25/50 | 25/50 | >100 | >100 | >100 | 100/>100 |
| 35409-13 | 25/50 | 25/100 | 100/>100 | >100 | 50/100 | 25/50 |
| Ciprofloxacin * | 0.008 | 0.016 | 0.016 | >0.256 | 0.016 | >0.256 |
| Gentamicin * | - | - | - | 2 | 1 | >16 |
| Ampicillin * | <0.016 | >16 | <0.016 | - | - | - |
| Peptide | Sequence | MIC (µM) against E. coli 25922 | ||
|---|---|---|---|---|
| MHB | Serum (without Pre-Incubation) | Serum (6 h Pre-Incubation) | ||
| 35409 | RYRRKKKMKKALQYIKLLKE | 25 | 25 | >100 |
| 35409-1 | RKKKMKKALQYIKLLKE | 25 | 12.5 | 50 |
| 35409-2 | KKKMKKALQYIKLLKE | 25 | 25 | >100 |
| 35409-4 | KMKKALQYIKLLKE | 100 | 100 | >100 |
| 35409-13 | KKMKKALQYIKLLK | 50 | 50 | >100 |
| Ciprofloxacin * | 0.008 | 0.008 | 0.004 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreto-Santamaría, A.; Rivera, Z.J.; García, J.E.; Curtidor, H.; Patarroyo, M.E.; Patarroyo, M.A.; Arévalo-Pinzón, G. Shorter Antibacterial Peptide Having High Selectivity for E. coli Membranes and Low Potential for Inducing Resistance. Microorganisms 2020, 8, 867. https://doi.org/10.3390/microorganisms8060867
Barreto-Santamaría A, Rivera ZJ, García JE, Curtidor H, Patarroyo ME, Patarroyo MA, Arévalo-Pinzón G. Shorter Antibacterial Peptide Having High Selectivity for E. coli Membranes and Low Potential for Inducing Resistance. Microorganisms. 2020; 8(6):867. https://doi.org/10.3390/microorganisms8060867
Chicago/Turabian StyleBarreto-Santamaría, Adriana, Zuly Jenny Rivera, Javier Eduardo García, Hernando Curtidor, Manuel Elkin Patarroyo, Manuel Alfonso Patarroyo, and Gabriela Arévalo-Pinzón. 2020. "Shorter Antibacterial Peptide Having High Selectivity for E. coli Membranes and Low Potential for Inducing Resistance" Microorganisms 8, no. 6: 867. https://doi.org/10.3390/microorganisms8060867