Nasal Microbiota in RSV Bronchiolitis

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Virus Detection and RSV Subtyping
2.3. Study of Nasal Microbiota
2.4. Statistical Analysis
3. Results
3.1. Virus Detection
3.2. Clinical Variables
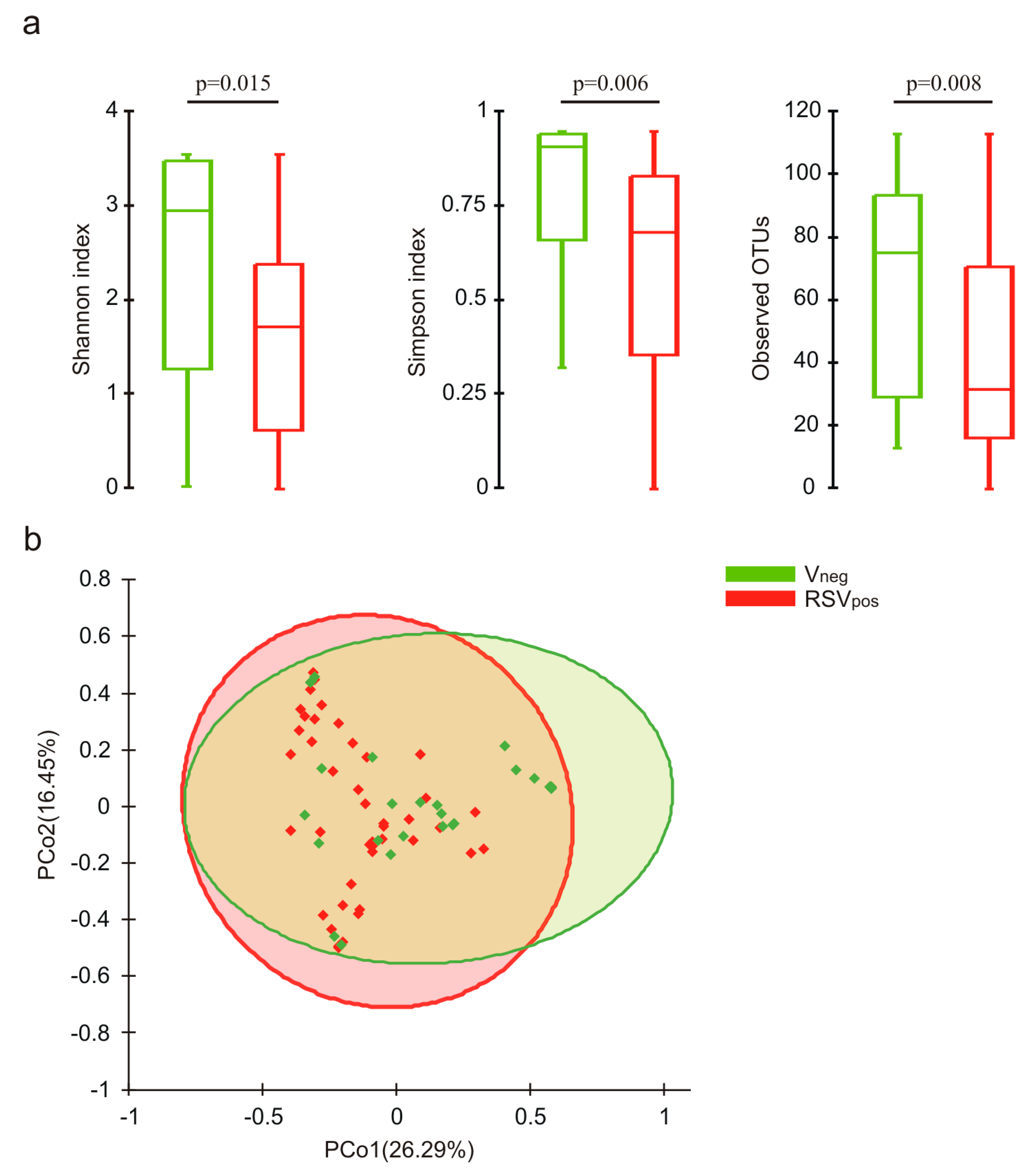
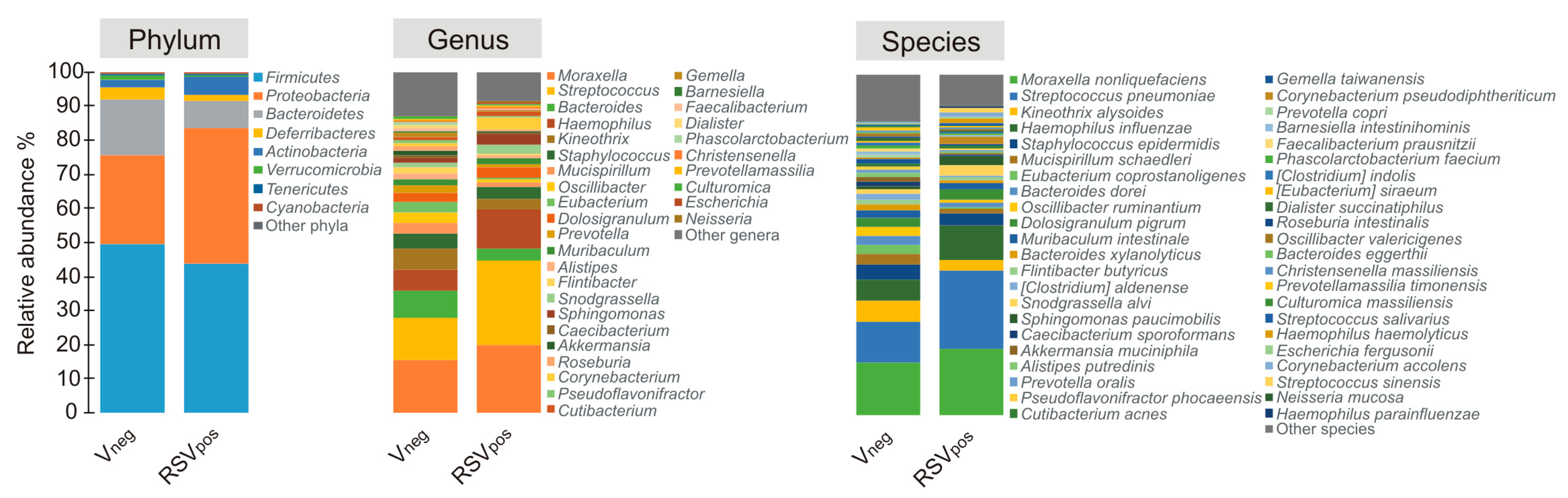
3.3. Nasal Microbiota Characterization
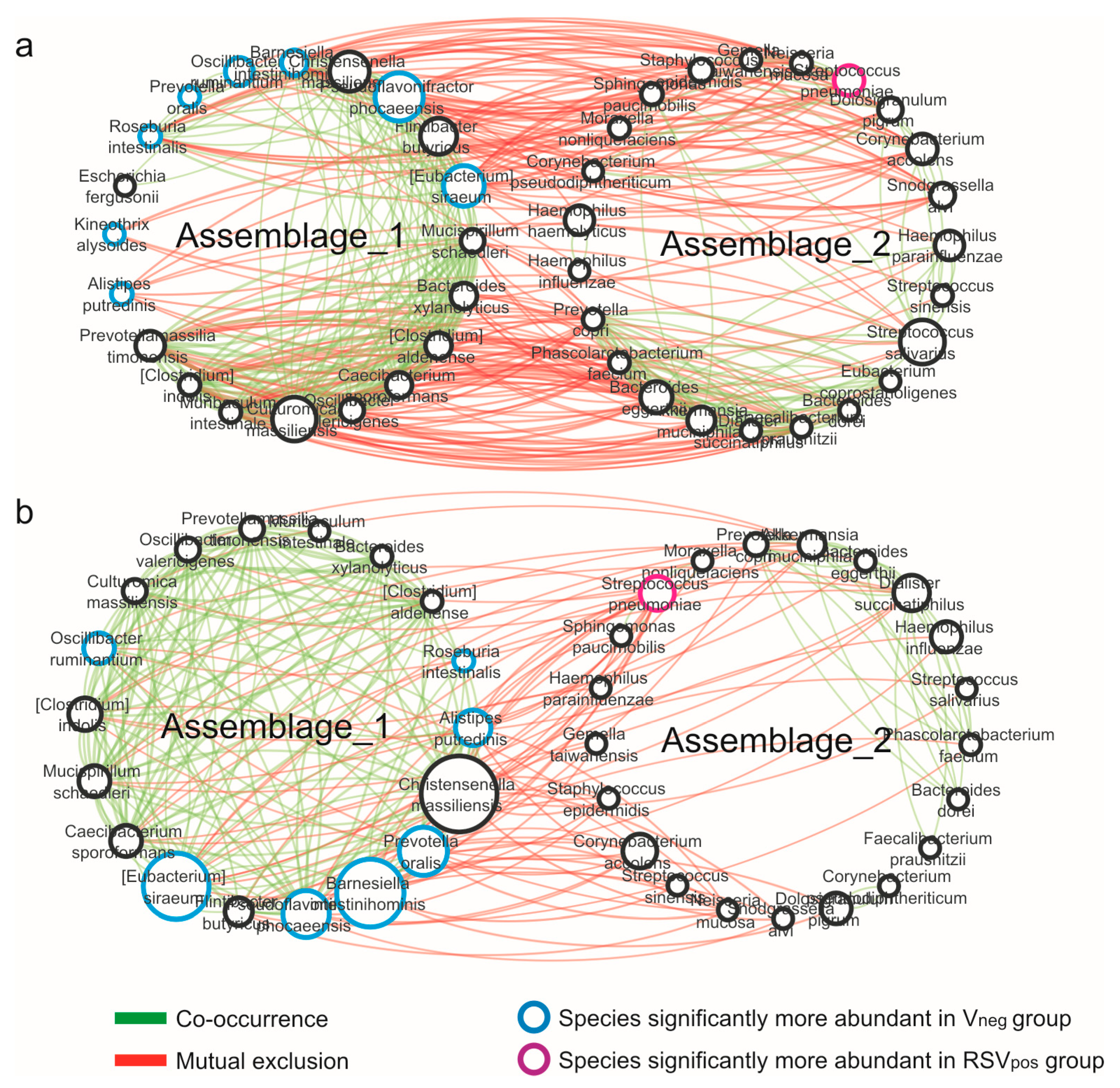
3.4. Network Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Piedra, P.A.; Mansbach, J.M.; Teach, S.J.; Sullivan, A.F.; Forgey, T.; Clark, S.; Espinola, J.A.; Camargo, C.A. for the MARC-30 investigators prospective multicenter study of viral etiology and hospital length of stay in children with severe bronchiolitis. Arch. Pediatr. Adolesc. Med. 2012, 166, 700–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midulla, F.; Nenna, R.; Scagnolari, C.; Petrarca, L.; Frassanito, A.; Viscido, A.; Arima, S.; Antonelli, G.; Pierangeli, A. How respiratory syncytial virus genotypes influence the clinical course in infants hospitalized for bronchiolitis. J. Infect. Dis. 2018, 219, 526–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesbroek, G.; Tsivtsivadze, E.; Sanders, E.A.M.; Montijn, R.; Veenhoven, R.H.; Keijser, B.J.F.; Bogaert, D. Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am. J. Respir. Crit. Care Med. 2014, 190, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Teo, S.M.; Mok, D.; Pham, K.; Kusel, M.; Serralha, M.; Troy, N.; Holt, B.J.; Hales, B.J.; Walker, M.L.; Hollams, E.; et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 2015, 17, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Hyde, E.R.; Petrosino, J.F.; Piedra, P.A.; Camargo, C.A.; Espinola, J.A.; Mansbach, J.M. Nasopharyngeal proteobacteria are associated with viral etiology and acute wheezing in children with severe bronchiolitis. J. Allergy Clin. Immunol. 2013, 133, 1220–1222. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Camargo, C.A. Airway microbiota and acute respiratory infection in children. Expert Rev. Clin. Immunol. 2015, 11, 789–792. [Google Scholar] [CrossRef] [Green Version]
- Passariello, C.; Schippa, S. Rhinovirus promote internalization of S.aureus into non-fully permissive cultered pneumocytes. Microbes Infect. 2006, 8, 758–766. [Google Scholar] [CrossRef]
- Rosas-Salazar, C.; Shilts, M.H.; Tovchigrechko, A.; Schobel, S.; Chappell, J.D.; Larkin, E.K.; Gebretsadik, T.; Halpin, R.A.; Nelson, K.E.; Moore, M.L.; et al. Nasopharyngeal Lactobacillus is associated with a reduced risk of childhood wheezing illnesses following acute respiratory syncytial virus infection in infancy. J. Allergy Clin. Immunol. 2018, 142, 1447–1456.e9. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Linnemann, R.W.; Mansbach, J.M.; Ajami, N.J.; Espinola, J.A.; Petrosino, J.F.; Piedra, P.A.; Stevenson, M.D.; Sullivan, A.F.; Thompson, A.D.; et al. The Fecal Microbiota Profile and Bronchiolitis in Infants. Pediatrics 2016, 138, e20160218. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Mansbach, J.M.; Ajami, N.J.; Espinola, J.A.; Henke, D.M.; Petrosino, J.F.; Piedra, P.A.; Shaw, C.A.; Sullivan, A.F.; Camargo, C.A.; et al. Association of nasopharyngeal microbiota profiles with bronchiolitis severity in infants hospitalised for bronchiolitis. Eur. Respir. J. 2016, 48, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Sonawane, A.R.; Tian, L.; Chu, C.-Y.; Qiu, X.; Wang, L.; Holden-Wiltse, J.; Grier, A.; Gill, S.R.; Caserta, M.T.; Falsey, A.R.; et al. Microbiome-transcriptome interactions related to severity of respiratory syncytial virus infection. Sci. Rep. 2019, 9, 13824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midulla, F.; Scagnolari, C.; Bonci, E.; Pierangeli, A.; Antonelli, G.; De Angelis, D.; Berardi, R.; Moretti, C. Respiratory syncytial virus, human bocavirus and rhinovirus bronchiolitis in infants. Arch. Dis. Child. 2009, 95, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.L.; Openshaw, P.J. Bronchiolitis. Lancet 2006, 368, 312–322. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Boil. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.B.; Hoffman, L.R.; Carroll, M.P.; Bruce, K.D. Interpreting infective microbiota: The importance of an ecological perspective. Trends Microbiol. 2013, 21, 271–276. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, S.L.; Leth, M.; Michalak, L.; Hansen, M.E.; Pudlo, N.A.; Glowacki, R.; Pereira, G.; Workman, C.T.; Arntzen, M.; Pope, P.B.; et al. The human gut Firmicute Roseburia intestinalis is a primary degrader of dietary β-mannans. Nat. Commun. 2019, 10, 905. [Google Scholar] [CrossRef]
- Zhu, C.; Song, K.; Shen, Z.; Quan, Y.; Tan, B.; Luo, W.; Wu, S.; Tang, K.; Yang, Z.; Wang, X. Roseburia intestinalis inhibits interleukin-17 excretion and promotes regulatory T cells differentiation in colitis. Mol. Med. Rep. 2018, 17, 7567–7574. [Google Scholar] [CrossRef]
- Dhaliwal, G. Alistipes: The influence of a commensal on anxiety and depression. Catalyst 2019, 3, 47–51. [Google Scholar]
- Brealey, J.C.; Chappell, K.J.; Galbraith, S.; Fantino, E.; Gaydon, J.; Tozer, S.; Young, P.; Holt, P.; Sly, P.D. Streptococcus pneumoniae colonization of the nasopharynx is associated with increased severity during respiratory syncytial virus infection in young children. Respirology 2017, 23, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.T.; Louwen, R.; Elberse, K.; Van Amerongen, G.; Yüksel, S.; Luijendijk, A.; Osterhaus, A.D.M.E.; Duprex, W.P.; De Swart, R.L. Streptococcus pneumoniae enhances human respiratory syncytial virus infection In Vitro and In Vivo. PLoS ONE 2015, 10, e0127098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.M.; Sandrini, S.; Datta, S.; Freestone, P.; Shafeeq, S.; Radhakrishnan, P.; Williams, G.; Glenn, S.M.; Kuipers, O.P.; Hirst, R.A.; et al. Respiratory syncytial virus increases the virulence of Streptococcus pneumoniae by binding to penicillin binding protein 1a. A new paradigm in respiratory infection. Am. J. Respir. Crit. Care Med. 2014, 190, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rynda-Apple, A.; Robinson, K.M.; Alcorn, J.F. Influenza and bacterial superinfection: Illuminating the immunologic mechanisms of disease. Infect. Immun. 2015, 83, 3764–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma-Chawla, N.; Sender, V.; Kershaw, O.; Gruber, A.D.; Volckmar, J.; Henriques-Normark, B.; Stegemann-Koniszewski, S.; Bruder, D. Influenza a virus infection predisposes hosts to secondary infection with different streptococcus pneumoniae serotypes with similar outcome but serotype-specific manifestation. Infect. Immun. 2016, 84, 3445–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Genet. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Piters, W.D.S.; Jochems, S.P.; Mitsi, E.; Rylance, J.; Pojar, S.; Nikolaou, E.; German, E.L.; Holloway, M.; Carniel, B.F.; Chu, M.L.J.N.; et al. Interaction between the nasal microbiota and S. pneumoniae in the context of live-attenuated influenza vaccine. Nat. Commun. 2019, 10, 2981. [Google Scholar] [CrossRef]
- Sharma-Chawla, N.; Stegemann-Koniszewski, S.; Christen, H.; Boehme, J.D.; Kershaw, O.; Schreiber, J.; Guzmán, C.A.; Bruder, D.; Vargas, E.A.H. In vivo neutralization of pro-inflammatory cytokines during secondary streptococcus pneumoniae infection post influenza a virus infection. Front. Immunol. 2019, 10, 1864. [Google Scholar] [CrossRef] [Green Version]
- Piters, W.D.S.; Heinonen, S.; Hasrat, R.; Bunsow, E.; Smith, B.; Suarez-Arrabal, M.-C.; Chaussabel, D.; Cohen, D.M.; Sanders, E.A.M.; Ramilo, O.; et al. Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am. J. Respir. Crit. Care Med. 2016, 194, 1104–1115. [Google Scholar] [CrossRef]
- Sande, C.J.; Njunge, J.M.; Ngoi, J.M.; Mutunga, M.N.; Chege, T.; Gicheru, E.T.; Gardiner, E.M.; Gwela, A.; Green, A.C.; Drysdale, S.B.; et al. Airway response to respiratory syncytial virus has incidental antibacterial effects. Nat. Commun. 2019, 10, 2218. [Google Scholar] [CrossRef]
- Mika, M.; Mack, I.; Korten, I.; Qi, W.; Aebi, S.; Frey, U.; Latzin, P.; Hilty, M. Dynamics of the nasal microbiota in infancy: A prospective cohort study. J. Allergy Clin. Immunol. 2015, 135, 905–912.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; Brandt, P.A.V.D.; Stobberingh, E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoetendal, E.G.; Ben-Amor, K.; Akkermans, A.D.; Abee, T.; De Vos, W.M. DNA isolation protocols affect the detection limit of pcrapproaches of bacteria in samples from the human gastrointestinal Tract. Syst. Appl. Microbiol. 2001, 24, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Howard, L.M.; Zhu, Y.; Griffin, M.R.; Edwards, K.M.; Williams, J.V.; Gil, A.I.; Vidal, J.E.; Klugman, K.P.; Lanata, C.F.; Grijalva, C.G. Nasopharyngeal pneumococcal density during asymptomatic respiratory virus infection and risk for subsequent acute respiratory illness. Emerg. Infect. Dis. 2019, 25, 2040–2047. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Salazar, C.; Shilts, M.H.; Tovchigrechko, A.; Chappell, J.D.; Larkin, E.K.; Nelson, K.E.; Moore, M.L.; Anderson, L.J.; Das, S.R.; Hartert, T.V. Nasopharyngeal microbiome in respiratory syncytial virus resembles profile associated with increased childhood asthma risk. Am. J. Respir. Crit. Care Med. 2016, 193, 1180–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Vneg (N = 28) | RSVpos (N = 48) | p Value |
|---|---|---|---|
| Male | 13 (46.4%) | 25 (52.1%) | 0.634 |
| Caesarean section | 12 (42.9%) | 27 (43.8%) | 0.940 |
| Age (days) | 65.7 ± 3 4.8 | 76.3 ± 34.5 | 0.139 |
| Weight | 5.0 ± 1.2 | 5.3 ± 1.0 | 0.291 |
| Hospitalization (days) | 3.7 ± 1.6 | 4.8 ± 2.4 | 0.071 |
| Severity score | 3.0 ± 1.6 | 3.9 ± 1.5 | 0.016 |
| O2 therapy | 3 (10.7%) | 18 (37.5%) | 0.012 |
| Admission to PICU | 0 (0.0%) | 3 (6.3%) | 0.018 |
| White blood cells | 11083.2 ± 3615.9 | 9784.4 ± 2965.8 | 0.189 |
| Lymphocytes absolute | 5683.2 ± 1689.1 | 4797.4 ± 2039.4 | 0.030 |
| Neutrophils absolute | 3323.7 ± 2464.9 | 3166.5 ± 1967.3 | 0.834 |
| Eosinophil absolute | 212.7 ± 192.9 | 136.7 ± 190.5 | 0.062 |
| Eosinophil > 400 | 4 (14.3%) | 2 (4.2%) | 0.115 |
| Breastfeeding | 15 (55.61%) | 31 (64.6%) | 0.441 |
| Properties | Vneg | RSVpos |
|---|---|---|
| Nodes | 43 | 39 |
| Edges | 221 | 124 |
| Edges/node ratio | 5.14 | 3.18 |
| Synergisms | 121 | 85 |
| Exclusions | 100 | 39 |
| Syn/Escl ratio | 1.21 | 2.17 |
| Average number of neighbors | 10.28 | 6.359 |
| Network density | 0.245 | 0.167 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schippa, S.; Frassanito, A.; Marazzato, M.; Nenna, R.; Petrarca, L.; Neroni, B.; Bonfiglio, G.; Guerrieri, F.; Frasca, F.; Oliveto, G.; et al. Nasal Microbiota in RSV Bronchiolitis. Microorganisms 2020, 8, 731. https://doi.org/10.3390/microorganisms8050731
Schippa S, Frassanito A, Marazzato M, Nenna R, Petrarca L, Neroni B, Bonfiglio G, Guerrieri F, Frasca F, Oliveto G, et al. Nasal Microbiota in RSV Bronchiolitis. Microorganisms. 2020; 8(5):731. https://doi.org/10.3390/microorganisms8050731
Chicago/Turabian StyleSchippa, Serena, Antonella Frassanito, Massimiliano Marazzato, Raffaella Nenna, Laura Petrarca, Bruna Neroni, Giulia Bonfiglio, Francesca Guerrieri, Federica Frasca, Giuseppe Oliveto, and et al. 2020. "Nasal Microbiota in RSV Bronchiolitis" Microorganisms 8, no. 5: 731. https://doi.org/10.3390/microorganisms8050731