Plasticity of Mitochondrial DNA Inheritance and Its Impact on Nuclear Gene Transcription in Yeast Hybrids


Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Media
2.2. Construction of Hybrids
2.3. Whole Genome DNA Extraction
2.4. DNA Content Analysis by Flow Cytometry
2.5. Analysis of Mitochondrial DNA by Sanger Sequencing
2.6. Growth Assays
2.7. RNA Extraction and Sequencing
2.8. Reverse Transcription and Real-Time Quantitative PCR
2.9. Assembly, annotation, and DE analysis
2.10. Functional Annotation and Co-Expression Analysis
3. Results and Discussion
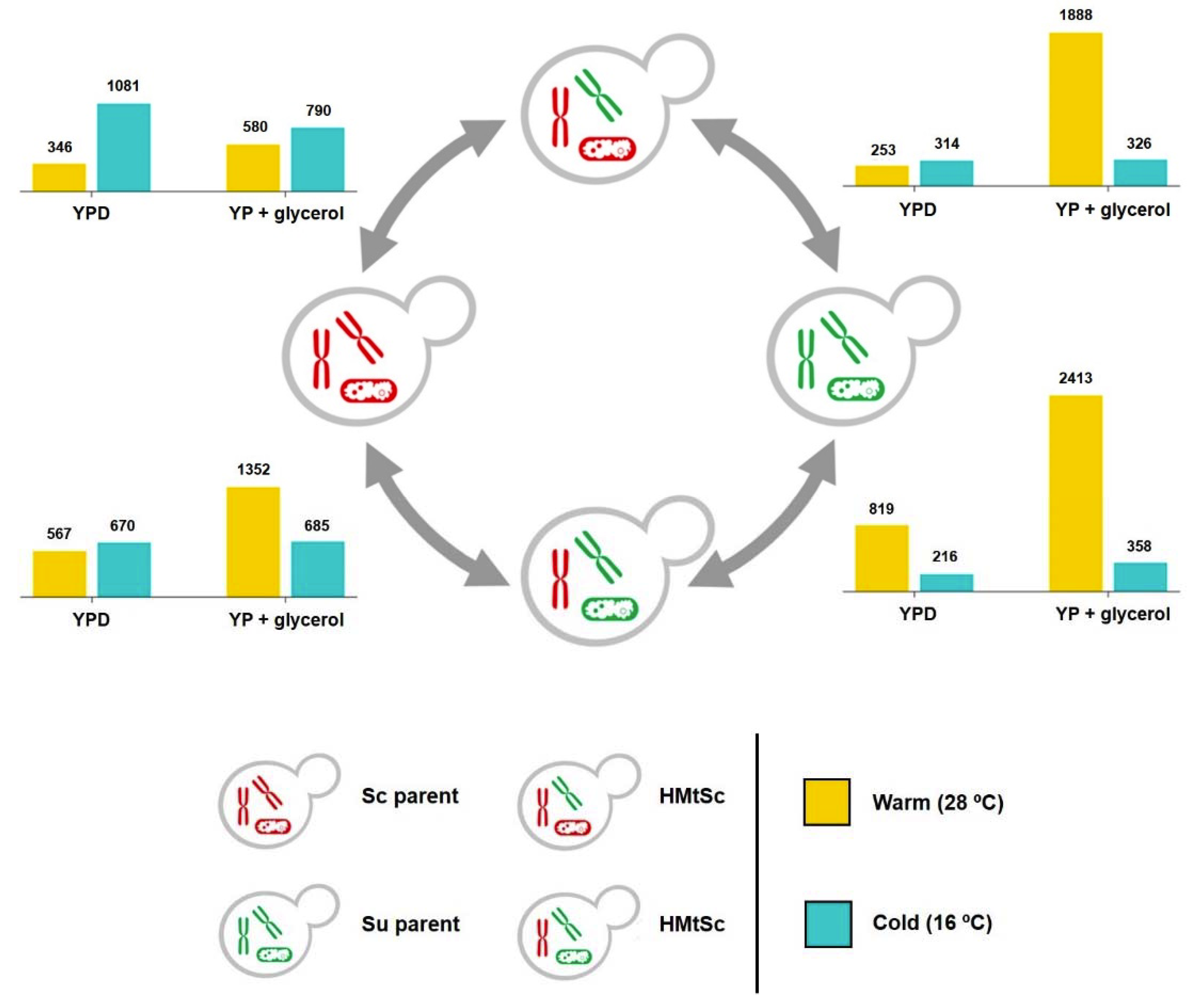
3.1. Different Mitochondrial DNA is Retained in Saccharomyces Hybrids Depending on the Environments Where They Have Been Formed
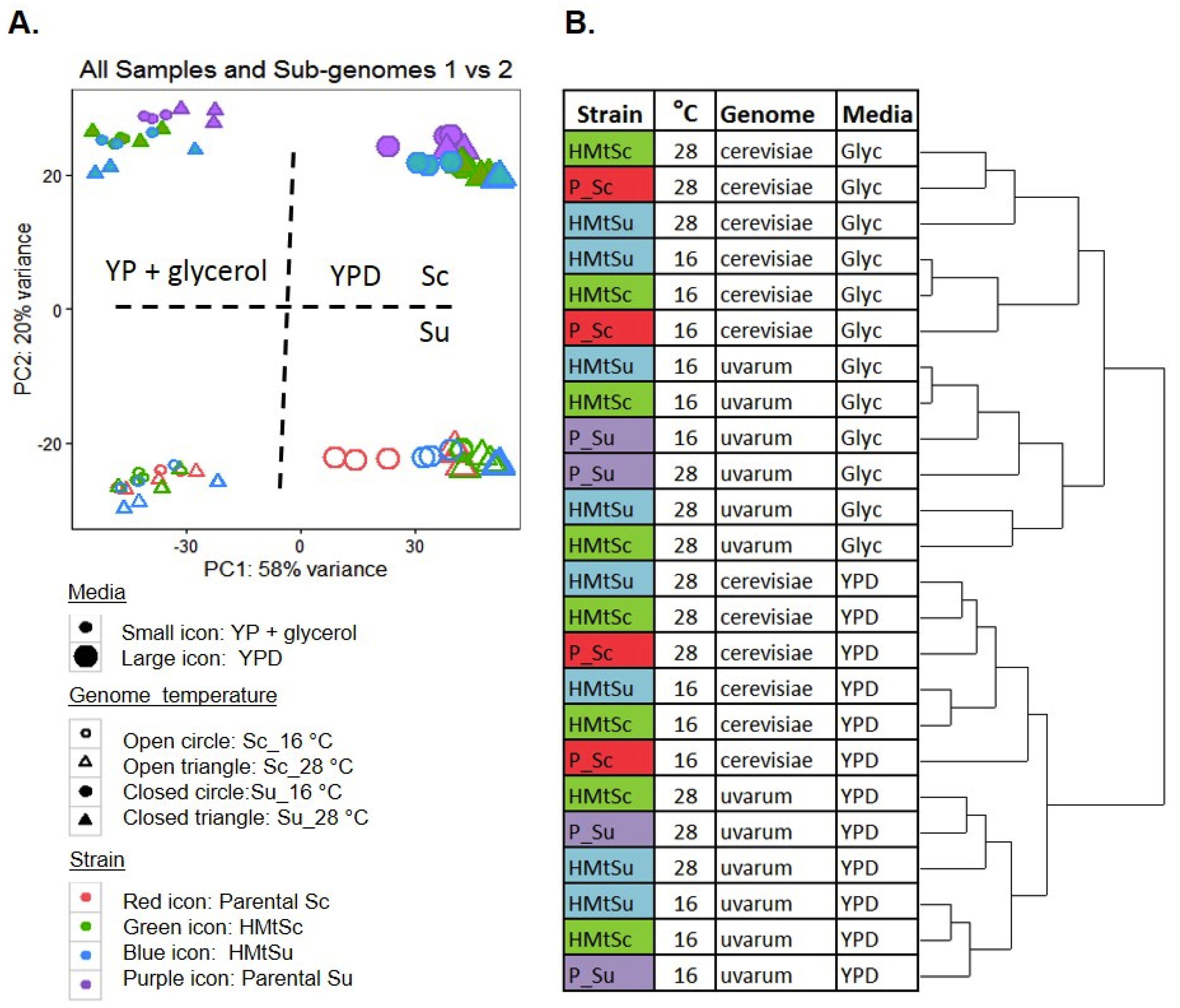
3.2. RNA Sequencing Strategy and Hierarchy of Factors Affecting Transcriptional Responses
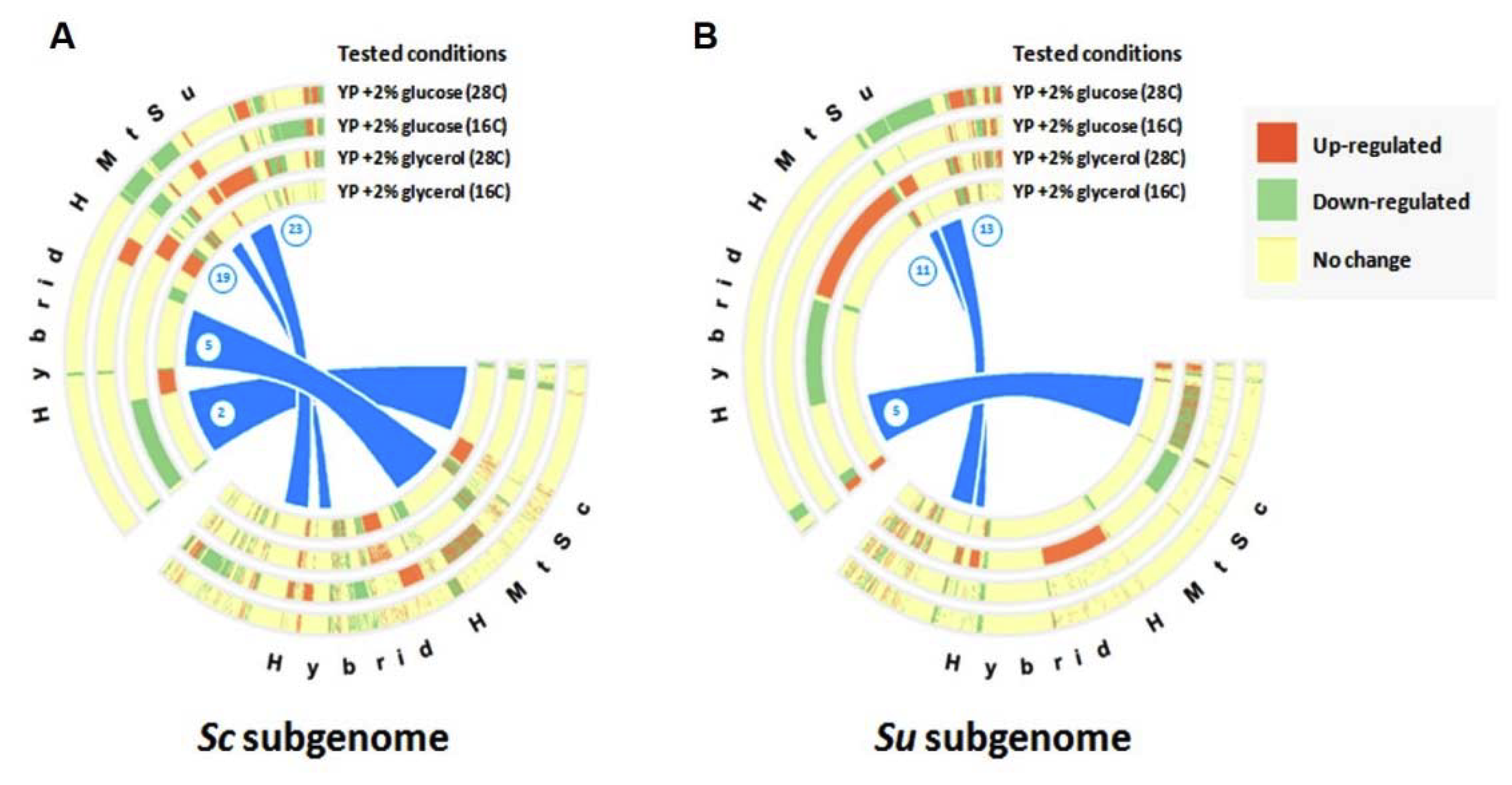
3.3. Plasticity of Allele Specific Expression in Sc/Su Hybrids
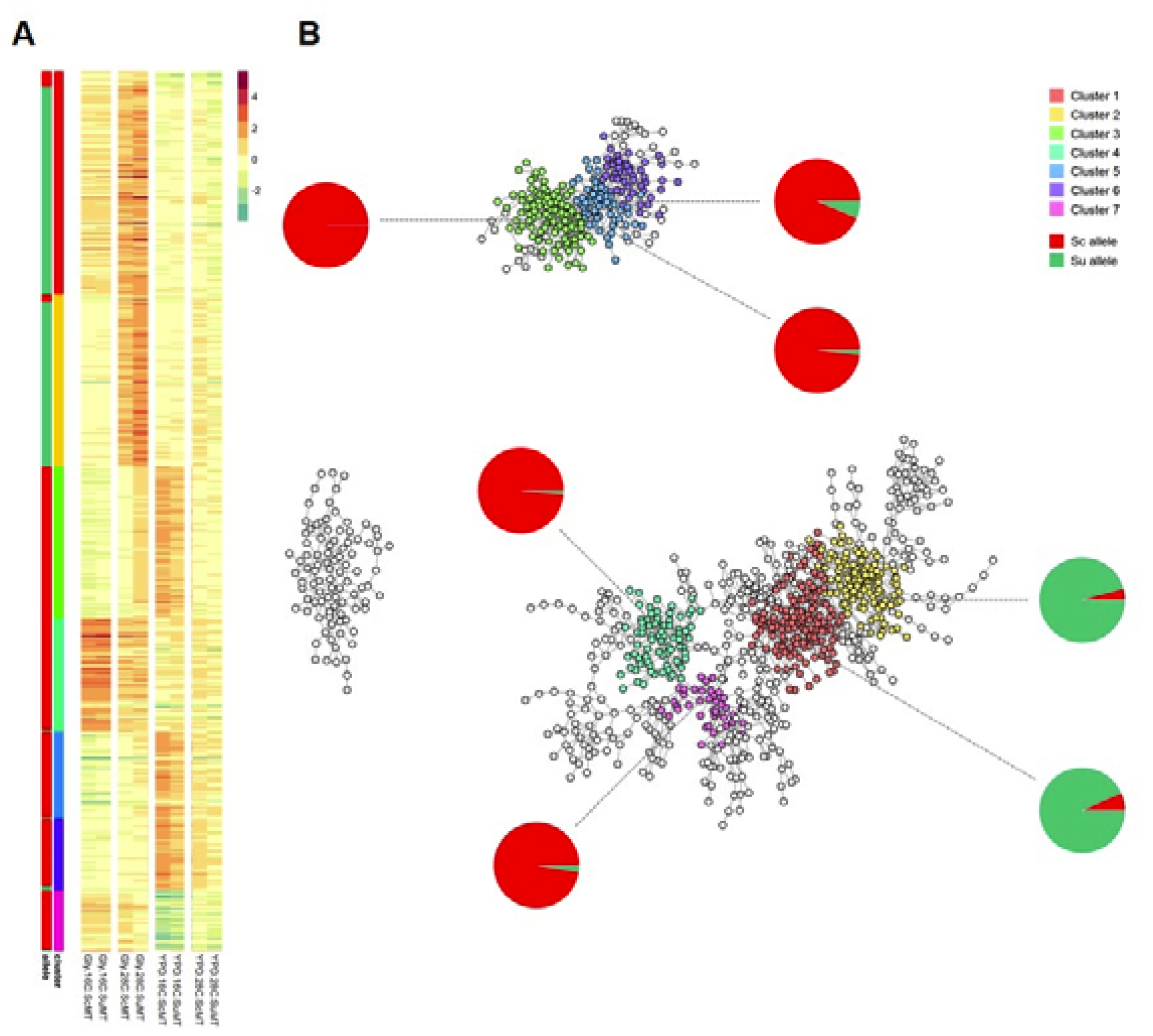
3.4. Co-Expression Analysis in HMtSc and HMtSu Hybrids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Libkind, D.; Hittinger, C.T.; Valerio, E.; Goncalves, C.; Dover, J.; Johnston, M.; Goncalves, P.; Sampaio, J.P. Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 14539–14544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, B.; Sherlock, G. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 2008, 18, 1610–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peris, D.; Sylvester, K.; Libkind, D.; Goncalves, P.; Sampaio, J.P.; Alexander, W.G.; Hittinger, C.T. Population structure and reticulate evolution of Saccharomyces eubayanus and its lager-brewing hybrids. Mol. Ecol. 2014, 23, 2031–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainieri, S.; Kodama, Y.; Nakao, Y.; Pulvirenti, A.; Giudici, P. The inheritance of mtDNA in lager brewing strains. FEMS Yeast Res. 2008, 8, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.S.; Barrio, E.; Gafner, J.; Querol, A. Natural hybrids from Saccharomyces cerevisiae, Saccharomyces bayanus and Saccharomyces kudriavzevii in wine fermentations. FEMS Yeast Res. 2006, 6, 1221–1234. [Google Scholar] [CrossRef] [Green Version]
- Masneuf, I.; Hansen, J.; Groth, C.; Piskur, J.; Dubourdieu, D. New hybrids between Saccharomyces sensu stricto yeast species found among wine and cider production strains. Appl. Environ. Microbiol. 1998, 64, 3887–3892. [Google Scholar] [CrossRef] [Green Version]
- Salvado, Z.; Arroyo-Lopez, F.N.; Guillamon, J.M.; Salazar, G.; Querol, A.; Barrio, E. Temperature adaptation markedly determines evolution within the genus Saccharomyces. Appl. Environ. Microbiol. 2011, 77, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, J.P.; Goncalves, P. Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl. Environ. Microbiol. 2008, 74, 2144–2152. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, S.K.; Donaldson, I.J.; Lovell, S.C.; Delneri, D. Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse. PLoS ONE 2014, 9, e92203. [Google Scholar] [CrossRef] [Green Version]
- Piatkowska, E.M.; Naseeb, S.; Knight, D.; Delneri, D. Chimeric protein complexes in hybrid species generate novel phenotypes. PLoS Genet. 2013, 9, e1003836. [Google Scholar] [CrossRef] [Green Version]
- Naseeb, S.; Carter, Z.; Minnis, D.; Donaldson, I.; Zeef, L.; Delneri, D. Widespread Impact of Chromosomal Inversions on Gene Expression Uncovers Robustness via Phenotypic Buffering. Mol. Biol. Evol. 2016, 33, 1679–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belloch, C.; Querol, A.; Garcia, M.D.; Barrio, E. Phylogeny of the genus Kluyveromyces inferred from the mitochondrial cytochrome-c oxidase II gene. Int. J. Syst. Evol. Microbiol. 2000, 50, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseeb, S.; Ames, R.M.; Delneri, D.; Lovell, S.C. Rapid functional and evolutionary changes follow gene duplication in yeast. Proc. Biol. Sci. 2017, 284. [Google Scholar] [CrossRef] [Green Version]
- Hooks, K.B.; Naseeb, S.; Parker, S.; Griffiths-Jones, S.; Delneri, D. Novel Intronic RNA Structures Contribute to Maintenance of Phenotype in Saccharomyces cerevisiae. Genetics 2016, 203, 1469–1481. [Google Scholar] [CrossRef] [Green Version]
- Scannell, D.R.; Zill, O.A.; Rokas, A.; Payen, C.; Dunham, M.J.; Eisen, M.B.; Rine, J.; Johnston, M.; Hittinger, C.T. The Awesome Power of Yeast Evolutionary Genetics: New Genome Sequences and Strain Resources for the Saccharomyces sensu stricto Genus. G3 2011, 1, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Remm, M.; Storm, C.E.; Sonnhammer, E.L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001, 314, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabasi, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar] [CrossRef] [Green Version]
- Albert, R. Scale-free networks in cell biology. J. Cell Sci. 2005, 118, 4947–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thieffry, D.; Romero, D. The modularity of biological regulatory networks. BioSystems 1999, 50, 49–59. [Google Scholar] [CrossRef]
- Albertin, W.; da Silva, T.; Rigoulet, M.; Salin, B.; Masneuf-Pomarede, I.; de Vienne, D.; Sicard, D.; Bely, M.; Marullo, P. The mitochondrial genome impacts respiration but not fermentation in interspecific Saccharomyces hybrids. PLoS ONE 2013, 8, e75121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Sun, S.; Shahid, M.; Xu, J. Environment factors can influence mitochondrial inheritance in the fungus Cryptococcus neoformans. Fungal Genet. Biol. 2007, 44, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.P.; Peris, D.; Moriarty, R.V.; Li, X.C.; Fay, J.C.; Hittinger, C.T. Mitochondrial DNA and temperature tolerance in lager yeasts. Sci. Adv. 2019, 5, eaav1869. [Google Scholar] [CrossRef] [Green Version]
- Li, X.C.; Peris, D.; Hittinger, C.T.; Sia, E.A.; Fay, J.C. Mitochondria-encoded genes contribute to evolution of heat and cold tolerance in yeast. Sci. Adv. 2019, 5, eaav1848. [Google Scholar] [CrossRef] [Green Version]
- Belloch, C.; Perez-Torrado, R.; Gonzalez, S.S.; Perez-Ortin, J.E.; Garcia-Martinez, J.; Querol, A.; Barrio, E. Chimeric genomes of natural hybrids of Saccharomyces cerevisiae and Saccharomyces kudriavzevii. Appl. Environ. Microbiol. 2009, 75, 2534–2544. [Google Scholar] [CrossRef] [Green Version]
- Andersen, S.L.; Petes, T.D. Reciprocal uniparental disomy in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, 9947–9952. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, N.M.V.; Watson, R.A.; Argueso, J.L. Controlled Reduction of Genomic Heterozygosity in an Industrial Yeast Strain Reveals Wide Cryptic Phenotypic Variation. Front. Genet. 2019, 10, 782. [Google Scholar] [CrossRef]
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef] [Green Version]
- De Meaux, J.; Pop, A.; Mitchell-Olds, T. Cis-regulatory evolution of chalcone-synthase expression in the genus Arabidopsis. Genetics 2006, 174, 2181–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Yang, S.; Rupe, M.; Hu, B.; Bickel, D.R.; Arthur, L.; Smith, O. Genome-wide allele-specific expression analysis using Massively Parallel Signature Sequencing (MPSS) reveals cis- and trans-effects on gene expression in maize hybrid meristem tissue. Plant Mol. Biol. 2008, 66, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, A.; Pir, P.; Castrillo, J.I.; Charles, P.D.; Lilley, K.S.; Oliver, S.G. Nutrient control of eukaryote cell growth: A systems biology study in yeast. BMC Biol. 2010, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regenberg, B.; Grotkjaer, T.; Winther, O.; Fausboll, A.; Akesson, M.; Bro, C.; Hansen, L.K.; Brunak, S.; Nielsen, J. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006, 7, R107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegarty, M.J.; Barker, G.L.; Brennan, A.C.; Edwards, K.J.; Abbott, R.J.; Hiscock, S.J. Changes to gene expression associated with hybrid speciation in plants: Further insights from transcriptomic studies in Senecio. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3055–3069. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Merino, R.A.; Kuanyshev, N.; Byrne, K.P.; Varela, J.A.; Morrissey, J.P.; Porro, D.; Wolfe, K.H.; Branduardi, P. Transcriptional Response to Lactic Acid Stress in the Hybrid Yeast Zygosaccharomyces parabailii. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Sun, Y.; Shen, K.; Sun, S.; Wang, J.; Jiang, T.; Cao, S.; Josiah, S.M.; Pang, J.; Lin, X.; et al. Immediate Genetic and Epigenetic Changes in F1 Hybrids Parented by Species with Divergent Genomes in the Rice Genus (Oryza). PLoS ONE 2015, 10, e0132911. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Sun, Y.; Wang, X.; Lin, X.; Sun, S.; Shen, K.; Wang, J.; Jiang, T.; Zhong, S.; Xu, C.; et al. Transcriptome shock in an interspecific F1 triploid hybrid of Oryza revealed by RNA sequencing. J. Integr. Plant Biol. 2016, 58, 150–164. [Google Scholar] [CrossRef]
- Boldogh, I.R.; Pon, L.A. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 2006, 1763, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Moseley, J.B.; Goode, B.L. The yeast actin cytoskeleton: From cellular function to biochemical mechanism. Microbiol. Mol. Biol. Rev. 2006, 70, 605–645. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamanoi, F. Ras signaling in yeast. Genes Cancer 2011, 2, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, J.I.; Zeef, L.A.; Hoyle, D.C.; Zhang, N.; Hayes, A.; Gardner, D.C.; Cornell, M.J.; Petty, J.; Hakes, L.; Wardleworth, L.; et al. Growth control of the eukaryote cell: A systems biology study in yeast. J. Biol. 2007, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zhang, X.; Hu, J.Y.; Turck, F.; Dong, X.; Goebel, U.; Borevitz, J.O.; de Meaux, J. Widespread interspecific divergence in cis-regulation of transposable elements in the Arabidopsis genus. Mol. Biol. Evol. 2012, 29, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Moritz, M.; Pulaski, B.A.; Woolford, J.L., Jr. Assembly of 60S ribosomal subunits is perturbed in temperature-sensitive yeast mutants defective in ribosomal protein L16. Mol. Cell. Biol. 1991, 11, 5681–5692. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.Y.; Hung, Y.S.; Lin, K.H.; Lee, H.Y.; Leu, J.Y. Multiple molecular mechanisms cause reproductive isolation between three yeast species. PLoS Biol. 2010, 8, e1000432. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Chou, J.Y.; Cheong, L.; Chang, N.H.; Yang, S.Y.; Leu, J.Y. Incompatibility of nuclear and mitochondrial genomes causes hybrid sterility between two yeast species. Cell 2008, 135, 1065–1073. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature | Media | Total Number of Crosses Made | Total Number of Hybrids Made | No. of Hybrids with Sc MtDNA | No. of Hybrids with Su MtDNA |
|---|---|---|---|---|---|
| 28 °C | YPD | 22 | 21 | 10 | 11 |
| YP+Glycerol | 22 | 19 | 19 ** | 0 ** | |
| 16 °C | YPD | 26 | 19 | 13 | 6 |
| YP+Glycerol | 22 | 5 | 5 * | 0 | |
| 10 °C | YPD | 48 | 17 | 5 ** | 12 ** |
| YP+Glycerol | 62 | 1 | 0 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hewitt, S.K.; Duangrattanalert, K.; Burgis, T.; Zeef, L.A.H.; Naseeb, S.; Delneri, D. Plasticity of Mitochondrial DNA Inheritance and Its Impact on Nuclear Gene Transcription in Yeast Hybrids. Microorganisms 2020, 8, 494. https://doi.org/10.3390/microorganisms8040494
Hewitt SK, Duangrattanalert K, Burgis T, Zeef LAH, Naseeb S, Delneri D. Plasticity of Mitochondrial DNA Inheritance and Its Impact on Nuclear Gene Transcription in Yeast Hybrids. Microorganisms. 2020; 8(4):494. https://doi.org/10.3390/microorganisms8040494
Chicago/Turabian StyleHewitt, Sarah K., Kobchai Duangrattanalert, Tim Burgis, Leo A.H. Zeef, Samina Naseeb, and Daniela Delneri. 2020. "Plasticity of Mitochondrial DNA Inheritance and Its Impact on Nuclear Gene Transcription in Yeast Hybrids" Microorganisms 8, no. 4: 494. https://doi.org/10.3390/microorganisms8040494