
A Machine Learning-Based Diagnostic Model for Crohn’s Disease and Ulcerative Colitis Utilizing Fecal Microbiome Analysis
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Research Cohorts and Sample Collection
2.2. Sample Preparation and 16S rRNA Gene Sequencing
2.3. Data Processing and Downstream Analysis
2.4. Machine Learning for Disease Prediction Model
3. Results
3.1. Processing of 16S rRNA Gene Amplicon Sequencing Data
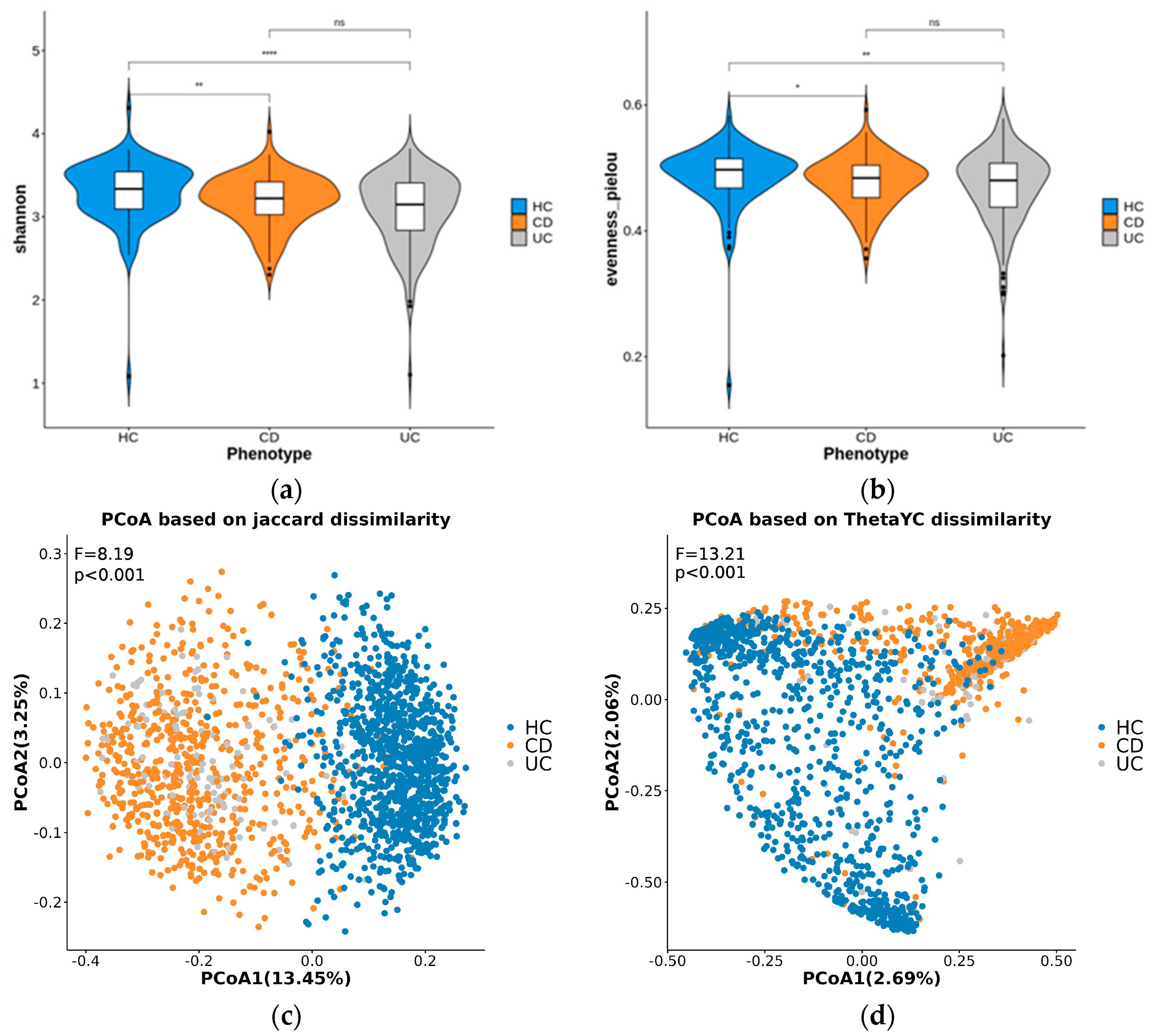
3.2. Diversity Analysis
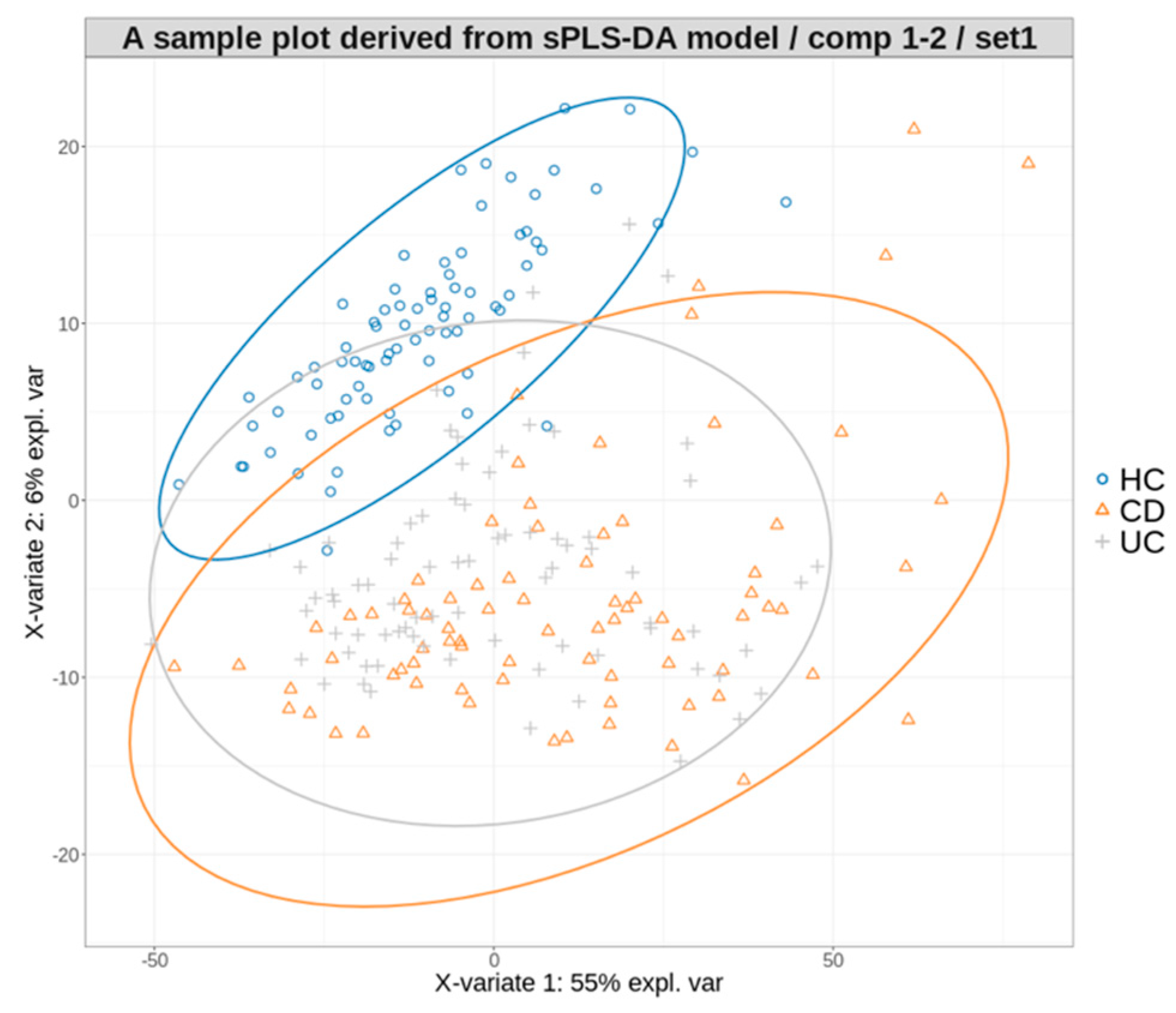
3.3. Multiclass Disease Prediction Model
3.4. Hierarchical Disease Prediction Model
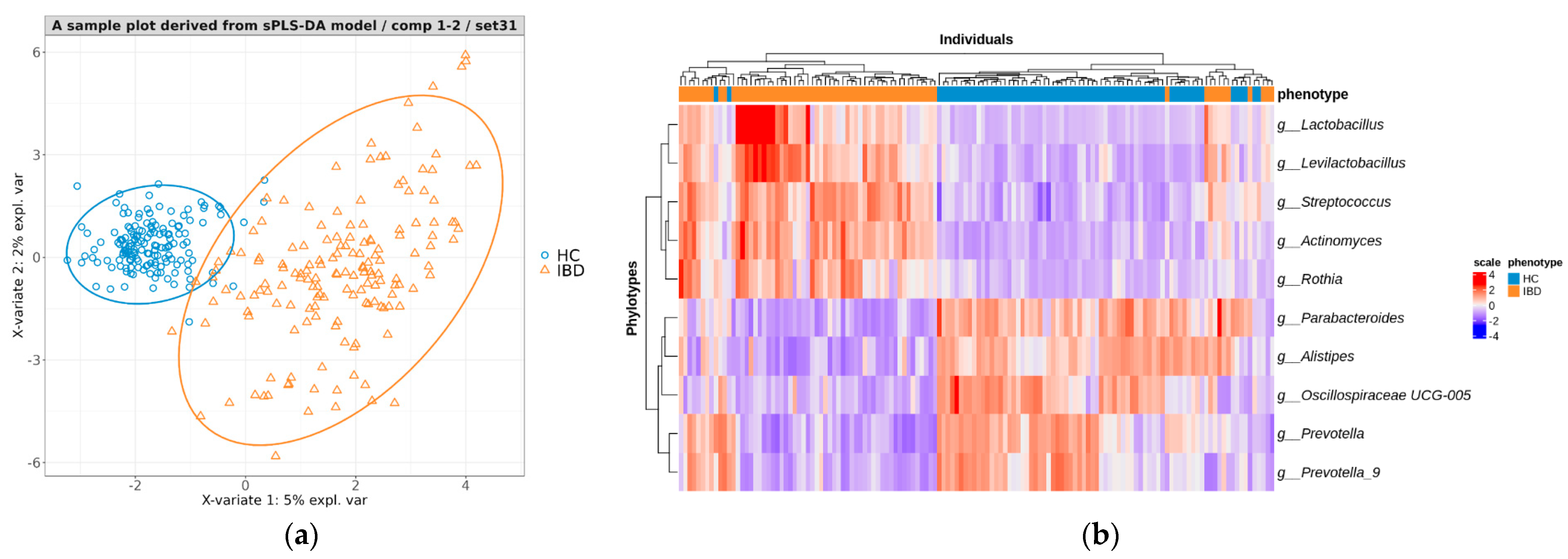
3.4.1. Creating a Predictive Model for Distinguishing between IBD and HC
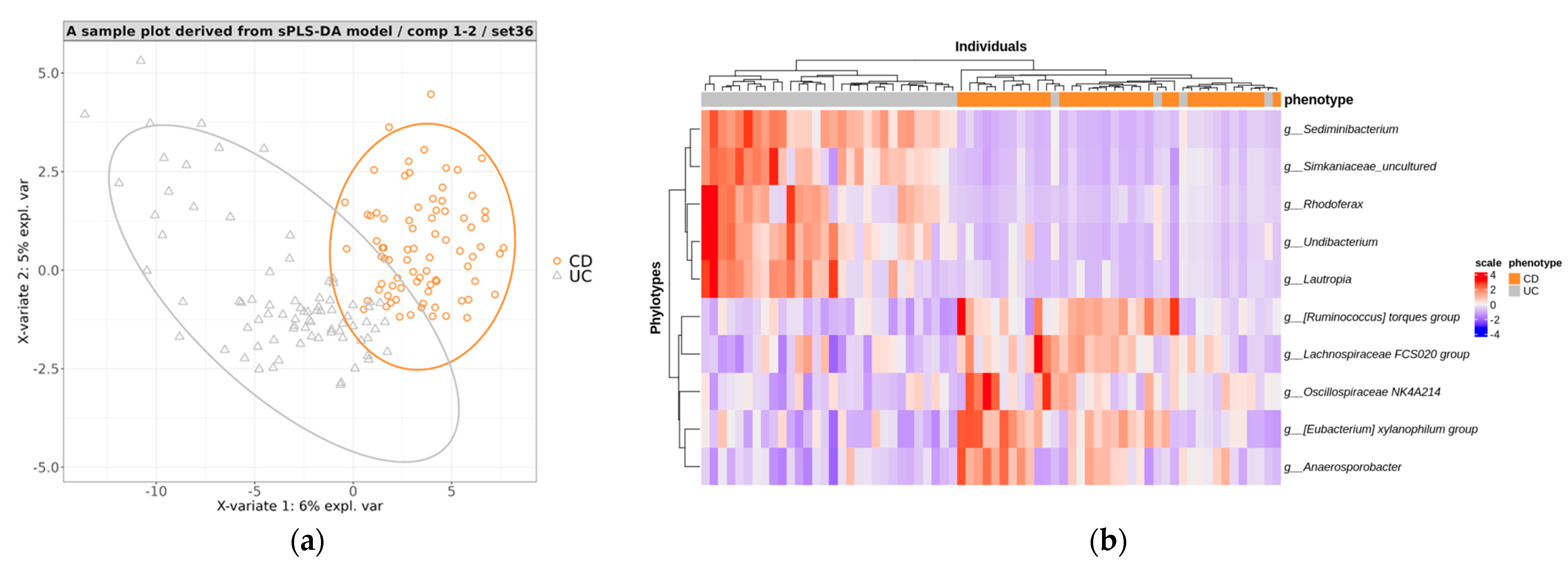
3.4.2. Creating a Predictive Model for Distinguishing between CD and UC
3.4.3. Performance Evaluation of Models in Hierarchical Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Massironi, S.; Viganò, C.; Palermo, A.; Pirola, L.; Mulinacci, G.; Allocca, M.; Peyrin-Biroulet, L.; Danese, S. Inflammation and malnutrition in inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2023, 8, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohn’s Colitis 2019, 13, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Gomollón, F.; Dignass, A.; Annese, V.; Tilg, H.; Van Assche, G.; Lindsay, J.O.; Peyrin-Biroulet, L.; Cullen, G.J.; Daperno, M.; Kucharzik, T.; et al. 3rd European Evidence-based Consensus on the Diagnosis and Management of Crohn’s Disease 2016: Part 1: Diagnosis and Medical Management. J. Crohn’s Colitis 2017, 11, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileoanal Pouch Disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Shivashankar, R.; Lichtenstein, G.R. Lichtenstein, Mimics of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 2315–2321. [Google Scholar] [CrossRef] [PubMed]
- Jayasooriya, N.; Baillie, S.; Blackwell, J.; Bottle, A.; Petersen, I.; Creese, H.; Saxena, S.; Pollok, R.C. Systematic review with meta-analysis: Time to diagnosis and the impact of delayed diagnosis on clinical outcomes in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2023, 57, 635–652. [Google Scholar] [CrossRef]
- Lee, D.W.; Koo, J.S.; Choe, J.W.; Suh, S.J.; Kim, S.Y.; Hyun, J.J.; Jung, S.W.; Jung, Y.K.; Yim, H.J.; Lee, S.W. Diagnostic delay in inflammatory bowel disease increases the risk of intestinal surgery. World J. Gastroenterol. 2017, 23, 6474–6481. [Google Scholar] [CrossRef]
- Nguyen, V.Q.; Jiang, D.; Hoffman, S.N.; Guntaka, S.; Mays, J.L.; Wang, A.; Gomes, J.; Sorrentino, D. Impact of Diagnostic Delay and Associated Factors on Clinical Outcomes in a U.S. Inflammatory Bowel Disease Cohort. Inflamm. Bowel Dis. 2017, 23, 1825–1831. [Google Scholar] [CrossRef]
- Gubatan, J.; Boye, T.L.; Temby, M.; Sojwal, R.S.; Holman, D.R.; Sinha, S.R.; Rogalla, S.R.; Nielsen, O.H. Gut Microbiome in Inflammatory Bowel Disease: Role in Pathogenesis, Dietary Modulation, and Colitis-Associated Colon Cancer. Microorganisms 2022, 10, 1371. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Nishida, A.; Nishino, K.; Sakai, K.; Owaki, Y.; Noda, Y.; Imaeda, H. Can control of gut microbiota be a future therapeutic option for inflammatory bowel disease? World J. Gastroenterol. 2021, 27, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Sun, Q.; Zhang, J.; Ng, S.C. The role of gut microbiome in inflammatory bowel disease diagnosis and prognosis. United Eur. Gastroenterol. J. 2022, 10, 1091–1102. [Google Scholar] [CrossRef]
- Chamorro, N.; Montero, D.A.; Gallardo, P.; Farfán, M.; Contreras, M.; De la Fuente, M.; Dubois, K.; Hermoso, M.A.; Quera, R.; Pizarro-Guajardo, M.; et al. Landscapes and bacterial signatures of mucosa-associated intestinal microbiota in Chilean and Spanish patients with inflammatory bowel disease. Microb. Cell (Graz Austria) 2021, 8, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, I.; Alimadadi, A.; Aryal, S.; Munroe, P.B.; Joe, B.; Cheng, X. Gut microbiome-based supervised machine learning for clinical diagnosis of inflammatory bowel diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G328–G337. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Sarrabayrouse, G.; Elias, A.; Yáñez, F.; Mayorga, L.; Varela, E.; Bartoli, C.; Casellas, F.; Borruel, N.; Herrera de Guise, C.; Machiels, K.; et al. Fungal and Bacterial Loads: Noninvasive Inflammatory Bowel Disease Biomarkers for the Clinical Setting. mSystems 2021, 6, e01277-20. [Google Scholar] [CrossRef]
- Kubinski, R.; Djamen-Kepaou, J.Y.; Zhanabaev, T.; Hernandez-Garcia, A.; Bauer, S.; Hildebrand, F.; Korcsmaros, T.; Karam, S.; Jantchou, P.; Kafi, K.; et al. Benchmark of Data Processing Methods and Machine Learning Models for Gut Microbiome-Based Diagnosis of Inflammatory Bowel Disease. Front. Genet. 2022, 13, 784397. [Google Scholar] [CrossRef]
- Liñares-Blanco, J.; Fernandez-Lozano, C.; Seoane, J.A.; López-Campos, G. Machine Learning Based Microbiome Signature to Predict Inflammatory Bowel Disease Subtypes. Front. Microbiol. 2022, 13, 872671. [Google Scholar] [CrossRef]
- Su, Q.; Liu, Q.; Lau, R.I.; Zhang, J.; Xu, Z.; Yeoh, Y.K.; Leung, T.W.H.; Tang, W.; Zhang, L.; Liang, J.Q.Y.; et al. Faecal microbiome-based machine learning for multi-class disease diagnosis. Nat. Commun. 2022, 13, 6818. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; Boitard, S.; Besse, P. Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinform. 2011, 12, 253. [Google Scholar] [CrossRef]
- Park, S.K.; Kim, H.N.; Choi, C.H.; Im, J.P.; Cha, J.M.; Eun, C.S.; Kim, T.O.; Kang, S.B.; Bang, K.B.; Kim, H.G.; et al. Differentially Abundant Bacterial Taxa Associated with Prognostic Variables of Crohn’s Disease: Results from the IMPACT Study. J. Clin. Med. 2020, 9, 1748. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.Y.; Kim, H.N.; Lee, S.H.; Kim, S.J.; Chang, Y.; Ryu, S.; Shin, H.; Kim, H.L.; Lee, J.H. Association between Neutrophil-to-Lymphocyte Ratio and Gut Microbiota in a Large Population: A Retrospective Cross-Sectional Study. Sci. Rep. 2018, 8, 16031. [Google Scholar] [CrossRef]
- Shin, S.Y.; Kim, S.; Choi, J.W.; Kang, S.B.; Kim, T.O.; Seo, G.S.; Cha, J.M.; Chun, J.; Jung, Y.; Im, J.P.; et al. The Common and Unique Pattern of Microbiome Profiles among Saliva, Tissue, and Stool Samples in Patients with Crohn’s Disease. Microorganisms 2022, 10, 1467. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.; Choi, J.W.; Ahn, K.S.; Park, D.I.; Kim, S. A streamlined pipeline based on HmmUFOtu for microbial community profiling using 16S rRNA amplicon sequencing. Genom. Inf. 2023, 21, e40. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bartow-McKenney, C.; Meisel, J.S.; Grice, E.A. HmmUFOtu: An HMM and phylogenetic placement based ultra-fast taxonomic assignment and OTU picking tool for microbiome amplicon sequencing studies. Genome Biol. 2018, 19, 82. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.I.H.; Morgan, X.C. Searching for a Consensus Among Inflammatory Bowel Disease Studies: A Systematic Meta-Analysis. Inflamm. Bowel Dis. 2023, 29, 125–139. [Google Scholar] [CrossRef]
- Shin, S.Y.; Kim, Y.; Kim, W.S.; Moon, J.M.; Lee, K.M.; Jung, S.A.; Park, H.; Huh, E.Y.; Kim, B.C.; Lee, S.C.; et al. Compositional changes in fecal microbiota associated with clinical phenotypes and prognosis in Korean patients with inflammatory bowel disease. Intest. Res. 2023, 21, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Amos, G.C.A.; Murphy, A.R.J.; Murch, S.; Wellington, E.M.H.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Ng, S.C. The Gut Microbiota in the Pathogenesis and Therapeutics of Inflammatory Bowel Disease. Front. Microbiol. 2018, 9, 2247. [Google Scholar] [CrossRef] [PubMed]
- Vester-Andersen, M.K.; Mirsepasi-Lauridsen, H.C.; Prosberg, M.V.; Mortensen, C.O.; Träger, C.; Skovsen, K.; Thorkilgaard, T.; Nøjgaard, C.; Vind, I.; Krogfelt, K.A.; et al. Increased abundance of proteobacteria in aggressive Crohn’s disease seven years after diagnosis. Sci. Rep. 2019, 9, 13473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Wang, S.N.; Miao, C.Y. Influence of Microbiota on Intestinal Immune System in Ulcerative Colitis and Its Intervention. Front. Immunol. 2017, 8, 1674. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Siles, M.; Martinez-Medina, M.; Abellà, C.; Busquets, D.; Sabat-Mir, M.; Duncan, S.H.; Aldeguer, X.; Flint, H.J.; Garcia-Gil, L.J. Mucosa-associated Faecalibacterium prausnitzii phylotype richness is reduced in patients with inflammatory bowel disease. Appl. Environ. Microbiol. 2015, 81, 7582–7592. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, J.; Ran, Z.H. Association between Faecalibacterium prausnitzii Reduction and Inflammatory Bowel Disease: A Meta-Analysis and Systematic Review of the Literature. Gastroenterol. Res. Pract. 2014, 2014, 872725. [Google Scholar] [CrossRef]
- Guo, X.; Huang, C.; Xu, J.; Xu, H.; Liu, L.; Zhao, H.; Wang, J.; Huang, W.; Peng, W.; Chen, Y.; et al. Gut Microbiota Is a Potential Biomarker in Inflammatory Bowel Disease. Front. Nutr. 2021, 8, 818902. [Google Scholar] [CrossRef]
- Teofani, A.; Marafini, I.; Laudisi, F.; Pietrucci, D.; Salvatori, S.; Unida, V.; Biocca, S.; Monteleone, G.; Desideri, A. Intestinal Taxa Abundance and Diversity in Inflammatory Bowel Disease Patients: An Analysis including Covariates and Confounders. Nutrients 2022, 14, 260. [Google Scholar] [CrossRef]
- Mah, C.; Jayawardana, T.; Leong, G.; Koentgen, S.; Lemberg, D.; Connor, S.J.; Rokkas, T.; Grimm, M.C.; Leach, S.T.; Hold, G.L. Assessing the Relationship between the Gut Microbiota and Inflammatory Bowel Disease Therapeutics: A Systematic Review. Pathogens 2023, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Prideaux, L.; Kang, S.; Wagner, J.; Buckley, M.; Mahar, J.E.; De Cruz, P.; Wen, Z.; Chen, L.; Xia, B.; van Langenberg, D.R.; et al. Impact of ethnicity, geography, and disease on the microbiota in health and inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 2906–2918. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD (n = 671) | UC (n = 114) | HC (n = 1462) | |
|---|---|---|---|
| Age (year), mean ± SD | 35.9 ± 13.2 | 39.9 ± 15.9 | 45.9 ± 9.2 |
| Male, n (%) | 483 (71) | 84 (73.7) | 907 (62) |
| BMI (kg/m2), mean ± SD | 22.1 ± 3.8 | 23.2 ± 3 | 23.7 ± 3.1 |
| Smoking status, n (%) | |||
| Current | 93 (13.9) | 14 (12.3) | |
| Former | 26 (3.9) | 22 (19.3) | |
| Never | 454 (67.7) | 77 (67.2) | |
| Unknown | 98 (14.6) | 1 (0.0) | |
| Disease location, n (%) | Ileum, 186 (27.7) | Proctitis, 48 (42.1) | |
| Colon, 84 (12.5) | Distal, 38 (33.3) | ||
| Ileocolon, 337 (50.0) | Extensive, 27 (23.7) | ||
| Ileum + upper GI, 6 (0.9) | |||
| Colon + upper GI, 1 (0.0) | |||
| Ileocolon + upper GI, 17 (2.5) | |||
| Unknown, 40 (6.0) | Unknown, 1 (0.0) |
| OTUs/ Phylotypes | Samples | Total Reads (% of the Raw) | |
|---|---|---|---|
| Raw | 2255 | 164,539,577 | |
| fastp | 2255 | 157,961,202 (0.96) | |
| HmmUFOtu clustering | 88,927 | 2255 | 157,865,293 (0.9594) |
| Chimera removal | 83,562 | 2255 | 150,585,336 (0.9152) |
| Taxonomic assignments | 67,283 | 2255 | 150,549,827 (0.915) |
| Phylotyping | 2525 | 2255 | 150,549,827 (0.915) |
| Abundance table filtering process | |||
| Abundance > 20 k or counts > 10 | 1526 | 1853 | 145,380,866 (0.8836) |
| Non-bacterial phylotypes | 1518 | 1853 | 145,353,664 (0.8834) |
| Domain only | 1517 | 1853 | 140,691,441 (0.8551) |
| Abundance > 20 k | 1517 | 1846 | 140,552,068 (0.8542) |
| Accuray | CD Sens. | CD Prec. | UC Sens. | UC Prec. | HC Sens. | HC Prec. | AUC | |
|---|---|---|---|---|---|---|---|---|
| Min. | 0.539 | 0.177 | 0.292 | 0.235 | 0.353 | 0.677 | 0.566 | 0.539 |
| 1st Qu. | 0.608 | 0.324 | 0.443 | 0.441 | 0.486 | 0.934 | 0.756 | 0.608 |
| Median | 0.637 | 0.412 | 0.5 | 0.544 | 0.551 | 0.971 | 0.823 | 0.637 |
| Mean | 0.638 | 0.434 | 0.505 | 0.53 | 0.545 | 0.952 | 0.814 | 0.638 |
| 3rd Qu. | 0.667 | 0.529 | 0.559 | 0.618 | 0.591 | 1 | 0.95 | 0.667 |
| Max. | 0.755 | 0.824 | 0.793 | 0.765 | 0.735 | 1 | 0.944 | 0.755 |
| Accuracy | Sensitivity | Specificity | Precision | AUC | |
|---|---|---|---|---|---|
| Min. | 0.89 | 0.809 | 0.918 | 0.91 | 0.972 |
| 1st Qu. | 0.941 | 0.897 | 0.971 | 0.97 | 0.989 |
| Median | 0.949 | 0.919 | 0.985 | 0.984 | 0.993 |
| Mean | 0.95 | 0.918 | 0.982 | 0.981 | 0.992 |
| 3rd Qu. | 0.963 | 0.941 | 1 | 1 | 0.996 |
| Max. | 0.993 | 0.985 | 1 | 1 | 1 |
| Accuracy | Sensitivity | Specificity | Precision | AUC | |
|---|---|---|---|---|---|
| Min. | 0.853 | 0.794 | 0.824 | 0.846 | 0.947 |
| 1st Qu. | 0.927 | 0.912 | 0.934 | 0.93 | 0.984 |
| Median | 0.956 | 0.941 | 0.971 | 0.967 | 0.991 |
| Mean | 0.945 | 0.941 | 0.949 | 0.95 | 0.988 |
| 3rd Qu. | 0.971 | 0.971 | 0.971 | 0.971 | 0.997 |
| Max. | 1 | 1 | 1 | 1 | 1 |
| Accuracy | CD Sens. | CD Prec. | UC Sens. | UC Prec. | HC Sens. | HC Prec. | AUC | |
|---|---|---|---|---|---|---|---|---|
| Min. | 0.873 | 0.706 | 0.953 | 0.912 | 0.824 | 0.912 | 0.773 | 0.873 |
| 1st Qu. | 0.922 | 0.853 | 0.941 | 0.924 | 0.941 | 0.971 | 0.85 | 0.922 |
| Median | 0.931 | 0.882 | 0.969 | 0.941 | 0.97 | 1 | 0.895 | 0.931 |
| Mean | 0.936 | 0.888 | 0.965 | 0.933 | 0.964 | 0.956 | 0.891 | 0.936 |
| 3rd Qu. | 0.951 | 0.941 | 1 | 0.971 | 1 | 1 | 0.919 | 0.951 |
| Max. | 0.99 | 1 | 1 | 1 | 1 | 1 | 1 | 0.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Na, J.E.; Kim, S.; Kim, T.-O.; Park, S.-K.; Lee, C.-W.; Kim, K.O.; Seo, G.-S.; Kim, M.S.; Cha, J.M.; et al. A Machine Learning-Based Diagnostic Model for Crohn’s Disease and Ulcerative Colitis Utilizing Fecal Microbiome Analysis. Microorganisms 2024, 12, 36. https://doi.org/10.3390/microorganisms12010036
Kim H, Na JE, Kim S, Kim T-O, Park S-K, Lee C-W, Kim KO, Seo G-S, Kim MS, Cha JM, et al. A Machine Learning-Based Diagnostic Model for Crohn’s Disease and Ulcerative Colitis Utilizing Fecal Microbiome Analysis. Microorganisms. 2024; 12(1):36. https://doi.org/10.3390/microorganisms12010036
Chicago/Turabian StyleKim, Hyeonwoo, Ji Eun Na, Sangsoo Kim, Tae-Oh Kim, Soo-Kyung Park, Chil-Woo Lee, Kyeong Ok Kim, Geom-Seog Seo, Min Suk Kim, Jae Myung Cha, and et al. 2024. "A Machine Learning-Based Diagnostic Model for Crohn’s Disease and Ulcerative Colitis Utilizing Fecal Microbiome Analysis" Microorganisms 12, no. 1: 36. https://doi.org/10.3390/microorganisms12010036