

The Future Is Now: Unraveling the Expanding Potential of Human (Necro)Microbiome in Forensic Investigations

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Methods
3. Human Microbiota and Microbiome
3.1. Core Microbiome
3.2. The Necrobiome: Thanatomicrobiome and Epinecrotic Communities
3.2.1. Factors Triggering Microbial Invasion after Death
3.2.2. Body Decomposition and Microbial Succession after Death

{kind=link}
{kind=link}
| Body Site | After Death | ||
|---|---|---|---|
| Overall Changes a | Model Used and Timepoints | References | |
| Body | Richness ↑ Diversity↓ | Human (n = 6); 0–20 d; Human (n = 4) 0–30 d | [9,70] |
| Richness ↓ (except in the rectum) Actinomycetota and Bacteroidota ↓ Pseudomonadota ↑ | Human (n = 188); <48 h/>49 h (2 timepoints) | [72] | |
| S. aureus KUB7 5–7 d ↑and then decrease until no detection at 30 d S. aureus highest concentrations by culture on 5 d for surface sterilized mice S. aureus highest concentrations by culture on 7 d for non-surface sterilized mice | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] | |
| Dominance of Clostridium spp. in internal postmortem communities; Bacillota suggested as a stable biomarker Female: high abundance of Pseudomonas and Clostridiales Male: high abundance of Clostridiales and Streptococcus; exclusive presence of Rothia Clostridium and Prevotella species as predictive of different periods of decomposition | Human (n = 27); 3.5–240 h (66 timepoints) | [54] | |
| Richness ↓ Bacteroidaceae and Moraxellaceae were good indicators in the initial sampling; Bacillaceae/Clostridiales were significant after 5 d Pseudomonadota was dominant followed by Bacillota Pseudomonadota ↓ over time until 5 d Bacillota ↑ over time Moraxellaceae ↑ 0 d Aerococcaceae, Enterobacteriaceae ↑ 3 d and no presence after 5 d Planococcaceae, Clostridiales, Clostridiaceae—dominant at 5 d | Swine (n = 3); 0–5 d (4 timepoints) | [3] | |
| Ignatzschineria and Wohlfahrtimonas were common at bloat and purge and until tissues began to dehydrate Acinetobacter were common after dehydration and skeletonization Ignatzschineria dominated during the wettest phases and ↓ until skeletonization Ignatzschineria was less abundant and less persistent Wohlfahrtiimonas associated with myiasis | Human (n = 2); 1–20 d (10 timepoints) | [79] | |
| Skin | Bacteroidota (Sphingobacteriaceae), Alphaproteobacteria (Brucellaceae, Phyllobacteriaceae, and Hyphomicrobiaceae), and Betaproteobacteria (Alcaligenaceae) ↑ during the advanced decay. Taxa in Rhizobiales were among the most important predictive taxa at each sample site. | Mouse (n = 40); 0–48 days (8 timepoints) | [80] |
| Dominated by Pseudomonadota at first 2 d ↑ Bacillota, Actinomycetota during the later phases Pseudomonas and Acinetobacter were dominant before purging ↑ Ignatzschineria after purge and ↓ at dry stage Clostridium dominated in the later phases | Human (n = 2); 1–20 d (10 timepoints) | [79] | |
| Clostridium ↑ max. at 5 d and 7 d | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] | |
| Blood | At 5 min, 25% culture-positive to enterococci, lactobacilli, and/or Bacteroides/Prevotella spp. At 1 h, bacterial translocation rates were lowest (virtually no bacterial growth) Culture-positive until 30 min, ↓ at 1 h, ↑ to max. at 48 h and 72 h At 72 h, culture-positive for E. coli (100%), enterococci (75%) and lactobacilli (62.5%) | Mice; 0–72 h (10 timepoints) | [83] |
| Brain | Dominated by MLE1-12 (Candidatus Melainabacteria), Saprospirales and Burkholderiales ↑ Relative abundance in ASVs belonging to the order Clostridiales ↓ Relative abundance in ASVs belonging to the order MLE1-12 (not significant) | Human (n = 40); 24–432 h | [19] |
| Bacteroidota and Pseudomonadota showed different succession patterns At the genus level, Ochrobactrum and Sediminibacterium were dominant, and ↓ with PMI progression ↑ Acinetobacter, Cupriavidus, and Agrobacterium (were dominants) At the phylum level, Pseudomonadota was the most prevalent ↑ Deinococcota during 12 h At the order level, Rhizobiales was dominant ↓ Saprospirales, Caulobacterales and Thermales ↑ Burkholderiales and Pseudomonadales during 1 d ↑ Acinetobacter at 8 h; ↑ Cupriavidus and Agrobacterium after 8 h | Mice (n = 30); 0:30 h–1 d (5 timepoints) | [84] | |
| Eyes | ↑ Streptococcus early in PMI ranges (<24 h, 25–48 h) | Human (n = 188); <48 h/>49 h (timepoints) | [72] |
| Oral cavity/Mouth | ↑ Pseudomanodota followed by ↑ Bacillota Pseudomonas and Enterococcaceae dominated before purging Planococcaceae dominated after purging and then dropped off as ↑ Clostridium | Human (n = 2); 1–20 d (10 timepoints) | [79] |
| Pseudomonas was detected in pre-bloat but was not in any end-bloat At the end-bloat stage, Pseudomonas was replaced by common GI tract bacteria (Clostridia, Lactobacillus, etc.) Streptococcus, Prevotella, and Veillonella detected in pre-bloat swab and scrape Pre-bloat swab and end-bloat scrape was predominated by Bacillota Pre-bloat scrape was predominated by Pseudomonadota | Human (n = 2); 0–30 d (8 timepoints) | [46] | |
| Pseudomonadota showed a positive linear correlation with PMI ↓ Alpha diversity over decomposition time Pseudomanodota and Bacillota were dominant Pseudomanodota ↓ first and then ↑ Bacillota↑ first and then ↓ Actinomycetota and Bacteroidota ↓ At 0 h, abundance of Pseudomonadota (Acinetobacter, Pseudomonas, Phyllobacterium, Photobacterium, Vibrio, Arcobacter, Muribacter) and Actinomycetota (Propionibacterium, Rhodococcus), Bacillota (Ruminococcaceae_UCG-014, Clostridium sensu_stricto_1, Paeniclostridium, Lactobacillus, Christensenelaceae_R-7_Group), Bacteroidota (Alistipes, Prevotella _9, Marinitilum) and Fusobacteria (Fusobacterium, Psychrilyobacter). At 24 h, abundance of Bacillota (Blautia, Enterococcus, Streptococcus, Faecalbacterium), Pseudomonadota (Pasteurella), Bacteroidota (Bacteroides), Actinomycetota (Bifidobacterium). At 144 h, abundance of Actinomycetota (Staphylococcus, Subdoligranulum, Romboutsia) and Pseudomonadota (Morganella, Escherichia shigella, Enterobacter). At 240 h, abundance of Pseudomonadota (Citrobacter, Proteus) ↓ Alpha-proteobacteria and Bacteroidia ↑ Gammaproteobacteria Bacilli and Clostridia ↑ first and then ↓ ↑ Enterobacterales, ↑ Proteus ↓ Pasteurellales, Bacteroidales and Rhizobiales Lactobacillales ↑ first and then ↓ ↓ Pasteurellaceaeae and Phyllobacteriaceae Streptococcaceae, Ruminococcaceae, and Bacteroidaceae ↑ first and then ↓ Muribacter and Phyllobacterium ↑ first and then ↓ | Mice (n = 24); 0–240 h (4 timepoints) | [10] | |
| Microbial communities were similar in diversity over decomposition time ↓ Alpha diversity over decomposition time Haemophilus parainfluenzae and Streptococcus were most abundant at <24 h and 25–48 h Bacteroidota (e.g., Prevotella) during the earlier stages of decomposition Streptococcus was a predominant taxon during pre-bloat and during the first 4 d Streptococcus as a potential biomarker during the first 2 d H. parainfluenzae potential bioindicator <48 h after death | Human (n = 188); <48 h/>49 h (2 timepoints) | [72] | |
| Bacillota and Actinomycetota are the predominant phyla in the fresh stage Tenericutes’ presence corresponds to the bloat stage Peptostreptococcaceae and Bacteroidaceae were predominant families in the bloat stage Bacillota is the predominant phyla in advanced decay (different community from the fresh stage) The fresh stage was characterized by Lactobacillaceae, Staphylococcaceae, Gemellaceae, Carnobacteriaceae, Aerococcaceae, Veillonellaceae, Streptococcaceae, Campylobacteraceae, Micrococcaceae, Bifidobacteriaceae, Actinomycetaceae and Corynebacteriaceae. Bacillota and Actinomycetota predominant from 1 d to 5 d, but their relative abundances ↓ from 1 d to 5–6 d ↑ Bacillota 6–12 d (Clostridiales and Bacillaceae—representative Bacillota from bloat to advanced decay stages) ↑ Tenericutes transiently between 5 d and 7 d, just at the bloat stage ↑ Ignatzschineria and Clostridiales in the bloat stage Gammaproteobacteria, Pseudomonadaceae, Alcaligenaceae, and Planococcaceae are predominant families in advanced decay Bacillia nd Clostridia presence in skeletonization/dry stage | Human (n = 3); 1–12 d (7–8 timepoints) | [73] | |
| Buccal Cavity | ↑ Alpha diversity after death At 4 h, Bacillota and Actinomycetota were dominant Bacillota gradually ↓ At 1 d, ↑ Pseudomonadota (predominant phylum) and ↑ Moraxellaceae (predominant family) and gradually ↓ At 2 d, Enterobacteriaceae dramatically ↑ and ↓ at 4 d Xanthomonadaceae gradually ↑ (dominant taxon from 3 d) At 6 d, ↑ Pseudomonadaceae Streptococcaceae and Pasteurellaceae gradually ↓ | Rat (n = 18); 1–9 d (9 timepoints) | [11] |
| Heart | Dominated by MLE1-12 (Candidatus Melainabacteria), Saprospirales and Burkholderiales ↑ Relative abundance in ASVs belonging to the order Burkholderiales ↓ Relative abundance in ASVs belonging to the order MLE1-12 (not significant) | Human (n = 40); 24–432 h | [19] |
| S. aureus remained at 0 until 7 d, ↑ to max. after 14 d ↑ and ↓ to levels near zero at 30 d At 5 h, a sample showed 100% Escherichia and others have Candidatus Arthromitus, Parabacteroides, Anaerostipes, and Dorea At 7 d, Clostridium dominated (72.1%) with Lactobacillus and Peptostreptococcaceae spp. | Mice (n = 63); 1 h–30 d (7 timepoints) | [62] | |
| Varying numbers of Clostridium from 1 h to 24 h, that reached and remained at max. countable limits 5 d to 14 d; Clostridium isolates were also recovered at 30 d and 60 d | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] | |
| At the genus level, Thermus was more abundant ↓ Enhydrobacter and Caulobacter, belonging Alphaproteobacteria and Methyloversatilis during 1 d ↑ Pseudomonas at 8 h ↑ Sphingomonas and Cupriavidus to peak values at 12 h At the phylum level, Pseudomonadota and Deinococcota were dominant perimortem ↑ Bacillota and ↓ Actinomycetota during 1 d At the order level, Pseudomonadales, Thermales, and Burkholderiales were dominant ↑ Sphingomonadales to a peak value at 12 h ↑ Rhizobiales during 1 d ↑ Deinococcales at 12 h ↓ Rhodocyclales, Rhodospirillales, and Caulobacterales during 1 d | Mice (n = 30); 0:30 h–1 d (5 timepoints) | [84] | |
| Pericardial Fluid | Streptococcus sp. isolates found 5–7 d Clostridium sp. isolates found 1–3 d Clostridium sp., Enterobacter sp., Bifidobacterium sp., Bacteroides sp. ↑ | Human (n = 33); 1–7 d (3 timepoints) | [85] |
| Lungs | S. aureus at 5 h postmortem ↓ to 0, after 5 h ↑↑ to max. at 14 d and ↓ up to 30 d At 5 h PM, contained 100% Lactobacillus At 7 d, contained 44% Clostridium and 55% Staphylococcus | Mice (n = 63); 1 h–30 d (7 timepoints) | [62] |
| Varying numbers of Clostridium from the 1 h to 24 h, that reached and remained at max. countable limits 5 d to 14 d Clostridium isolates were also recovered at 30 d and 60 d | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] | |
| Abdominal cavity | Bacillota (Lactobacilaceae, e.g., Lactobacillus) and Bacteroidota (Bacteroidaceae, e.g., Bacteroides) ↑ during the bloating stage (6–9 d) Bacillota (Lactobacilaceae, e.g., Lactobacillus) and Bacteroidota (Bacteroidaceae, e.g., Bacteroides) ↓ after rupture occurs (∼9 d) Rhizobiales (Alphaproteobacteria) in the families Phyllobacteriaceae, Hyphomicrobiaceae, and Brucellaceae (e.g., Pseudochrobactrum and Ochrobactrum) dominate Serratia, Escherichia, Klebsiella, and Proteus become abundant after rupture | Mouse (n = 40); 0–48 days (8 timepoints) | [80] |
| Gut | Total bacteria load ↑ Relative abundances ↓ ↓ Bacteroides and Lactobacillus over time Bifidobacterium no significant change over the study | Human (n = 6); 0–20 d | [9] |
| Enterobacterales and Escherichia were detected in the lower GI tract for both pre-bloat and end-bloat Clostridium is abundant at the end of the bloat stage | Human (n = 2); 0–30 d (8 timepoints) | [46] | |
| Bacteroidales (Bacteroides, Parabacteroides) ↓ Clostridiales (Clostridium, Anaerosphaera) and Gammaproteobacteria, Ignatzschineria and Wohlfahrtiimonas ↑ Relative abundances and diversity ↓ Bacteroides, Parabacteroides and Lactobacillus ↓ | Human (n = 4); 0–30 d | [70] | |
| Total bacterial load ↑ 12 h and 24 h post sacrifice with high levels of enterobacteria and lactobacilli Total bacterial load ↓ 15 and 30 min post sacrifice with ↓ Enterobacteria, enterococci, bifidobacteria, and Clostridium spp. Enterobacteria, enterococci, bifidobacteria, and Clostridium spp. ↑ to de max. levels from 30 min until the end of the study Varying numbers of Clostridium from the 1 h to 24 h, that reached and remained at max. countable limits 5 d to 14 d Clostridium isolates were also recovered at 30 d and 60 d | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] | |
| Until 5 h postmortem Parabacteroides, Mucispirillum, and Lactobacillus dominated At 24 h ↓ relative abundance of Parabacteroides, disappearance of Mucispirillum and ↑ Lactobacillus At 7 d ↓ Lactobacillus and ↑ Anaerostipes, Clostridium, and Enterococcus Staphylococcus aureus—stable 1–5 h, ↓ at 24 h, ↑ to max. after 7 d and ↓↓ to min. at 14–30 d | Mice (n = 63); 1 h–30 d (7 timepoints) | [62] | |
| Lactobacillus, Dubosiella, Enterococcus, and Lachnospiraceae—proposed as significant biomarkers Bacillota (Lactobacillus reuteri/johnsonii, Clostridium tetani, Enterococcus faecalis), Bacteroidota, Actinomycetota- dominant Bacteroidota e Actinomycetota 2 d↑—2 d-4 d↓ Bacillota bacterium M10-2—appeared on 2 d and 2 d-4 d↑ Enterococcus faecalis—appeared on 2 d and 2 d-10 d↑ Tenericutes (bloat phase) Lactobacillus reuteri ↑—peak values 7 d and 15 d Clostridium tetani E88—appeared on 7 d until 15 d and then ↓ Lactobacillus johnsonii ↑ 1 week after death Helicobacter ↓ gradually during 15 d Gordonibacter, Bifidobacterium, Enterorhabdus, Lactococcus, Clostridium sensu stricto, Anaerosalibacter, Enterococcus, Dubosiella, Lactobacillus—remained at 15 d | Mice (n = 240); 6–10 w (10 timepoints) | [86] | |
| Colon | Total bacterial load ↓ between 3 h and 6 h with ↓ lactobacilli and Bacteroides/Prevotella spp. ↑ Enterococci between 6 h and 12 h and remain stable until 72 h Lactobacilli ↓ between mice alive and 72 h Escherichia coli remained stable at 0 until 72 h Bacteroides/Prevotella spp. ↓ 3–12 h | Mice; 0–72 h (10 timepoints) | [83] |
| Bifidobacterium detected at end-bloat | Human (n = 2); 0–30 d (8 timepoints) | [46] | |
| Ileum | ↑ Distinct in fastly replying aerobic species between 6 h and 24 h Total eubacterial loads ↑ 72 h with max. loads of enterobacteria, enterococci and lactobacilli Enterobacteria ↑ between 3 h and 12 h Enterococci ↑ between 6 h and 24 h Enterobacteriaceae 12 h–72 h↑ Enterococci 24–72 h↑ Lactobacilli significantly ↓ until 72 h Bacteroides/Prevotella spp. ↑3 h, ↓12 h, ↑72 h Clostridium coccoides and leptum groups ↑3 h, ↓12 h, ↑72 h Mouse Intestinal Bacteroides ↑3 h, ↓12 h, ↑72 h Bifidobacteria ↑6 h, ↓24 h | Mice; 0–72 h (10 timepoints) | [83] |
| Rectum | Taxon richness first ↓ and then ↑ Bacillota, Pseudomonadota, Bacteroidota, and Actinomycetota were found at all the timepoints At the phylum level, Pseudomonadota and Bacillota showed major shifts At the phylum level, bacterial richness ↓ from 0 h to 9 d and ↑ from 9 d to 15 d At the family level, Prevotellaceae, Muribaculaceae, and Lachnospiraceae ↓ at 0 h, 8 h, 16 h, 3 d, 7 d, 15 d At the family level, bacterial richness ↓ from 0 h to 9 d and ↑ from 9 d to 15 d At the genus level, Lactobacillus dominated at 1 d and Enterococcus from 3 d to 13 d Bacteroidota ↓↓ after death, but ↑ at 3 d and 15 d Actinomycetota relative abundances ↓ at 16 h, 7 d, and 15 d Bacillota and Pseudomonadota peak values at 8 h, 1 d, and 9 d Helicobacter was absent at 7 d, 9 d and 15 d ↑ Lactobacillaceae, Enterobacteriaceae, and Enterococcaceae represented the majority from 0 h to 15 d Enterococcus and Vagococcus relative abundances ↑ at 0 h, 8 h, 3 d, 7 d and 15 d Proteus was most abundant at 15 d At the species level, Enterococcus faecalis ↓ and Proteus mirabilis ↑ after 5 d Clostridium sporogenes ↓ abundance before 1 d and Falsiporphyromonas_endometrii after 3 d E. faecalis and P. mirabilis appeared during the whole 15 d | Rat (n = 8); alive-15 d (11 timepoints) | [74] |
| Bacteroidota and Bacillota were the predominant phyla until 2 d Prevotellaceae was the predominant family until 2 d Pseudomonadota was the most abundant phylum after 2 d Enterobacteriaceae was a predominant family after 2 d | Rat (n = 18); 1–9 d (9 timepoints) | [11] | |
| Feces | Bacteroidota and Bacillota were the most abundant phyla before purging Pseudomonadota dominated after purging until the drier phases ↑ Bacillota and Actinomycetota in dry phases Clostridiaceae, Bacteroides, and Porphyromonas presented before purging Corynebacterium was the most abundant at the dry stage Ignatzschineria ↑ to max. after purge and ↓ at the dry stage Clostridium became the most abundant at the dry stage Clostridiaceae were the most abundant at the dry stage | Human (n = 2); 1–20 d (10 timepoints) | [79] |
| Bacillota mainly dominated with very few Bacteroidota detected in a sample Pseudomonadota dominated in another sample Pseudomonas was detected in pre-bloat but was not in any end-bloat At the end-bloat stage, Pseudomonas was replaced by other GI tract bacteria (Clostridia, Lactobacillus, etc.) | Human (n = 2); 0–30 d (8 timepoints) | [46] | |
| Liver | Sterility up to 5 d After 5 d, Clostridium sp., Streptococcus sp., Enterobacter sp., Enterococcus sp., Escherichia sp., Staphylococcus sp. and Streptococcus sp. | Human (n = 33); 1–7 d (3 timepoints) | [85] |
| Dominated by MLE1-12 (Candidatus Melainabacteria), Saprospirales and Burkholderiales ↑ Relative abundance in ASVs belonging to the order Clostridiales ↓ Relative abundance in ASVs belonging to the order MLE1-12 (not significant) | Human (n = 40); 24–432 h | [19] | |
| Varying numbers of Clostridium from the 1 h to 24 h, that reached and remained at max. countable limits 5 d to 14 d Clostridium isolates were also recovered at 30 d and 60 d | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] | |
| At 1 h, bacterial translocation rates were lowest (virtually no bacterial growth) Culture-positive until 30 min, ↓ at 1 h, ↑ to max. at 48 h and 72 h. | Mice; 0–72 h (10 timepoints) | [83] | |
| At the genus level, Thermus and Cupriavidus were dominant ↓ Microbacterium to zero at 24 h ↑ Acinetobacter, Cupriavidus, and Pseudomonas over decomposition Genera Paracoccus and Cryocola were detected only at 0:30 h At the phylum level, Pseudomonadota and Deinococcota were dominant Actinomycetota, Bacillota, Bacteroidota, and Cyanobacteria showed relative abundances of > 1% ↓ Actinomycetota during 1 d At the order level, Burkholderiales, Pseudomonadales, and Thermales were dominant ↑ Clostridiales during 1 d ↓ Actinomycetales; ↓ Rhodobacterales during 4 h Comamonadaceae, a family of Betaproteobacteria, was also significantly enriched | Mice (n = 30); 0:30 h–1 d (5 timepoints) | [84] | |
| Spleen | Varying numbers of Clostridium from the 1 h to 24 h, that reached and remained at max. countable limits 5 d to 14 d Clostridium isolates were also recovered at 30 d and 60 d | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] |
| Dominated by MLE1-12 (Candidatus Melainabacteria), Saprospirales and Burkholderiales ↑ Relative abundance in ASVs belonging to the order Clostridiales ↓ Relative abundance in ASVs belonging to the order MLE1-12 (not significant) | Human (n = 40); 24–432 h | [19] | |
| At 1 h, bacterial translocation rates were lowest (virtually no bacterial growth) Culture-positive until 30 min, ↓ at 1 h, ↑ to max. at 48 h and 72 h. | Mice; 0–72 h (10 timepoints) | [83] | |
| Kidney | S. aureus KUB7 detected 1 h post sacrifice; not detected at 3 h, 5 h, 24 h post sacrifice of surface-sterilized mice and detected again 5 d through 14 d Surface sterilized mice—Clostridium ↑ max. at 5 d and 7 d and ↓ at 14 d, 30 d, and 60 d Non-surface sterilized mice—Clostridium ↑ max. at 7 d and 14 d and ↓ at 30 d and 60 d | Mice (n = 90); 1 h–60 d (9 timepoints) | [82] |
| At 1 h, bacterial translocation rates were lowest (virtually no bacterial growth) Culture-positive until 30 min, ↓ at 1 h, ↑ to max. at 48 h and 72 h. | Mice; 0–72 h (10 timepoints) | [83] | |
| At the genus level, Thermus was dominant ↑ Acinetobacter and Pseudomonas during 8 h; ↓ Methyloversatilis during 1 d At the phylum level, Pseudomonadota, Deinococcota and Bacillota were dominant ↓ Fusobacteria and Cyanobacteria during 1 day ↑ Pseudomonadota and Actinomycetota At the order level, Pseudomonadales and Thermales were dominant ↓ Streptophyta, Clostridiales, and Rhodocyclales during 1 d ↑ Burkholderiales, Rhizobiales, Bacteroidales and Actinomycetales | Mice (n = 30); 0:30 h–1 d (5 timepoints) | [84] | |
| Bone marrow | S. aureus after 3 h postmortem ↓ to 0, ↑ after 5 h until max. at 7 d and ↓ after 14 d until 0 at 30 d Until 24 h, Propionibacteriaceae, Staphylococcus, Propionibacterium, Enterococcus, Pseudomonas were detected; at 7 d, Clostridium dominated with Peptostreptococcaceae spp. and Pseudomonas | Mice (n = 63); 1 h–30 d (7 timepoints) | [62] |
| Mesenteric lymph node | ↑ Clostridium sp., Enterobacter sp., Bifidobacterium sp., Bacteroides sp. | Human (n = 33); 1–7 d (3 timepoints) | [85] |
| Culture-positive until 30 min, ↓ at 1 h, ↑ to max. at 48 h and 72 h. At 5 min, lactobacilli have translocated, ↑ until 30 min, ↓ at 1 h, and then ↑ At 12 h culture + for lactobacilli (high levels), E. coli, enterococci, Bacteroides/Prevotella spp., clostridia | Mice; 0–72 h (10 timepoints) | [83] | |
| Uterus | ↑ Alpha diversity; Dominated by Clostridiales and Lactobacillales ↓ Relative abundance of MLE1-12 (Candidatus Melainabacteria) | Human (n = 40); 24–432 h | [19] |
| Prostate | ↑ Alpha diversity Dominated by Clostridiales and Lactobacillales ↓ Relative abundance of MLE1-12 (Candidatus Melainabacteria) | Human (n = 40); 24–432 h | [19] |
Gastrointestinal Tract
Regarding Skin and Mouth
Brain, Heart, Liver, Spleen and Kidney
Other Cadaveric Samples
3.2.3. Factors Affecting Decomposition
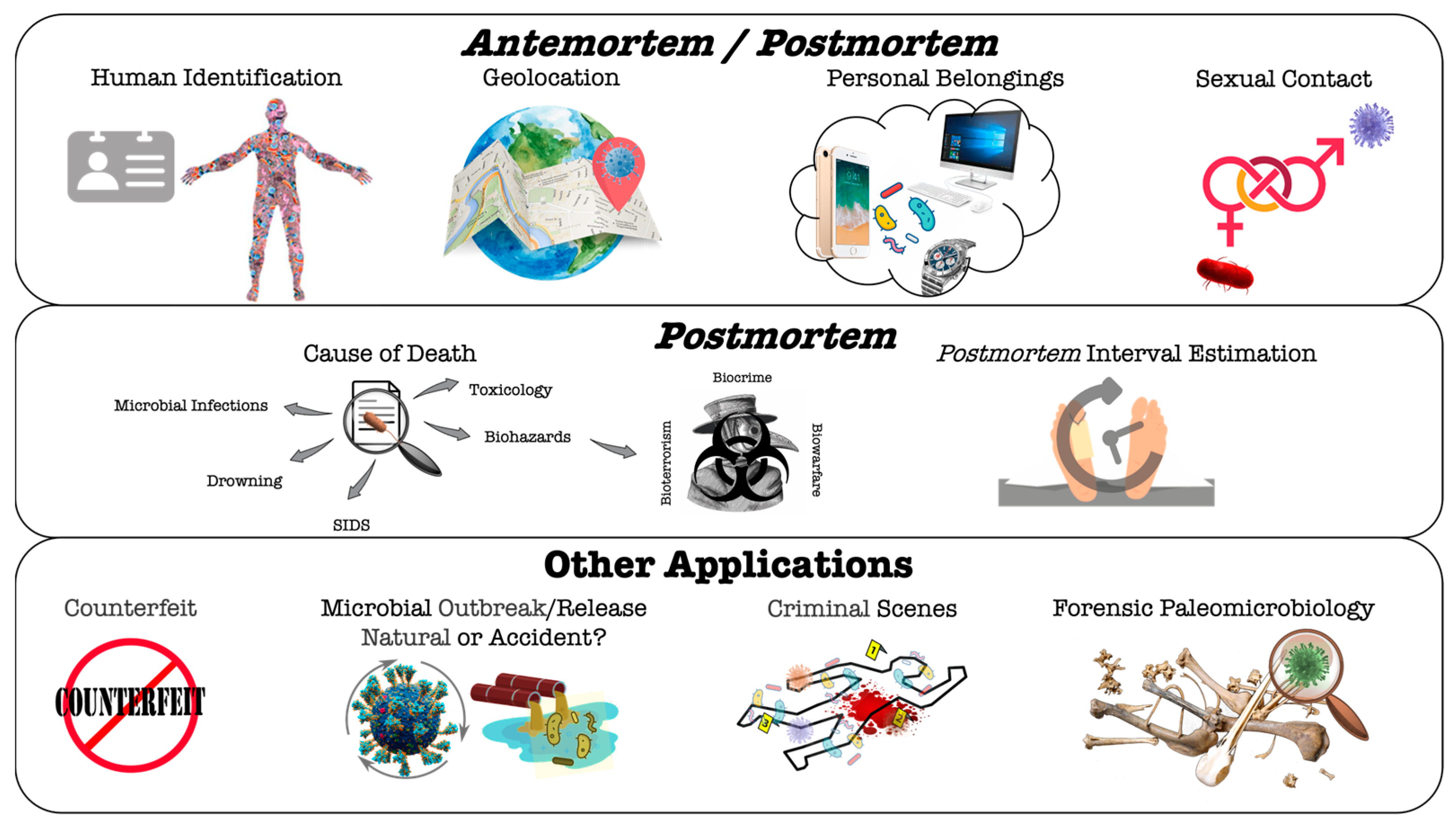
4. Microbiome-Based Analysis for Forensic Antemortem and/or Postmortem Applications
4.1. Microorganisms or Microbiome Analysis in Ante/Postmortem Forensic Studies
4.1.1. Human Identification
4.1.2. Geolocation
4.1.3. Personal Belongings
4.1.4. Sexual Contact
4.2. Microorganisms or Microbiome Analysis in Postmortem Forensic Studies
4.2.1. Cause of Death
Hospital/Community-Acquired Infections and Other Biorisks
Drowning
Sudden Infant Death Syndrome (SIDS)
Toxicological Effects Imposed by Microbial Metabolism
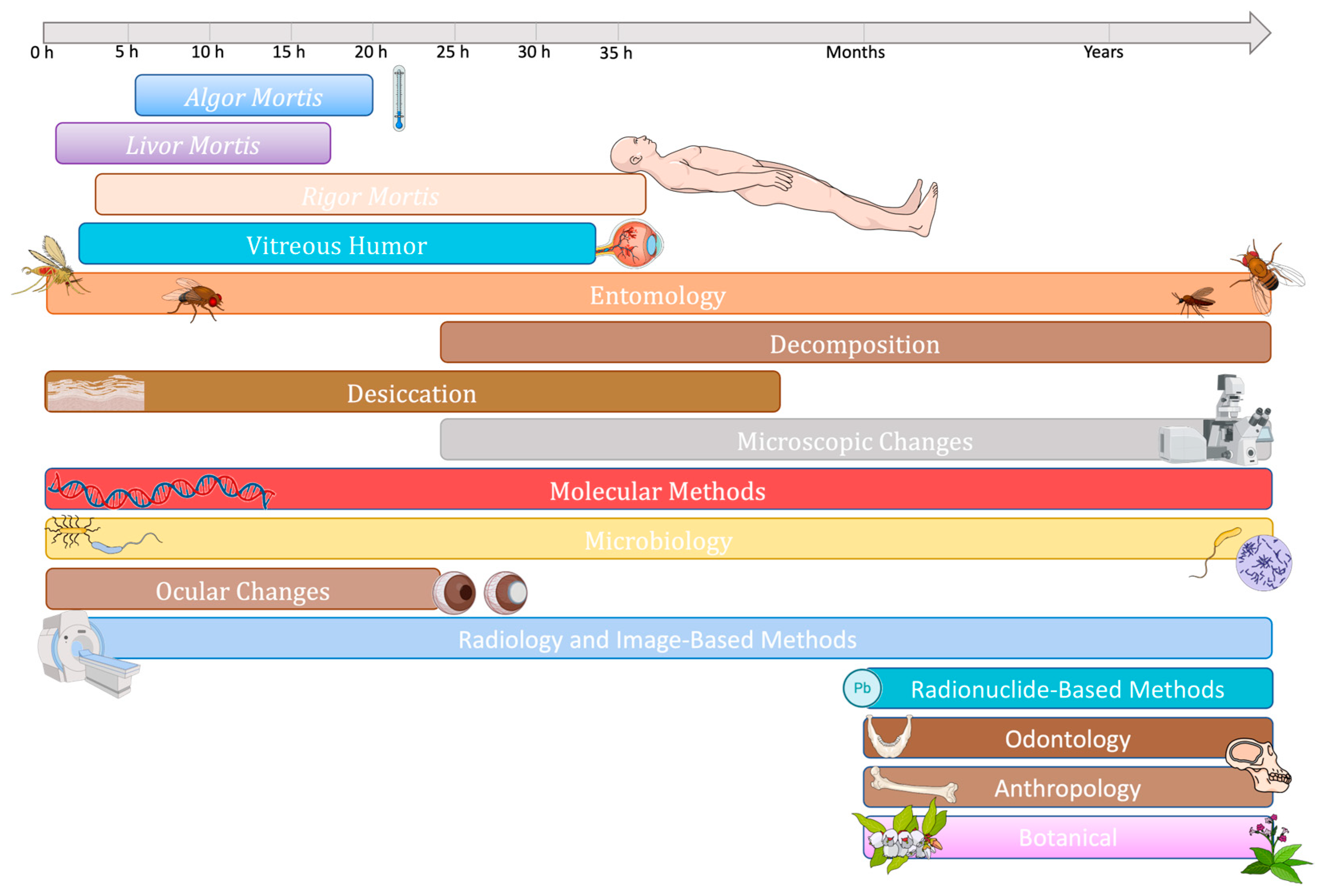
4.2.2. Estimation of Postmortem Interval
Evolution of Methods for PMI Estimation

Microbiome, Microbial Communities or Microbial Succession
5. Methods and Technical Issues
5.1. Culture-Dependent Methods
5.2. Culture-Independent Methods
6. Advantages and Limitations of Microbiome Analysis in Forensic Investigations
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Opal, S.M. A Brief History of Microbiology and Immunology. In Vaccines: A Biography; Springer: Berlin/Heidelberg, Germany, 2009; Chapter 3. [Google Scholar]
- Maccallum, W.G.; Hastings, T.W. A case of acute endocarditis caused by micrococcus zymogenes (nov. Spec.), with a description of the microorganism. J. Exp. Med. 1899, 4, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Pechal, J.L.; Crippen, T.L.; Benbow, M.E.; Tarone, A.M.; Dowd, S.; Tomberlin, J.K. The potential use of bacterial community succession in forensics as described by high throughput metagenomic sequencing. Int. J. Leg. Med. 2014, 128, 193–205. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Xu, Z.Z.; Weiss, S.; Lax, S.; Van Treuren, W.; Hyde, E.R.; Song, S.J.; Amir, A.; Larsen, P.; Sangwan, N.; et al. Microbial community assembly and metabolic function during mammalian corpse decomposition. Science 2016, 351, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, I. Microbial forensics: Next-generation sequencing as catalyst: The use of new sequencing technologies to analyze whole microbial communities could become a powerful tool for forensic and criminal investigations. EMBO Rep. 2016, 17, 1085–1087. [Google Scholar] [CrossRef]
- Carter, D.O.; Yellowlees, D.; Tibbett, M. Cadaver decomposition in terrestrial ecosystems. Naturwissenschaften 2007, 94, 12–24. [Google Scholar] [CrossRef]
- Carter, D.O.; Tomberlin, J.K.; Benbow, M.E.; Metcalf, J.L. Forensic science in focus. In Forensic Microbiology, 1st ed.; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Blondeau, L.D.; Rubin, J.E.; Deneer, H.; Kanthan, R.; Sanche, S.; Hamula, C.; Blondeau, J.M. Forensic, investigative and diagnostic microbiology: Similar technologies but different priorities. Future Microbiol. 2019, 14, 553–558. [Google Scholar] [CrossRef]
- Hauther, K.A.; Cobaugh, K.L.; Jantz, L.M.; Sparer, T.E.; Debruyn, J.M. Estimating Time Since Death from Postmortem Human Gut Microbial Communities. J. Forensic Sci. 2015, 60, 1234–1240. [Google Scholar] [CrossRef]
- Dong, K.; Xin, Y.; Cao, F.; Huang, Z.; Sun, J.; Peng, M.; Liu, W.; Shi, P. Succession of oral microbiota community as a tool to estimate postmortem interval. Sci. Rep. 2019, 9, 13063. [Google Scholar] [CrossRef]
- Guo, J.; Fu, X.; Liao, H.; Hu, Z.; Long, L.; Yan, W.; Ding, Y.; Zha, L.; Guo, Y.; Yan, J.; et al. Potential use of bacterial community succession for estimating post-mortem interval as revealed by high-throughput sequencing. Sci. Rep. 2016, 6, 24197. [Google Scholar] [CrossRef]
- Roy, D.; Tomo, S.; Purohit, P.; Setia, P. Microbiome in Death and Beyond: Current Vistas and Future Trends. Front. Ecol. Evol. 2021, 9, 630397. [Google Scholar] [CrossRef]
- Burcham, Z.M.; Jordan, H.R. History, current, and future use of microorganisms as physical evidence. In Forensic Microbiology; Wiley: Hoboken, NJ, USA, 2017; pp. 25–55. [Google Scholar]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Hoyles, L. Human microbiome myths and misconceptions. Nat. Microbiol. 2023, 8, 1392–1396. [Google Scholar] [CrossRef]
- Flores, G.E.; Caporaso, J.G.; Henley, J.B.; Rideout, J.R.; Domogala, D.; Chase, J.; Leff, J.W.; Vázquez-Baeza, Y.; Gonzalez, A.; Knight, R.; et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.H.; Gomez, A.; Singh, H.; Nelson, K.E.; Brinkac, L.M. Integrating the microbiome as a resource in the forensics toolkit. Forensic Sci. Int. Genet. 2017, 30, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.; Vangelatos, A.; Gottel, N.; Osculati, A.; Visona, S.; Finley, S.J.; Gilbert, J.A.; Javan, G.T. Effects of Extended Postmortem Interval on Microbial Communities in Organs of the Human Cadaver. Front. Microbiol. 2020, 11, 569630. [Google Scholar] [CrossRef]
- Gouello, A.; Dunyach-Remy, C.; Siatka, C.; Lavigne, J.-P. Analysis of Microbial Communities: An Emerging Tool in Forensic Sciences. Diagnostics 2021, 12, 1. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Neu, A.T.; Allen, E.E.; Roy, K. Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. USA 2021, 118, e2104429118. [Google Scholar] [CrossRef]
- Wernroth, M.-L.; Peura, S.; Hedman, A.M.; Hetty, S.; Vicenzi, S.; Kennedy, B.; Fall, K.; Svennblad, B.; Andolf, E.; Pershagen, G.; et al. Development of gut microbiota during the first 2 years of life. Sci. Rep. 2022, 12, 9080. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Sharon, I.; Quijada, N.M.; Pasolli, E.; Fabbrini, M.; Vitali, F.; Agamennone, V.; Dötsch, A.; Selberherr, E.; Grau, J.H.; Meixner, M.; et al. The Core Human Microbiome: Does It Exist and How Can We Find It? A Critical Review of the Concept. Nutrients 2022, 14, 2872. [Google Scholar] [CrossRef] [PubMed]
- Dekaboruah, E.; Suryavanshi, M.V.; Chettri, D.; Verma, A.K. Human microbiome: An academic update on human body site specific surveillance and its possible role. Arch. Microbiol. 2020, 202, 2147–2167. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef]
- Shetty, S.A.; Hugenholtz, F.; Lahti, L.; Smidt, H.; de Vos, W.M. Intestinal microbiome landscaping: Insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol. Rev. 2017, 41, 182–199. [Google Scholar] [CrossRef]
- Gopalakrishna, K.P.; Hand, T.W. Influence of Maternal Milk on the Neonatal Intestinal Microbiome. Nutrients 2020, 12, 823. [Google Scholar] [CrossRef]
- Deo, P.N.; Deshmukh, R. Oral microbiome: Unveiling the fundamentals. J. Oral Maxillofac. Pathol. 2019, 23, 122–128. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef]
- Ohta, J.; Sakurada, K. Oral gram-positive bacterial DNA-based identification of saliva from highly degraded samples. Forensic Sci. Int. Genet. 2019, 42, 103–112. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; NISC Comparative Sequencing Program; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Oliveira, M.; Amorim, A. Microbial forensics: New breakthroughs and future prospects. Appl. Microbiol. Biotechnol. 2018, 102, 10377–10391. [Google Scholar] [CrossRef]
- Ahannach, S.; Spacova, I.; Decorte, R.; Jehaes, E.; Lebeer, S. At the Interface of Life and Death: Post-mortem and Other Applications of Vaginal, Skin, and Salivary Microbiome Analysis in Forensics. Front. Microbiol. 2021, 12, 694447. [Google Scholar] [CrossRef] [PubMed]
- Thomas-White, K.; Brady, M.; Wolfe, A.J.; Mueller, E.R. The bladder is not sterile: History and current discoveries on the urinary microbiome. Curr. Bladder Dysfunct. Rep. 2016, 11, 18–24. [Google Scholar] [CrossRef]
- Perovic, S.U.; Ksiezarek, M.; Rocha, J.; Cappelli, E.A.; Sousa, M.; Ribeiro, T.G.; Grosso, F.; Peixe, L. Urinary Microbiome of Reproductive-Age Asymptomatic European Women. Microbiol. Spectr. 2022, 10, e0130822. [Google Scholar]
- Diop, K.; Dufour, J.-C.; Levasseur, A.; Fenollar, F. Exhaustive repertoire of human vaginal microbiota. Hum. Microbiome J. 2019, 11, 100051. [Google Scholar] [CrossRef]
- Baud, D.; Pattaroni, C.; Vulliemoz, N.; Castella, V.; Marsland, B.J.; Stojanov, M. Sperm Microbiota and Its Impact on Semen Parameters. Front. Microbiol. 2019, 10, 234. [Google Scholar] [CrossRef] [PubMed]
- López, C.D.; González, D.M.; Haas, C.; Vidaki, A.; Kayser, M. Microbiome-based body site of origin classification of forensically relevant blood traces. Forensic Sci. Int. Genet. 2020, 47, 102280. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Ko, K.K.; Chen, H.; Liu, J.; Loh, M. No evidence for a common blood microbiome based on a population study of 9,770 healthy humans. Nat. Microbiol. 2023, 8, 973–985. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Vieira, D.N.; Magalhães, T. Guidelines for Collection of Biological Samples for Clinical and Forensic Toxicological Analysis. Forensic Sci. Res. 2016, 1, 42–51. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Magalhães, T. Forensic toxicology in drug-facilitated sexual assault. Toxicol. Mech. Methods 2013, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Tridico, S.R.; Murray, D.C.; Addison, J.; Kirkbride, K.P.; Bunce, M. Metagenomic analyses of bacteria on human hairs: A qualitative assessment for applications in forensic science. Investig. Genet. 2014, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Hyde, E.R.; Haarmann, D.P.; Lynne, A.M.; Bucheli, S.R.; Petrosino, J.F. The living dead: Bacterial community structure of a cadaver at the onset and end of the bloat stage of decomposition. PLoS ONE 2013, 8, e77733. [Google Scholar] [CrossRef]
- American Society for Microbiology. FAQ: Human Microbiome; American Society for Microbiology: Washington, DC, USA, 2013. [Google Scholar]
- Benbow, M.E.; Barton, P.S.; Ulyshen, M.D.; Beasley, J.C.; DeVault, T.L.; Strickland, M.S.; Tomberlin, J.K.; Jordan, H.R.; Pechal, J.L. Necrobiome framework for bridging decomposition ecology of autotrophically and heterotrophically derived organic matter. Ecol. Monogr. 2019, 89, e01331. [Google Scholar] [CrossRef]
- Tomberlin, J.K.; Benbow, M.E.; Barnes, K.M.; Jordan, H.R. Arthropod–microbe interactions on vertebrate remains: Potential applications in the forensic sciences. In Forensic Microbiology, 1st ed.; Wiley: Hoboken, NJ, USA, 2017; Chapter 11; pp. 274–311. [Google Scholar]
- Speruda, M.; Piecuch, A.; Borzęcka, J.; Kadej, M.; Ogórek, R. Microbial traces and their role in forensic science. J. Appl. Microbiol. 2021, 132, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Javan, G.T.; Finley, S.J.; Abidin, Z.; Mulle, J.G. The Thanatomicrobiome: A Missing Piece of the Microbial Puzzle of Death. Front. Microbiol. 2016, 7, 225. [Google Scholar] [CrossRef]
- Zhou, W.; Bian, Y. Thanatomicrobiome composition profiling as a tool for forensic investigation. Forensic Sci. Res. 2018, 3, 105–110. [Google Scholar] [CrossRef]
- Can, I.; Javan, G.T.; Pozhitkov, A.E.; Noble, P.A. Distinctive thanatomicrobiome signatures found in the blood and internal organs of humans. J. Microbiol. Methods 2014, 106, 1–7. [Google Scholar] [CrossRef]
- Javan, G.T.; Finley, S.J.; Can, I.; Wilkinson, J.E.; Hanson, J.D.; Tarone, A.M. Human Thanatomicrobiome Succession and Time Since Death. Sci. Rep. 2016, 6, 29598. [Google Scholar] [CrossRef]
- Donaldson, A.E.; Lamont, I.L. Biochemistry changes that occur after death: Potential markers for determining post-mortem interval. PLoS ONE 2013, 8, e82011. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.L.; Carter, D.O.; Knight, R. Microbiology of death. Curr. Biol. 2016, 26, R561–R563. [Google Scholar] [CrossRef]
- Morris, J.A.; Harrison, L.M.; Partridge, S.M. Postmortem bacteriology: A re-evaluation. J. Clin. Pathol. 2006, 59, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Harrison, L.M.; Partridge, S.M. Practical and theoretical aspects of postmortem bacteriology. Curr. Diagn. Pathol. 2007, 13, 65–74. [Google Scholar] [CrossRef]
- Balzan, S.; de Almeida Quadros, C.; De Cleva, R.; Zilberstein, B.; Cecconello, I. Bacterial translocation: Overview of mechanisms and clinical impact. J. Gastroenterol. Hepatol. 2007, 22, 464–471. [Google Scholar] [CrossRef]
- Mesli, V.; Neut, C.; Hedouin, V. Postmortem bacterial translocation. In Forensic Anthropology and Medicine; Springer: Berlin/Heidelberg, Germany, 2017; Chapter 8; pp. 192–211. [Google Scholar]
- João, P. Decay process of a cadaver. In Forensic Anthropology and Medicine; Springer: Berlin/Heidelberg, Germany, 2006; Chapter 5. [Google Scholar]
- Burcham, Z.M.; Pechal, J.L.; Schmidt, C.J.; Bose, J.L.; Rosch, J.W.; Benbow, M.E.; Jordan, H.R. Bacterial Community Succession, Transmigration, and Differential Gene Transcription in a Controlled Vertebrate Decomposition Model. Front. Microbiol. 2019, 10, 745. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Metcalf, J.L.; Keepers, K.; Ackermann, G.; Carter, D.O.; Knight, R. Vertebrate decomposition is accelerated by soil microbes. Appl. Environ. Microbiol. 2014, 80, 4920–4929. [Google Scholar] [CrossRef]
- Metcalf, J.L. Estimating the postmortem interval using microbes: Knowledge gaps and a path to technology adoption. Forensic Sci. Int. Genet. 2019, 38, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Adserias-Garriga, J.; Hernandez, M.; Quijada, N.M.; Rodriguez Lazaro, D.; Steadman, D.; Garcia-Gil, J. Daily thanatomicrobiome changes in soil as an approach of postmortem interval estimation: An ecological perspective. Forensic Sci. Int. 2017, 278, 388–395. [Google Scholar] [CrossRef]
- Tarone, A.M.; Mann, A.E.; Zhang, Y.; Zascavage, R.R.; Mitchell, E.A.; Morales, E.; Rusch, T.W.; Allen, M.S. The devil is in the details: Variable impacts of season, BMI, sampling site temperature, and presence of insects on the post-mortem microbiome. Front. Microbiol. 2022, 13, 1064904. [Google Scholar]
- Hyde, E.R.; Metcalf, J.L.; Bucheli, S.R.; Lynne, A.M.; Knight, R. Microbial communities associated with decomposing corpses. In Forensic Microbiology; Wiley: Hoboken, NJ, USA, 2017; Chapter 10; pp. 245–273. [Google Scholar]
- Eden, R.E.; Brooke, T. Algor Mortis; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Javan, G.T.; Finley, S.J.; Tuomisto, S.; Hall, A.; Benbow, M.E.; Mills, D. An interdisciplinary review of the thanatomicrobiome in human decomposition. Forensic Sci. Med. Pathol. 2019, 15, 75–83. [Google Scholar] [CrossRef] [PubMed]
- DeBruyn, J.M.; Hauther, K.A. Postmortem succession of gut microbial communities in deceased human subjects. PeerJ 2017, 5, e3437. [Google Scholar] [CrossRef]
- Campobasso, C.P.; Mastroianni, G.; Feola, A.; Mascolo, P.; Carfora, A.; Liguori, B.; Zangani, P.; Dell’Annunziata, F.; Folliero, V.; Petrillo, A.; et al. MALDI-TOF Mass Spectrometry Analysis and Human Post-Mortem Microbial Community: A Pilot Study. Int. J. Environ. Res. Public Health 2022, 19, 4354. [Google Scholar] [CrossRef] [PubMed]
- Pechal, J.L.; Schmidt, C.J.; Jordan, H.R.; Benbow, M.E. A large-scale survey of the postmortem human microbiome, and its potential to provide insight into the living health condition. Sci. Rep. 2018, 8, 5724. [Google Scholar]
- Adserias-Garriga, J.; Quijada, N.M.; Hernandez, M.; Rodriguez Lazaro, D.; Steadman, D.; Garcia-Gil, L.J. Dynamics of the oral microbiota as a tool to estimate time since death. Mol. Oral Microbiol. 2017, 32, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, S.; Liu, R.; Yuan, L.; Wu, D.; Yang, E.; Yang, H.; Ullah, S.; Ishaq, H.M.; Liu, H.; et al. Potential use of molecular and structural characterization of the gut bacterial community for postmortem interval estimation in Sprague Dawley rats. Sci. Rep. 2021, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Dash, H.R.; Das, S. Thanatomicrobiome and epinecrotic community signatures for estimation of post-mortem time interval in human cadaver. Appl. Microbiol. Biotechnol. 2020, 104, 9497–9512. [Google Scholar]
- Carter, D.O.; Yellowlees, D.; Tibbett, M. Temperature Affects Microbial Decomposition of Cadavers (Rattus Rattus) in Contrasting Soils. Appl. Soil Ecol. 2008, 40, 129–137. [Google Scholar]
- Vass, A.A. Beyond the Grave—Understanding Human Decomposition. Microbiol. Today 2001, 28, 190–192. [Google Scholar]
- Janaway, R.C.; Percival, S.L.; Wilson, A.S. Decomposition of Human Remains. In Microbiology and Aging; Springer: Berlin/Heidelberg, Germany, 2009; pp. 313–334. [Google Scholar]
- Hyde, E.R.; Haarmann, D.P.; Petrosino, J.F.; Lynne, A.M.; Bucheli, S.R. Initial insights into bacterial succession during human decomposition. Int. J. Leg. Med. 2015, 129, 661–671. [Google Scholar]
- Metcalf, J.L.; Wegener Parfrey, L.; Gonzalez, A.; Lauber, C.L.; Knights, D.; Ackermann, G.; Humphrey, G.C.; Gebert, M.J.; Van Treuren, W.; Berg-Lyons, D.; et al. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. eLife 2013, 2, e01104. [Google Scholar] [CrossRef]
- Kakizaki, E.; Ogura, Y.; Kozawa, S.; Nishida, S.; Uchiyama, T.; Hayashi, T.; Yukawa, N. Detection of diverse aquatic microbes in blood and organs of drowning victims: First metagenomic approach using high-throughput 454-pyrosequencing. Forensic Sci. Int. 2012, 220, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Burcham, Z.; Hood, J.; Pechal, J.; Krausz, K.; Bose, J.; Schmidt, C.; Benbow, M.; Jordan, H. Fluorescently labeled bacteria provide insight on post-mortem microbial transmigration. Forensic Sci. Int. 2016, 264, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Heimesaat, M.M.; Boelke, S.; Fischer, A.; Haag, L.-M.; Loddenkemper, C.; Kühl, A.A.; Göbel, U.B.; Bereswill, S. Comprehensive postmortem analyses of intestinal microbiota changes and bacterial translocation in human flora associated mice. PLoS ONE 2012, 7, e40758. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, K.; Li, H.; Sun, Q.; Wei, X.; Li, H.; Zhang, S.; Fan, S.; Wang, Z. Dissecting the microbial community structure of internal organs during the early postmortem period in a murine corpse model. BMC Microbiol. 2023, 23, 38. [Google Scholar] [CrossRef]
- Tuomisto, S.; Karhunen, P.J.; Vuento, R.; Aittoniemi, J.; Pessi, T. Evaluation of postmortem bacterial migration using culturing and real-time quantitative PCR. J. Forensic Sci. 2013, 58, 910–916. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Q.; Zhang, K.; Wu, H.; Wang, G.; Cai, W.; Yu, K.; Sun, Q.; Fan, S.; Wang, Z. Analysis of Postmortem Intestinal Microbiota Successional Patterns with Application in Postmortem Interval Estimation. Microb. Ecol. 2022, 84, 1087–1102. [Google Scholar] [CrossRef]
- Harrison, L.; Kooienga, E.; Speights, C.; Tomberlin, J.; Lashley, M.; Barton, B.; Jordan, H. Microbial succession from a subsequent secondary death event following mass mortality. BMC Microbiol. 2020, 20, 309. [Google Scholar] [CrossRef]
- Huang, S.; Haiminen, N.; Carrieri, A.P.; Hu, R.; Jiang, L.; Parida, L.; Russell, B.; Allaband, C.; Zarrinpar, A.; Vázquez-Baeza, Y.; et al. Human Skin, Oral, and Gut Microbiomes Predict Chronological Age. mSystems 2020, 5, e00630-19. [Google Scholar] [CrossRef]
- Gevers, W. Biochemical aspects of cell death. Forensic Sci. 1975, 6, 25–29. [Google Scholar] [CrossRef]
- Dell’Annunziata, F.; Martora, F.; Della Pepa, M.E.; Folliero, V.; Luongo, L.; Bocelli, S.; Guida, F.; Mascolo, P.; Campobasso, C.P.; Maione, S.; et al. Postmortem interval assessment by MALDI-TOF mass spectrometry analysis in murine cadavers. J. Appl. Microbiol. 2022, 132, 707–714. [Google Scholar] [CrossRef]
- Emmons, A.L.; Mundorff, A.Z.; Hoeland, K.M.; Davoren, J.; Keenan, S.W.; Carter, D.O.; Campagna, S.R.; DeBruyn, J.M. Postmortem Skeletal Microbial Community Composition and Function in Buried Human Remains. mSystems 2022, 7, e0004122. [Google Scholar] [CrossRef]
- Javan, G.T.; Wells, T.; Allen, J.; Visona, S.; Moretti, M.; Tipton, C.; Scott, L.; Finley, S.J. Correlation between postmortem microbial signatures and substance abuse disorders. PLoS ONE 2022, 17, e0274401. [Google Scholar] [CrossRef]
- Mansour, S.; Moustafa, M.; Saad, B.; Hamed, R.; Moustafa, A.-R. Impact of diet on human gut microbiome and disease risk. New Microbes New Infect. 2021, 41, 100845. [Google Scholar] [CrossRef] [PubMed]
- Mondor, E.B.; Tremblay, M.N.; Tomberlin, J.K.; Benbow, E.M.; Tarone, A.M.; Crippen, T.L. The Ecology of Carrion Decomposition. Nat. Educ. Knowl. 2012, 3, 21. [Google Scholar]
- Matuszewski, S.; Konwerski, S.; Frątczak, K.; Szafałowicz, M. Effect of body mass and clothing on decomposition of pig carcasses. Int. J. Leg. Med. 2014, 128, 1039–1048. [Google Scholar] [CrossRef]
- Swayambhu, M.; Kümmerli, R.; Arora, N. Microbiome-Based Stain Analyses in Crime Scenes. Appl. Environ. Microbiol. 2023, 89, e0132522. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Mason-Buck, G.; Ballard, D.; Branicki, W.; Amorim, A. Biowarfare, bioterrorism and biocrime: A historical overview on microbial harmful applications. Forensic Sci. Int. 2020, 314, 110366. [Google Scholar] [CrossRef]
- Lax, S.; Hampton-Marcell, J.T.; Gibbons, S.M.; Colares, G.B.; Smith, D.; Eisen, J.A.; Gilbert, J.A. Forensic analysis of the microbiome of phones and shoes. Microbiome 2015, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, D.; Leung, M.H.Y.; Lee, P.K.H. Microbiota fingerprints lose individually identifying features over time. Microbiome 2017, 5, 1. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.J.; Lee, J.-A.; Kim, J.W.; Kim, S.B. Microbial forensic analysis of human-associated bacteria inhabiting hand surface. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e510–e512. [Google Scholar] [CrossRef]
- Schmedes, S.E.; Woerner, A.E.; Novroski, N.M.; Wendt, F.R.; King, J.L.; Stephens, K.M.; Budowle, B. Targeted sequencing of clade-specific markers from skin microbiomes for forensic human identification. Forensic Sci. Int. Genet. 2018, 32, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Schmedes, S.E.; Sajantila, A.; Budowle, B. Expansion of Microbial Forensics. J. Clin. Microbiol. 2016, 54, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Yoon, H.K.; An, S.; Lee, J.W.; Ahn, E.-R.; Kim, Y.-J.; Park, H.-C.; Lee, K.; Hwang, J.H.; Lim, S.-K. Rapid oral bacteria detection based on real-time PCR for the forensic identification of saliva. Sci. Rep. 2018, 8, 10852. [Google Scholar] [CrossRef]
- Ossowicki, A.; Raaijmakers, J.M.; Garbeva, P. Disentangling soil microbiome functions by perturbation. Environ. Microbiol. Rep. 2021, 13, 582–590. [Google Scholar] [CrossRef]
- Haarkötter, C.; Saiz, M.; Gálvez, X.; Medina-Lozano, M.I.; Álvarez, J.C.; Lorente, J.A. Usefulness of Microbiome for Forensic Geolocation: A Review. Life 2021, 11, 1322. [Google Scholar] [CrossRef]
- García, M.G.; Pérez-Cárceles, M.D.; Osuna, E.; Legaz, I. Impact of the Human Microbiome in Forensic Sciences: A Systematic Review. Appl. Environ. Microbiol. 2020, 86, e01451-20. [Google Scholar] [CrossRef] [PubMed]
- Escobar, J.S.; Klotz, B.; Valdes, B.E.; Agudelo, G.M. The gut microbiota of Colombians differs from that of Americans, Europeans and Asians. BMC Microbiol. 2014, 14, 311. [Google Scholar] [CrossRef]
- Brinkac, L.; Clarke, T.H.; Singh, H.; Greco, C.; Gomez, A.; Torralba, M.G.; Frank, B.; Nelson, K.E. Spatial and Environmental Variation of the Human Hair Microbiota. Sci. Rep. 2018, 8, 9017. [Google Scholar] [CrossRef]
- Phan, K.; Barash, M.; Spindler, X.; Gunn, P.; Roux, C. Retrieving forensic information about the donor through bacterial profiling. Int. J. Leg. Med. 2020, 134, 21–29. [Google Scholar] [CrossRef]
- Kodama, W.A.; Xu, Z.; Metcalf, J.L.; Song, S.J.; Harrison, N.; Knight, R.; Carter, D.O.; Happy, C.B. Trace Evidence Potential in Postmortem Skin Microbiomes: From Death Scene to Morgue. J. Forensic Sci. 2019, 64, 791–798. [Google Scholar] [CrossRef]
- Jauréguy, F.; Chariot, P.; Vessières, A.; Picard, B. Prevalence of Chlamydia trachomatis and Neisseria gonorrhoeae infections detected by real-time PCR among individuals reporting sexual assaults in the Paris, France area. Forensic Sci. Int. 2016, 266, 130–133. [Google Scholar] [CrossRef]
- Francés-Cuesta, C.; de la Caba, I.; Idigoras, P.; Fernández-Rodríguez, A.; Pérez, D.d.V.; Marimón, J.M.; González-Candelas, F. Whole-genome sequencing of Neisseria gonorrhoeae in a forensic transmission case. Forensic Sci. Int. Genet. 2019, 42, 141–146. [Google Scholar] [CrossRef]
- Williams, D.W.; Gibson, G. Individualization of pubic hair bacterial communities and the effects of storage time and temperature. Forensic Sci. Int. Genet. 2017, 26, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.; Egan, S.; Hughes, S.; Chapman, B. The Sexome—A proof of concept study into microbial transfer between heterosexual couples after sexual intercourse. Forensic Sci. Int. 2023, 348, 111711. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Gibson, G. Classification of individuals and the potential to detect sexual contact using the microbiome of the pubic region. Forensic Sci. Int. Genet. 2019, 41, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Torralba, M.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 2012, 56, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.R.; Wilkinson, J.E.; Robertson, B.K.; Javan, G.T. Sex-related differences in the thanatomicrobiome in postmortem heart samples using bacterial gene regions V1-2 and V4. Lett. Appl. Microbiol. 2018, 67, 144–153. [Google Scholar] [CrossRef] [PubMed]
- D’argenio, V.; Torino, M.; Precone, V.; Casaburi, G.; Esposito, M.V.; Iaffaldano, L.; Malapelle, U.; Troncone, G.; Coto, I.; Cavalcanti, P.; et al. The Cause of Death of a Child in the 18th Century Solved by Bone Microbiome Typing Using Laser Microdissection and Next Generation Sequencing. Int. J. Mol. Sci. 2017, 18, 109. [Google Scholar] [CrossRef]
- Piette, M.H.; De Letter, E.A. Drowning: Still a difficult autopsy diagnosis. Forensic Sci. Int. 2006, 163, 1–9. [Google Scholar] [CrossRef]
- Uchiyama, T.; Kakizaki, E.; Kozawa, S.; Nishida, S.; Imamura, N.; Yukawa, N. A new molecular approach to help conclude drowning as a cause of death: Simultaneous detection of eight bacterioplankton species using real-time PCR assays with TaqMan probes. Forensic Sci. Int. 2012, 222, 11–26. [Google Scholar] [CrossRef]
- Kakizaki, E.; Kozawa, S.; Imamura, N.; Uchiyama, T.; Nishida, S.; Sakai, M.; Yukawa, N. Detection of marine and freshwater bacterioplankton in immersed victims: Post-mortem bacterial invasion does not readily occur. Forensic Sci. Int. 2011, 211, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Azparren, J.; Fernandez-Rodriguez, A.; Vallejo, G. Diagnosing death by drowning in fresh water using blood strontium as an indicator. Forensic Sci. Int. 2003, 137, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Goldwater, P.N. Gut Microbiota and Immunity: Possible Role in Sudden Infant Death Syndrome. Front. Immunol. 2015, 6, 269. [Google Scholar] [CrossRef] [PubMed]
- Leong, L.E.; Taylor, S.L.; Shivasami, A.; Goldwater, P.N.; Rogers, G.B. Intestinal Microbiota Composition in Sudden Infant Death Syndrome and Age-Matched Controls. J. Pediatr. 2017, 191, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Highet, A.R.; Berry, A.M.; Bettelheim, K.A.; Goldwater, P.N. Gut microbiome in sudden infant death syndrome (SIDS) differs from that in healthy comparison babies and offers an explanation for the risk factor of prone position. Int. J. Med. Microbiol. 2014, 304, 735–741. [Google Scholar] [CrossRef]
- Praveen, V.; Praveen, S. Microbiome-Gut-Brain Axis: A Pathway for Improving Brainstem Serotonin Homeostasis and Successful Autoresuscitation in SIDS-A Novel Hypothesis. Front. Pediatr. 2016, 4, 136. [Google Scholar] [CrossRef]
- Castle, J.W.; Butzbach, D.M.; Walker, G.S.; Lenehan, C.E.; Reith, F.; Kirkbride, K.P. Microbial impacts in postmortem toxicology. In Forensic Microbiology; Wiley: Hoboken, NJ, USA, 2017; Chapter 9; pp. 212–244. [Google Scholar]
- Dinis-Oliveira, R.J. The Auto-Brewery Syndrome: A Perfect Metabolic “Storm” with Clinical and Forensic Implications. J. Clin. Med. 2021, 10, 4637. [Google Scholar] [CrossRef]
- Pearson, A.L.; Rzotkiewicz, A.; Pechal, J.L.; Schmidt, C.J.; Jordan, H.R.; Zwickle, A.; Benbow, M.E. Initial Evidence of the Relationships between the Human Postmortem Microbiome and Neighborhood Blight and Greening Efforts. Ann. Am. Assoc. Geogr. 2019, 119, 958–978. [Google Scholar] [CrossRef]
- Franceschetti, L.; Amadasi, A.; Bugelli, V.; Bolsi, G.; Tsokos, M. Estimation of Late Postmortem Interval: Where Do We Stand? A Literature Review. Biology 2023, 12, 783. [Google Scholar] [CrossRef]
- Pittner, S.; Bugelli, V.; Weitgasser, K.; Zissler, A.; Sanit, S.; Lutz, L.; Monticelli, F.; Campobasso, C.P.; Steinbacher, P.; Amendt, J. A field study to evaluate PMI estimation methods for advanced decomposition stages. Int. J. Leg. Med. 2020, 134, 1361–1373. [Google Scholar] [CrossRef]
- Sampaio-Silva, F.; Magalhaes, T.; Carvalho, F.; Dinis-Oliveira, R.J.; Silvestre, R. Profiling of RNA degradation for estimation of post mortem interval. PLoS ONE 2013, 8, e56507. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.; Carvalho, F.; Magalhães, T.; Guedes de Pinho, P.; Silvestre, R.; Dinis-Oliveira, R.J. Promising blood-derived biomarkers for estimation of the postmorteminterval. Toxicol. Res. 2015, 4, 1443–1452. [Google Scholar] [CrossRef]
- Brooks, J.W. Postmortem Changes in Animal Carcasses and Estimation of the Postmortem Interval. Vet. Pathol. 2016, 53, 929–940. [Google Scholar] [CrossRef]
- Cordeiro, C.; Ordóñez-Mayán, L.; Lendoiro, E.; Febrero-Bande, M.; Vieira, D.N.; Muñoz-Barús, J.I. A reliable method for estimating the postmortem interval from the biochemistry of the vitreous humor, temperature and body weight. Forensic Sci. Int. 2019, 295, 157–168. [Google Scholar] [CrossRef]
- Amendt, J.; Krettek, R.; Zehner, R. Forensic entomology. Naturwissenschaften 2004, 91, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Ubelaker, D.H.; Wu, Y. Fragment analysis in forensic anthropology. Forensic Sci. Res. 2020, 5, 260–265. [Google Scholar] [CrossRef]
- Amadasi, A.; Cappella, A.; Cattaneo, C.; Cofrancesco, P.; Cucca, L.; Merli, D.; Milanese, C.; Pinto, A.; Profumo, A.; Scarpulla, V.; et al. Determination of the post mortem interval in skeletal remains by the comparative use of different physico-chemical methods: Are they reliable as an alternative to. Homo 2017, 68, 213–221. [Google Scholar] [CrossRef]
- Mohammed, F.; Fairozekhan, A.T.; Bhat, S.; Menezes, R.G. Forensic Odontology; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Coyle, H.M.; Lee, C.-L.; Lin, W.-Y.; Lee, H.C.; Palmbach, T.M. Forensic botany: Using plant evidence to aid in forensic death investigation. Croat. Med. J. 2005, 46, 606–612. [Google Scholar]
- Deva, R.; Rattan, S.I.S. Microbiology and Aging: Clinical Manifestations; Percival, S.L., Ed.; Biogerontology; Humana: Totowa, NJ, USA, 2009; Volume 10, pp. 535–536. [Google Scholar]
- Zhang, J.; Liu, W.; Simayijiang, H.; Hu, P.; Yan, J. Application of Microbiome in Forensics. Genom. Proteom. Bioinf. 2022, 21, 97–107. [Google Scholar] [CrossRef]
- Na, J.-Y.; Park, J.H.; Kim, S.-H.; Park, J.-T. Bacteria as Normal Flora in Postmortem Body Fluid Samples. Korean J. Leg. Med. 2017, 41, 87–93. [Google Scholar] [CrossRef]
- Jo, J.; Oh, J.; Park, C. Microbial community analysis using high-throughput sequencing technology: A beginner’s guide for microbiologists. J. Microbiol. 2020, 58, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Adams, R.I.; Bateman, A.C.; Bik, H.M.; Meadow, J.F. Microbiota of the indoor environment: A meta-analysis. Microbiome 2015, 3, 49. [Google Scholar] [CrossRef]
- Tu, Q.; He, Z.; Zhou, J. Strain/species identification in metagenomes using genome-specific markers. Nucleic Acids Res. 2014, 42, e67. [Google Scholar] [CrossRef] [PubMed]
- Zolfo, M.; Asnicar, F.; Manghi, P.; Pasolli, E.; Tett, A.; Segata, N. Profiling microbial strains in urban environments using metagenomic sequencing data. Biol. Direct. 2018, 13, 9. [Google Scholar] [CrossRef]
- Tu, Q.; Li, J.; Shi, Z.; Chen, Y.; Lin, L.; Li, J.; Wang, H.; Yan, J.; Zhou, Q.; Li, X.; et al. HuMiChip2 for strain level identification and functional profiling of human microbiomes. Appl. Microbiol. Biotechnol. 2017, 101, 423–435. [Google Scholar] [CrossRef]
- Zhang, Y.; Pechal, J.L.; Schmidt, C.J.; Jordan, H.R.; Wang, W.W.; Benbow, M.E.; Sze, S.-H.; Tarone, A.M. Machine learning performance in a microbial molecular autopsy context: A cross-sectional postmortem human population study. PLoS ONE 2019, 14, e0213829. [Google Scholar] [CrossRef]
- Duong, V.-A.; Park, J.-M.; Lim, H.-J.; Lee, H. Proteomics in Forensic Analysis: Applications for Human Samples. Appl. Sci. 2021, 11, 3393. [Google Scholar] [CrossRef]
- Burcham, Z.M.; Cowick, C.A.; Baugher, C.N.; Pechal, J.L.; Schmidt, C.J.; Rosch, J.W.; Benbow, M.E.; Jordan, H.R. Total RNA Analysis of Bacterial Community Structural and Functional Shifts Throughout Vertebrate Decomposition. J. Forensic Sci. 2019, 64, 1707–1719. [Google Scholar] [CrossRef]
- Li, C.; Ma, D.; Deng, K.; Chen, Y.; Huang, P.; Wang, Z. Application of MALDI-TOF MS for Estimating the Postmortem Interval in Rat Muscle Samples. J. Forensic Sci. 2017, 62, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Soni, S.; White, J.; Can, G.; Javan, G.T. Evaluation of DNA degradation using flow cytometry: Promising tool for postmortem interval determination. Am. J. Forensic Med. Pathol. 2015, 36, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Nearing, J.T.; Comeau, A.M.; Langille, M.G.I. Identifying biases and their potential solutions in human microbiome studies. Microbiome 2021, 9, 113. [Google Scholar] [CrossRef]
- Benbow, M.E.; Pechal, J. Approaches and considerations for forensic microbiology decomposition research. In Forensic Microbiology; Wiley: Hoboken, NJ, USA, 2017; Chapter 3; pp. 56–71. [Google Scholar]
- Yuan, H.; Wang, Z.; Wang, Z.; Zhang, F.; Guan, D.; Zhao, R. Trends in forensic microbiology: From classical methods to deep learning. Front. Microbiol. 2023, 14, 1163741. [Google Scholar] [CrossRef]
- Li, P.; Luo, H.; Ji, B.; Nielsen, J. Machine learning for data integration in human gut microbiome. Microb. Cell Fact. 2022, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, F.; Wang, L.; Yuan, H.; Guan, D.; Zhao, R. Advances in artificial intelligence-based microbiome for PMI estimation. Front. Microbiol. 2022, 13, 1034051. [Google Scholar] [CrossRef] [PubMed]
- Saegeman, V.; Cohen, M.C.; Burton, J.L.; Martinez, M.J.; Rakislova, N.; Offiah, A.C.; Fernandez-Rodriguez, A. Microbiology in minimally invasive autopsy: Best techniques to detect infection. ESGFOR (ESCMID study group of forensic and post-mortem microbiology) guidelines. Forensic Sci. Med. Pathol. 2021, 17, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Riedel, S. The value of postmortem microbiology cultures. J. Clin. Microbiol. 2014, 52, 1028–1033. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, A.; Burton, J.L.; Andreoletti, L.; Alberola, J.; Fornes, P.; Merino, I.; Martínez, M.J.; Castillo, P.; Sampaio-Maia, B.; Caldas, I.M.; et al. Post-mortem microbiology in sudden death: Sampling protocols proposed in different clinical settings. Clin. Microbiol. Infect. 2019, 25, 570–579. [Google Scholar] [CrossRef]
- Jaquet-Chiffelle, D.-O.; Casey, E. A formalized model of the Trace. Forensic Sci. Int. 2021, 327, 110941. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cláudia-Ferreira, A.; Barbosa, D.J.; Saegeman, V.; Fernández-Rodríguez, A.; Dinis-Oliveira, R.J.; Freitas, A.R.; on behalf of the ESCMID Study Group of Forensic and Post-Mortem Microbiology (ESGFOR). The Future Is Now: Unraveling the Expanding Potential of Human (Necro)Microbiome in Forensic Investigations. Microorganisms 2023, 11, 2509. https://doi.org/10.3390/microorganisms11102509
Cláudia-Ferreira A, Barbosa DJ, Saegeman V, Fernández-Rodríguez A, Dinis-Oliveira RJ, Freitas AR, on behalf of the ESCMID Study Group of Forensic and Post-Mortem Microbiology (ESGFOR). The Future Is Now: Unraveling the Expanding Potential of Human (Necro)Microbiome in Forensic Investigations. Microorganisms. 2023; 11(10):2509. https://doi.org/10.3390/microorganisms11102509
Chicago/Turabian StyleCláudia-Ferreira, Ana, Daniel José Barbosa, Veroniek Saegeman, Amparo Fernández-Rodríguez, Ricardo Jorge Dinis-Oliveira, Ana R. Freitas, and on behalf of the ESCMID Study Group of Forensic and Post-Mortem Microbiology (ESGFOR). 2023. "The Future Is Now: Unraveling the Expanding Potential of Human (Necro)Microbiome in Forensic Investigations" Microorganisms 11, no. 10: 2509. https://doi.org/10.3390/microorganisms11102509