


Bacterial Microbiota of Ostreobium, the Coral-Isolated Chlorophyte Ectosymbiont, at Contrasted Salinities
 , and
, and 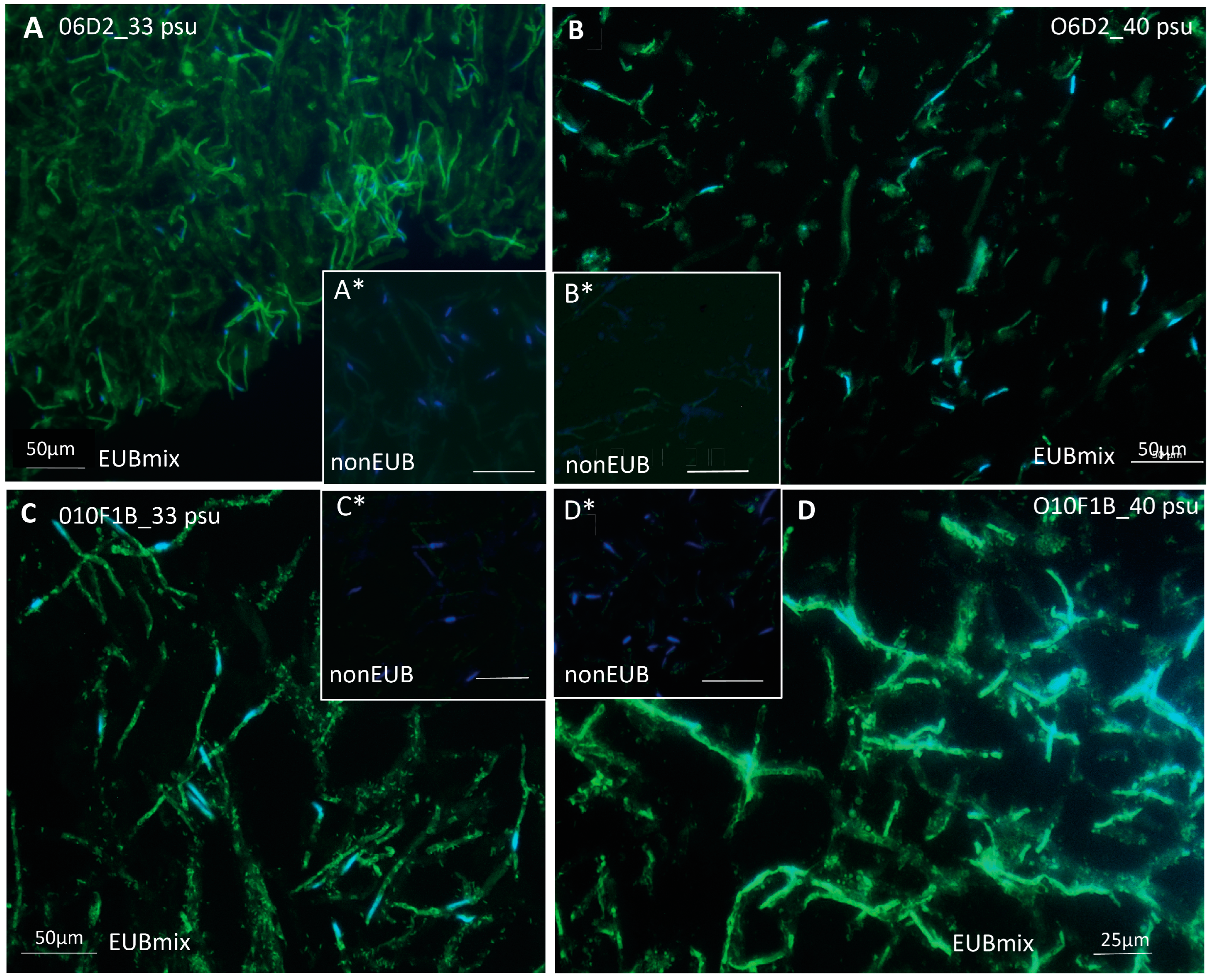{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ostreobium Strains and Acclimatation to Three Salinities
2.2. Catalyzed Reporter Deposition Fluorescence In Situ Hybridization (CARD-FISH)
2.3. Sampling and DNA Extraction
2.4. PCR Amplification and Illumina Sequencing
2.5. Sequence Dataset Analysis
2.6. Data Availability
3. Results
3.1. In Situ Localization of Bacteria Associated to Cultured Ostreobium Siphons
3.2. Quality of Illumina Sequencing Targeting Bacterial 16S rRNA V5–V7 Gene Fragment
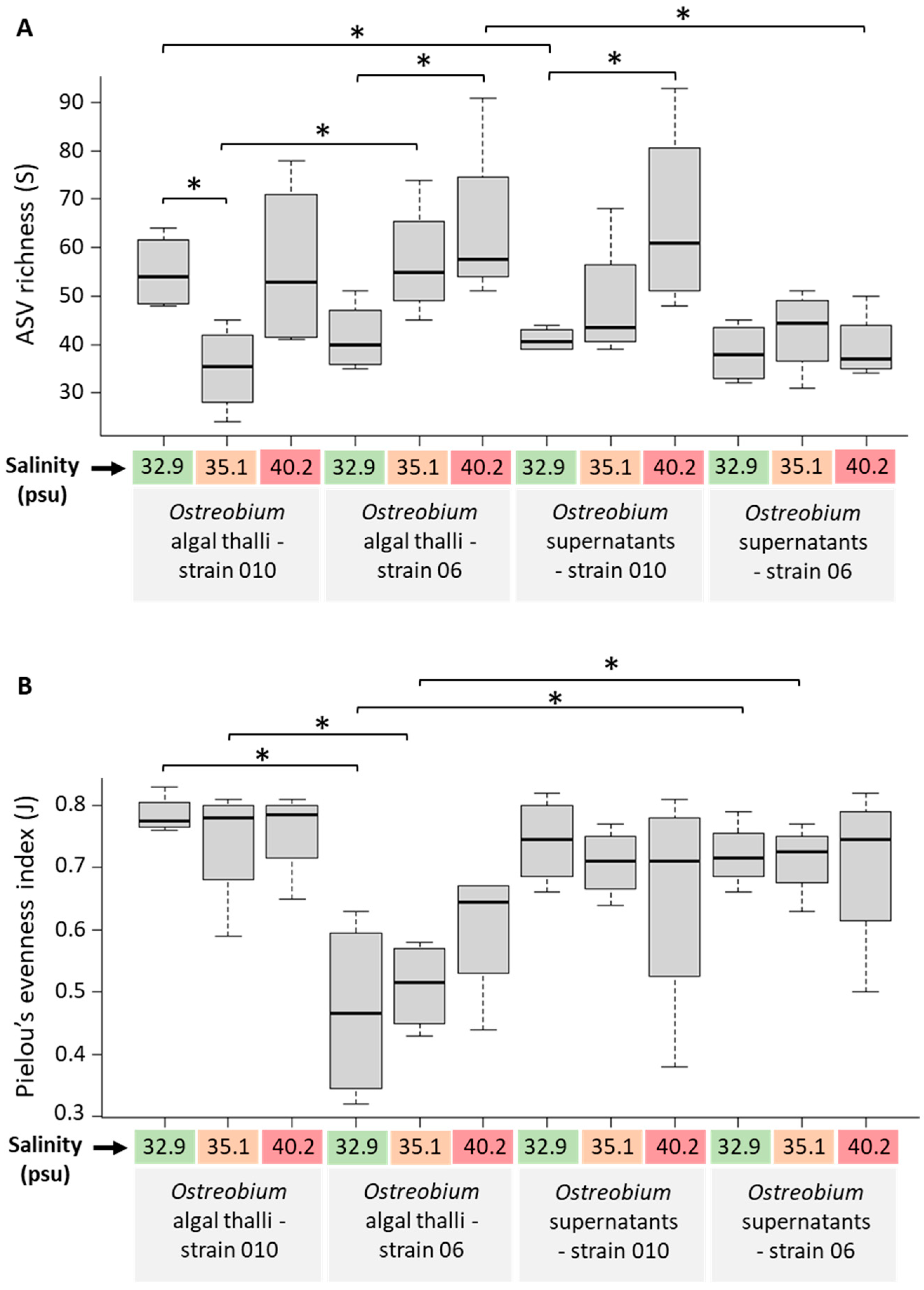
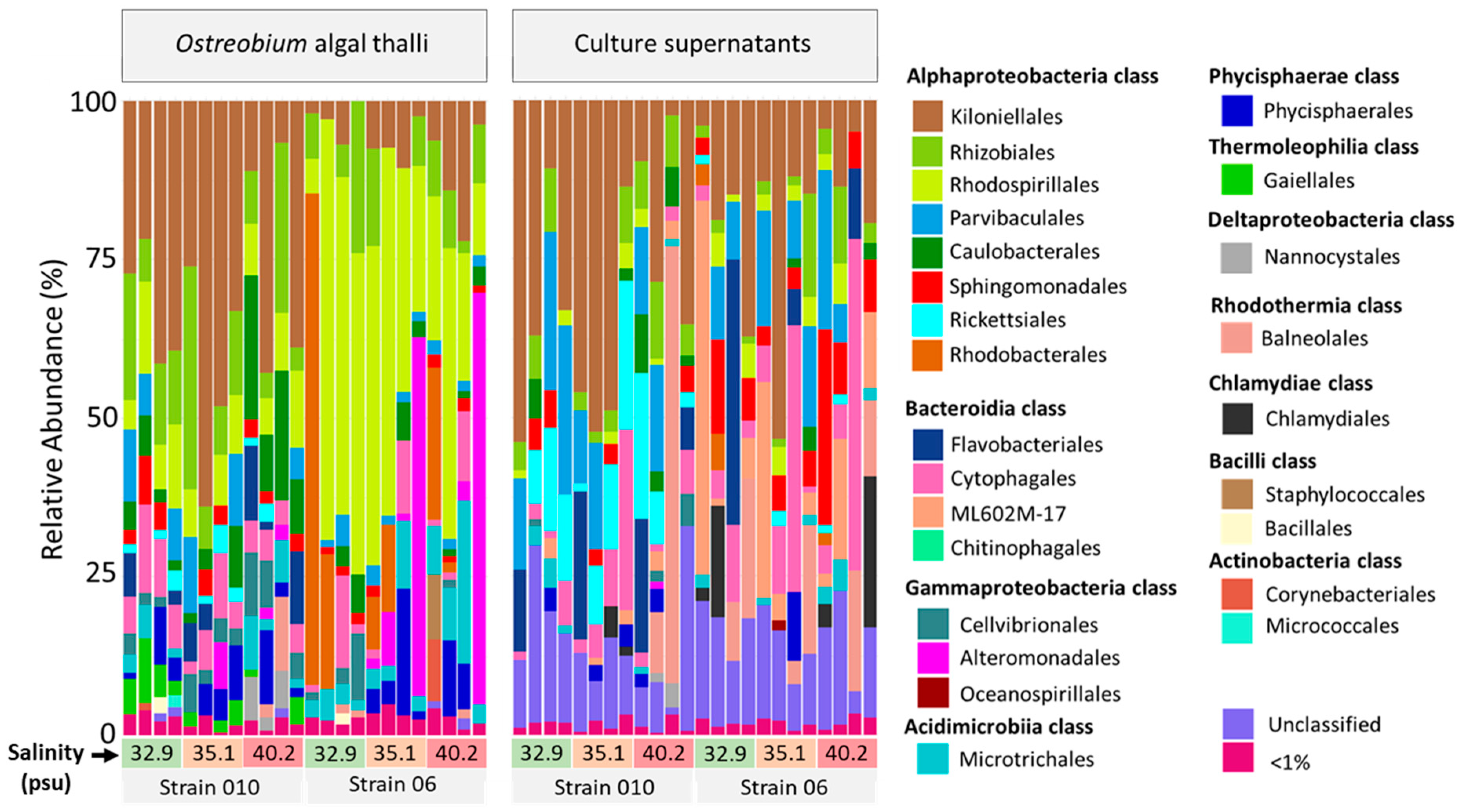
3.3. Diversity and Structure of Bacterial Communities in Cultured Ostreobium
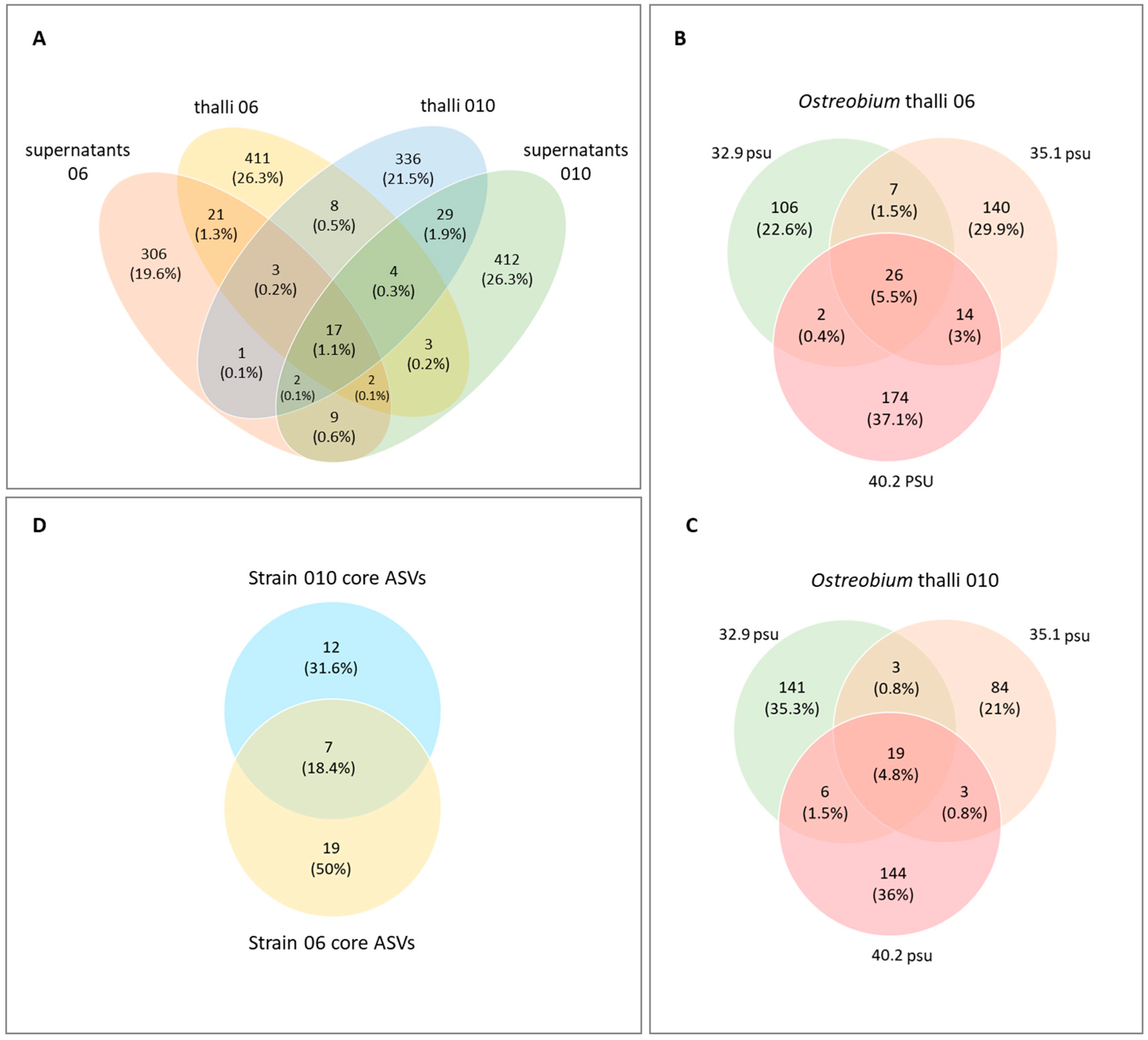
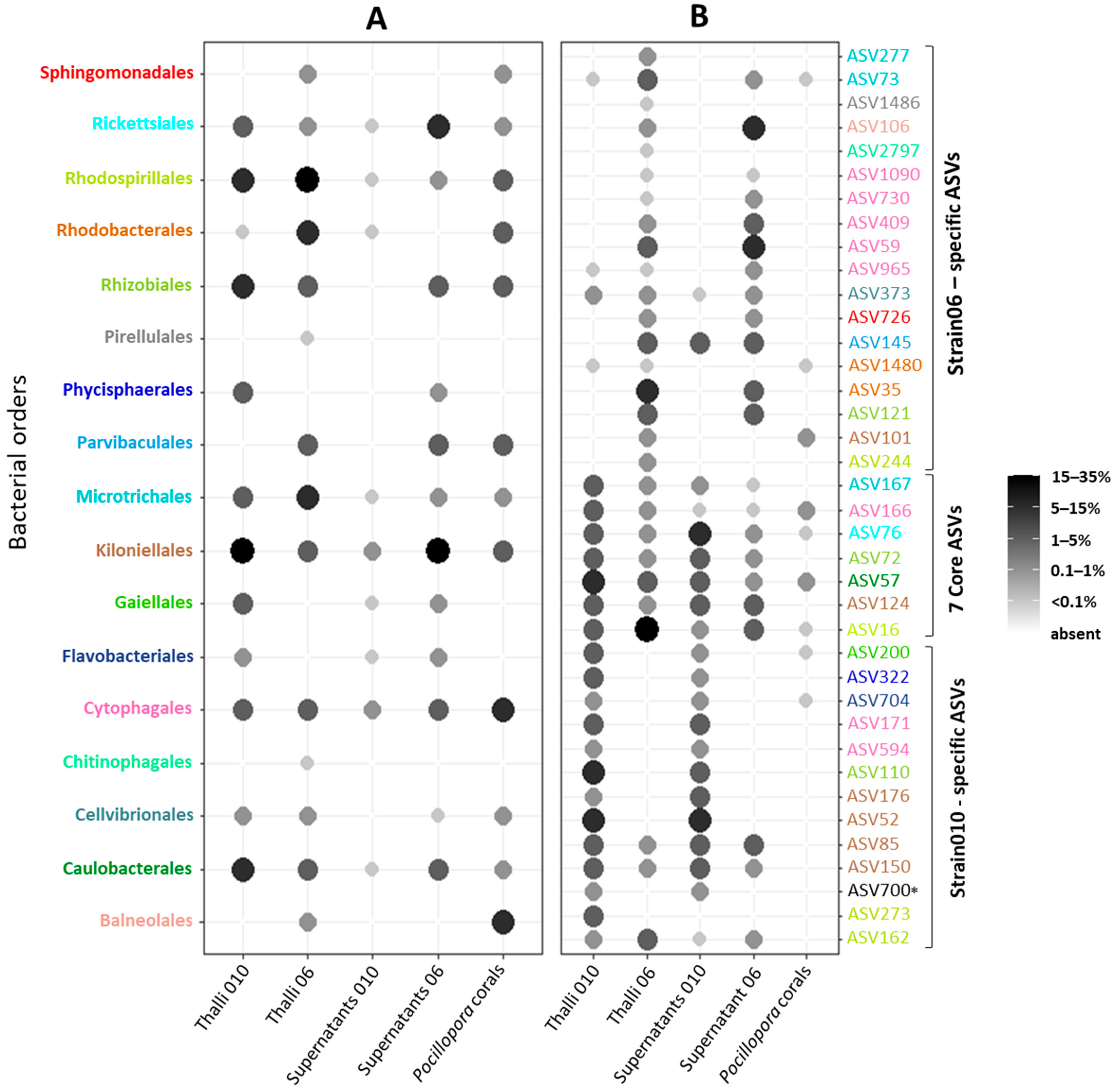
3.4. Taxonomic Composition: Identification of a Core Ostreobium Bacterial Microbiota
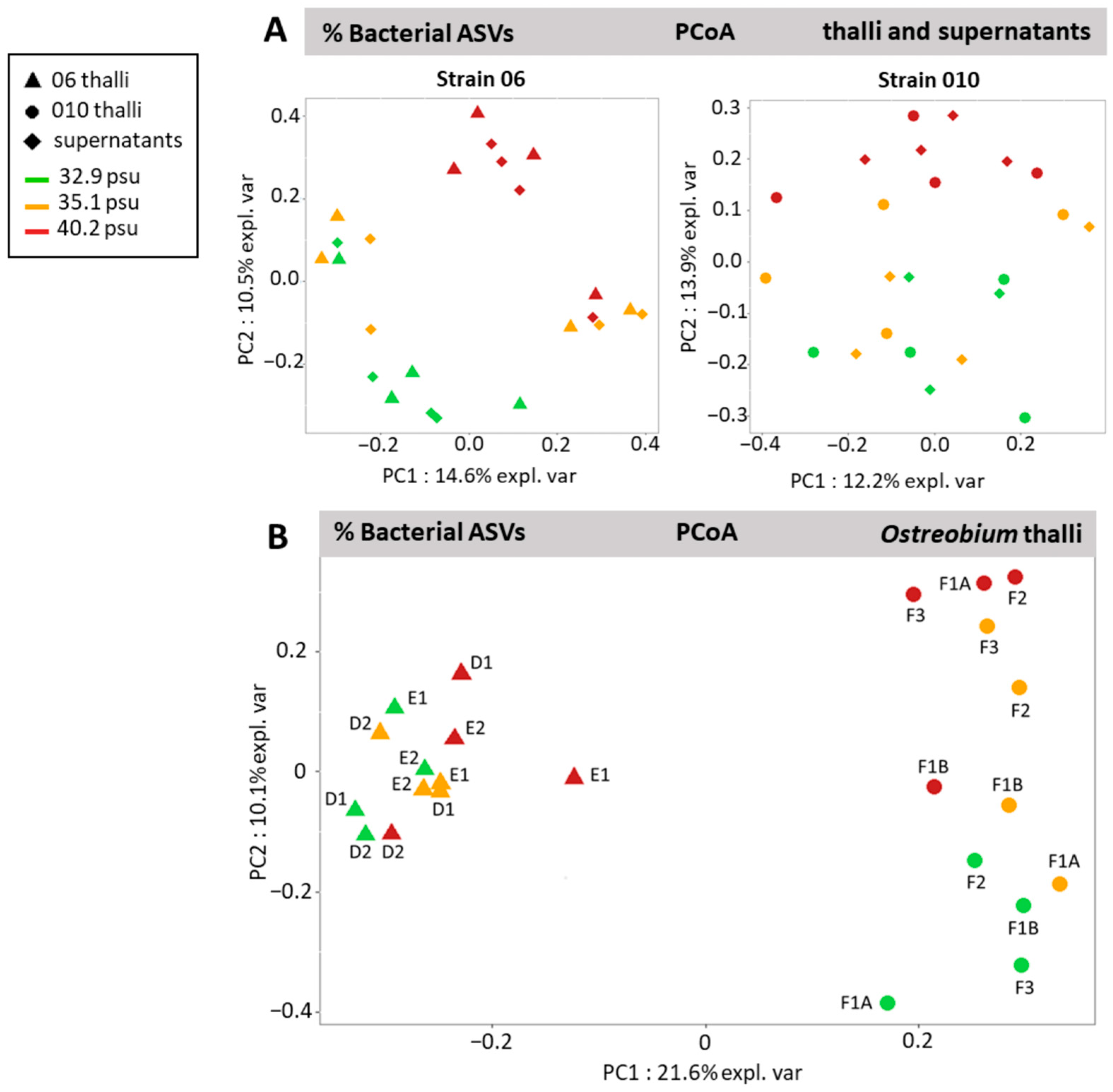
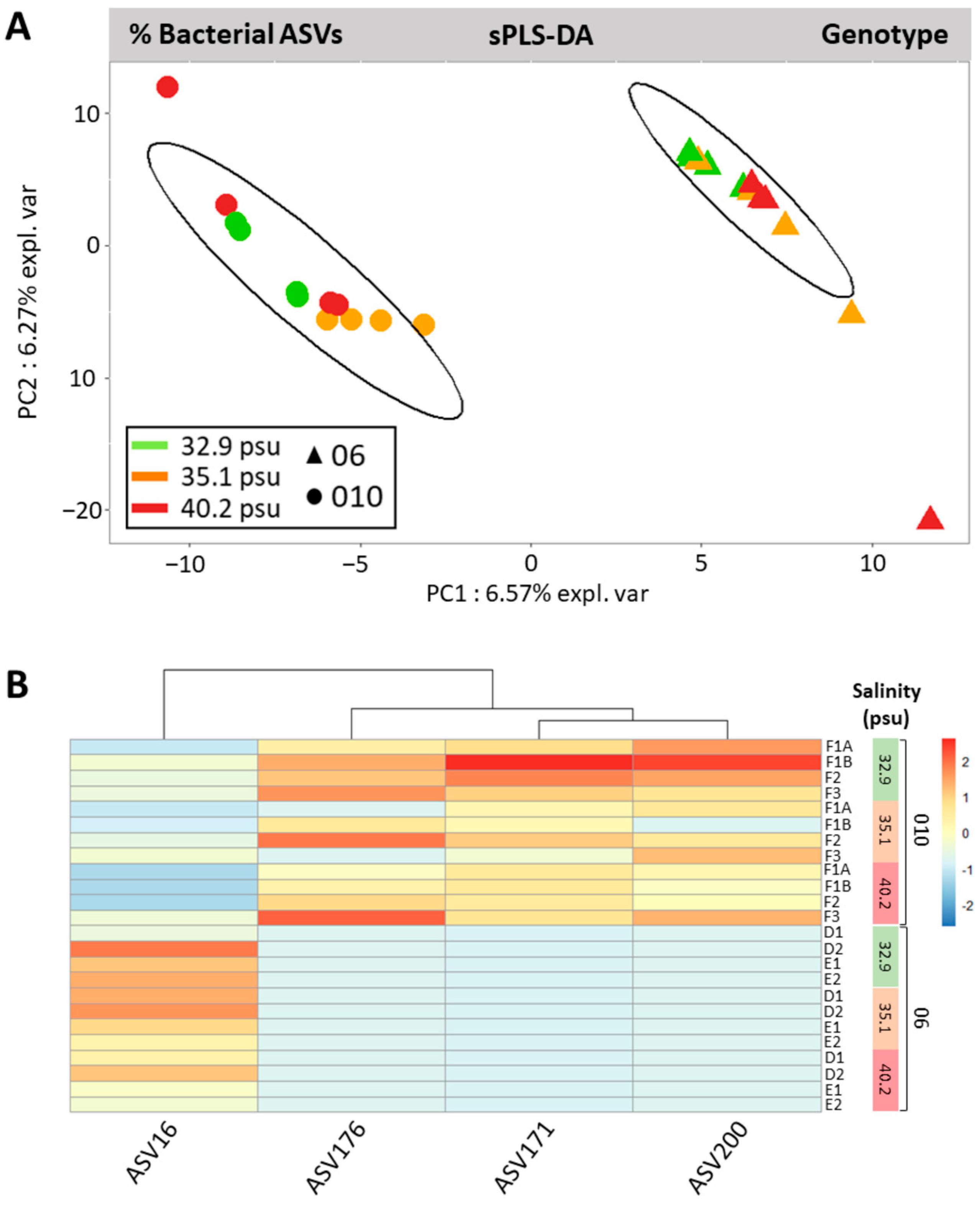
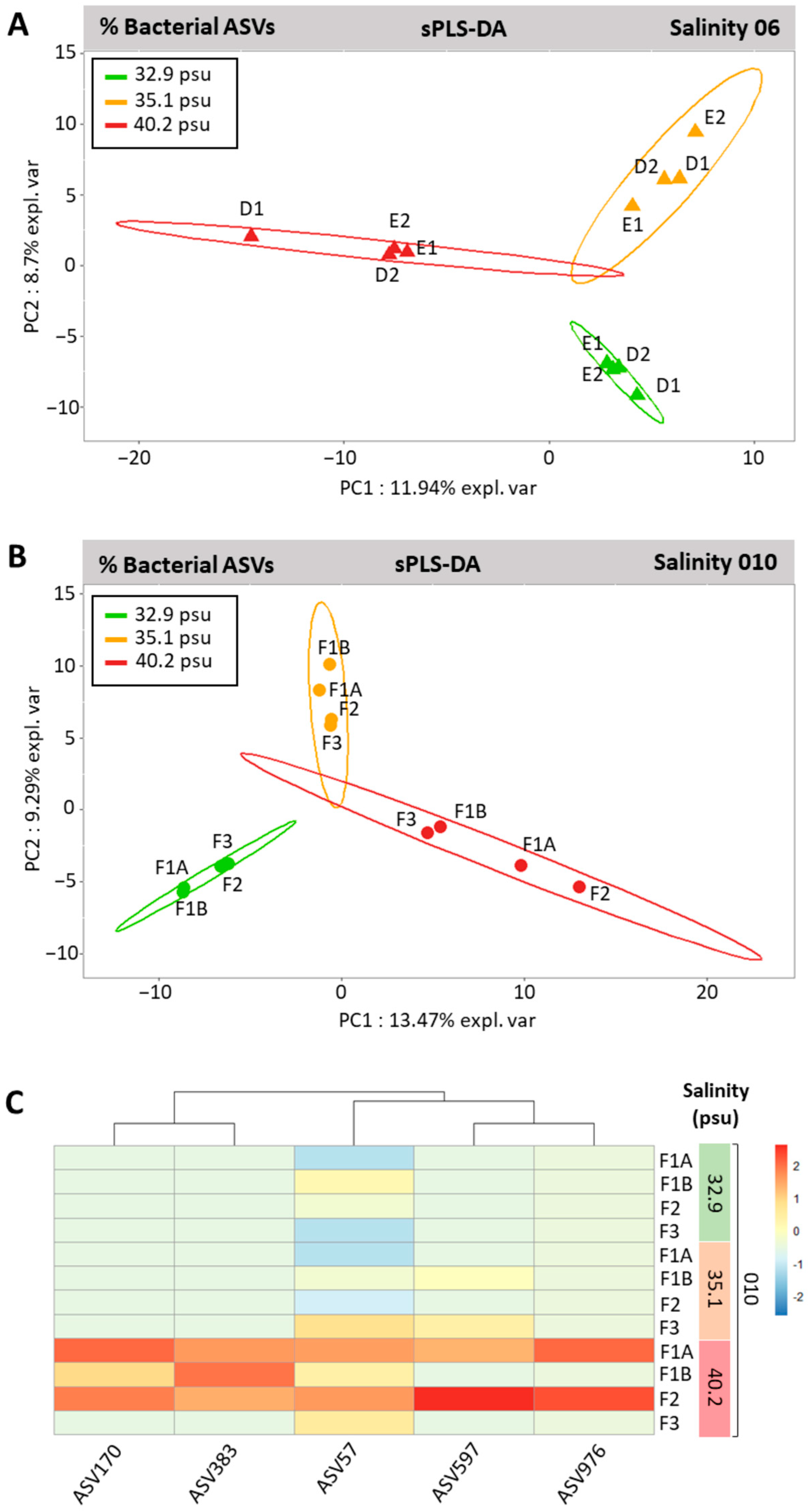
3.5. Genotype-Driven Structuration of Ostreobium Microbiota, with Limited Salinity Effect
4. Discussion
4.1. Small-Sized Core Ostreobium Bacterial Microbiota, Structured by Algal Genotype
4.2. Potential Intracellular Symbionts of Ostreobium
4.3. Adjustments of Ostreobium Bacterial Community to Salinity Increase
4.4. Epiphytic vs. Endophytic Lifestyle of Ostreobium-Associated Bacteria
4.5. Partial Overlap of Bacteria Detected in Domesticated vs. Wild Ostreobium (within Corals)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tribollet, A. The boring microflora in modern coral reef ecosystems: A review of its roles. In Current Developments in Bioerosion; Wisshak, M., Tapanila, L., Eds.; Erlangen Earth Conference Series; Springer: Berlin/Heidelberg, Germany, 2008; pp. 67–94. [Google Scholar] [CrossRef]
- Littler, M.M.; Littler, D.S.; Blair, S.M.; Norris, J.N. Deepest Known Plant Life Discovered on an Uncharted Seamount. Science 1985, 227, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Zapata, F.L.; Gómez-Osorio, S.; Sánchez, J.A. Conspicuous endolithic algal associations in a mesophotic reef-building coral. Coral Reefs 2018, 37, 705–709. [Google Scholar] [CrossRef]
- Rouzé, H.; Under The Pole Consortium; Galand, P.E.; Medina, M.; Bongaerts, P.; Pichon, M.; Pérez-Rosales, G.; Torda, G.; Moya, A.; Raina, J.-B.; et al. Symbiotic associations of the deepest recorded photosynthetic scleractinian coral (172 m depth). ISME J. 2021, 15, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, T.; Schmidt, W.E.; Suda, S.; Fredericq, S. A metabarcoding framework for facilitated survey of endolithic phototrophs with tufA. BMC Ecol. 2016, 16, 8. [Google Scholar] [CrossRef]
- Verbruggen, H.; Marcelino, V.R.; Guiry, M.D.; Cremen, M.C.M.; Jackson, C.J. Phylogenetic position of the coral symbiont Ostreobium (Ulvophyceae) inferred from chloroplast genome data. J. Phycol. 2017, 53, 790–803. [Google Scholar] [CrossRef]
- Massé, A.; Tribollet, A.; Meziane, T.; Bourguet-Kondracki, M.; Yéprémian, C.; Sève, C.; Thiney, N.; Longeon, A.; Couté, A.; Domart-Coulon, I. Functional diversity of microboring Ostreobium algae isolated from corals. Environ. Microbiol. 2020, 22, 4825–4846. [Google Scholar] [CrossRef]
- Pasella, M.M.; Lee, M.-F.E.; Marcelino, V.R.; Willis, A.; Verbruggen, H. Ten Ostreobium (Ulvophyceae) strains from Great Barrier Reef corals as a resource for algal endolith biology and genomics. Phycologia 2022, 61, 452–458. [Google Scholar] [CrossRef]
- Cocquyt, E.; Verbruggen, H.; Leliaert, F.; De Clerck, O. Evolution and Cytological Diversification of the Green Seaweeds (Ulvophyceae). Mol. Biol. Evol. 2010, 27, 2052–2061. [Google Scholar] [CrossRef]
- Ricci, F.; Fordyce, A.; Leggat, W.; Blackall, L.L.; Ainsworth, T.; Verbruggen, H. Multiple techniques point to oxygenic phototrophs dominating the Isopora palifera skeletal microbiome. Coral Reefs 2021, 40, 275–282. [Google Scholar] [CrossRef]
- Lukas, K.J. Two species of the chlorophyte genus Ostreobium from skeletons of Atlantic and Caribbean reef corals. J. Phycol. 1974, 10, 331–335. [Google Scholar] [CrossRef]
- Le Campion-Alsumard, T.; Golubic, S.; Hutchings, P. Microbial endoliths in skeletons of live and dead corals: Porites Iobata (Moorea, French Polynesia). Mar. Ecol. Prog. Ser. 1995, 117, 149–157. [Google Scholar] [CrossRef]
- Godinot, C.; Tribollet, A.; Grover, R.; Ferrier-Pagès, C. Bioerosion by euendoliths decreases in phosphate-enriched skeletons of living corals. Biogeosciences 2012, 9, 2377–2384. [Google Scholar] [CrossRef]
- Massé, A.; Domart-Coulon, I.; Golubic, S.; Duché, D.; Tribollet, A. Early skeletal colonization of the coral holobiont by the microboring Ulvophyceae Ostreobium sp. Sci. Rep. 2018, 8, 2293. [Google Scholar] [CrossRef] [PubMed]
- Fordyce, A.J.; Ainsworth, T.D.; Leggat, W. Light Capture, Skeletal Morphology, and the Biomass of Corals’ Boring Endoliths. Msphere 2021, 6, e00060-21. [Google Scholar] [CrossRef]
- Leggat, W.; Camp, E.F.; Suggett, D.J.; Heron, S.; Fordyce, A.; Gardner, S.; Deakin, L.; Turner, M.; Beeching, L.; Kuzhiumparambil, U.; et al. Rapid Coral Decay Is Associated with Marine Heatwave Mortality Events on Reefs. Curr. Biol. 2019, 29, 2723–2730.e4. [Google Scholar] [CrossRef] [PubMed]
- Pernice, M.; Raina, J.-B.; Rädecker, N.; Cárdenas, A.; Pogoreutz, C.; Voolstra, C.R. Down to the bone: The role of overlooked endolithic microbiomes in reef coral health. ISME J. 2019, 14, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Marcelino, V.R.; Blackall, L.L.; Kühl, M.; Medina, M.; Verbruggen, H. Beneath the surface: Community assembly and functions of the coral skeleton microbiome. Microbiome 2019, 7, 159. [Google Scholar] [CrossRef]
- Van Oppen, M.J.H.; Blackall, L.L. Coral microbiome dynamics, functions and design in a changing world. Nat. Rev. Genet. 2019, 17, 557–567. [Google Scholar] [CrossRef]
- Schlichter, D.; Zscharnack, B.; Krisch, H. Transfer of photoassimilates from endolithic algae to coral tissue. Naturwissenschaften 1995, 82, 561–564. [Google Scholar] [CrossRef]
- Fine, M.; Loya, Y. Endolithic algae: An alternative source of photoassimilates during coral bleaching. Proc. R. Soc. B Boil. Sci. 2002, 269, 1205–1210. [Google Scholar] [CrossRef]
- Sangsawang, L.; Casareto, B.E.; Ohba, H.; Vu, H.M.; Meekaew, A.; Suzuki, T.; Yeemin, T.; Suzuki, Y. 13C and 15N assimilation and organic matter translocation by the endolithic community in the massive coral Porites lutea. R. Soc. Open Sci. 2017, 4, 171201. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, A.; Raina, J.-B.; Pogoreutz, C.; Rädecker, N.; Bougoure, J.; Guagliardo, P.; Pernice, M.; Voolstra, C.R. Greater functional diversity and redundancy of coral endolithic microbiomes align with lower coral bleaching susceptibility. ISME J. 2022, 16, 2406–2420. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Martínez, C.T.; Weber, M.; Avila-Magaña, V.; Enríquez, S.; Kitano, H.; Medina, M.; Iglesias-Prieto, R. The role of the endolithic alga Ostreobium spp. during coral bleaching recovery. Sci. Rep. 2022, 12, 2977. [Google Scholar] [CrossRef]
- Marcelino, V.R.; van Oppen, M.; Verbruggen, H. Highly structured prokaryote communities exist within the skeleton of coral colonies. ISME J. 2017, 12, 300–303. [Google Scholar] [CrossRef]
- Marcelino, V.R.; Morrow, K.M.; Oppen, M.J.H.; Bourne, D.G.; Verbruggen, H. Diversity and stability of coral endolithic microbial communities at a naturally high p CO2 reef. Mol. Ecol. 2017, 26, 5344–5357. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumara, B.L.D.U.; Tandon, K.; Willis, A.; Verbruggen, H. The Bacterial Microbiome of the Coral Skeleton Algal Symbiont Ostreobium Shows Preferential Associations and Signatures of Phylosymbiosis. bioRxiv 2023, 1–21. [Google Scholar] [CrossRef]
- Kleypas, J.A.; McManus, J.W.; Meñez, L.A.B. Environmental Limits to Coral Reef Development: Where Do We Draw the Line? Am. Zool. 1999, 39, 146–159. [Google Scholar] [CrossRef]
- Lipizer, M.; Partescano, E.; Rabitti, A.; Giorgetti, A.; Crise, A. Qualified temperature, salinity and dissolved oxygen climatologies in a changing Adriatic Sea. Ocean Sci. 2014, 10, 771–797. [Google Scholar] [CrossRef]
- Durack, P.J.; Wijffels, S.E.; Matear, R.J. Ocean Salinities Reveal Strong Global Water Cycle Intensification During 1950 to 2000. Science 2012, 336, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.L.; Paytan, A.; Rahav, E.; Levy, O.; Silverman, J.; Barzel, O.; Potts, D.; Bar-Zeev, E. Impact of brine and antiscalants on reef-building corals in the Gulf of Aqaba—Potential effects from desalination plants. Water Res. 2018, 144, 183–191. [Google Scholar] [CrossRef]
- Ferrier-Pagès, C.; Gattuso, J.-P.; Jaubert, J. Effect of small variations in salinity on the rates of photosynthesis and respiration of the zooxanthellate coral Stylophora pistillata. Mar. Ecol. Prog. Ser. 1999, 181, 309–314. [Google Scholar] [CrossRef]
- Röthig, T.; Ochsenkühn, M.A.; Roik, A.; van der Merwe, R.; Voolstra, C.R. Long-term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Mol. Ecol. 2016, 25, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Tandon, K.; Pasella, M.M.; Iha, C.; Ricci, F.; Hu, J.; O’kelly, C.J.; Medina, M.; Kühl, M.; Verbruggen, H. Every refuge has its price: Ostreobium as a model for understanding how algae can live in rock and stay in business. Semin. Cell Dev. Biol. 2023, 134, 27–36. [Google Scholar] [CrossRef]
- Hamlaoui, S.; Yéprémian, C.; Duval, C.; Marie, B.; Djédiat, C.; Piquet, B.; Bernard, C.; Duperron, S. The Culture Collection of Cyanobacteria and Microalgae at the French National Museum of Natural History: A Century Old But Still Alive and Kicking! Including in Memoriam: Professor Alain Couté. Cryptogam. Algologie 2022, 43, 41–83. [Google Scholar] [CrossRef]
- Provasoli, L. Media and prospects for the cultivation of marine algae. In Proceedings of the US-Japan Conference, Hakone, Japan, 12–15 September 1966; Cultures and Collections of Algae. Watanake, A., Hahari, A., Eds.; The Japanese Society of Plant Physiologists: Matsue, Japan, 1968; pp. 63–75. [Google Scholar]
- Domart-Coulon, I.; Blanchoud, S. From primary cell and tissue cultures to aquatic invertebrate cell lines: An updated overview. In Advances in Aquatic Invertebrate Stem Cell Research; Rinkevich, B., Hobmayer, B., Ballarin, L., Eds.; MDPI: Basel, Switzerland, 2022; pp. 1–64. [Google Scholar]
- Amann, R.I.; Binder, B.J.; Olson, R.J.; Chisholm, S.W.; Devereux, R.; Stahl, D.A. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 1990, 56, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Daims, H.; Brühl, A.; Amann, R.; Schleifer, K.-H.; Wagner, M. The Domain-specific Probe EUB338 is Insufficient for the Detection of all Bacteria: Development and Evaluation of a more Comprehensive Probe Set. Syst. Appl. Microbiol. 1999, 22, 434–444. [Google Scholar] [CrossRef]
- Vieira, C.; Engelen, A.; Guentas, L.; Aires, T.; Houlbreque, F.; Gaubert, J.; Serrao, E.; De Clerck, O.; Payri, C. Species Specificity of Bacteria Associated to the Brown Seaweeds Lobophora (Dictyotales, Phaeophyceae) and Their Potential for Induction of Rapid Coral Bleaching in Acropora muricata. Front. Microbiol. 2016, 7, 316. [Google Scholar] [CrossRef] [PubMed]
- Tourneroche, A.; Lami, R.; Burgaud, G.; Domart-Coulon, I.; Li, W.; Gachon, C.; Gèze, M.; Boeuf, D.; Prado, S. The Bacterial and Fungal Microbiota of Saccharina latissima (Laminariales, Phaeophyceae). Front. Mar. Sci. 2020, 7, 587566. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M.; Lozano, L.; Martínez-Romero, E. Current phylogeny of Rhodospirillaceae: A multi-approach study. Mol. Phylogenetics Evol. 2019, 139, 106546. [Google Scholar] [CrossRef]
- Miller, I.J.; Weyna, T.R.; Fong, S.S.; Lim-Fong, G.E.; Kwan, J.C. Single sample resolution of rare microbial dark matter in a marine invertebrate metagenome. Sci. Rep. 2016, 6, srep34362. [Google Scholar] [CrossRef] [PubMed]
- Piampiano, E.; Pini, F.; Biondi, N.; Pastorelli, R.; Giovannetti, L.; Viti, C. Analysis of microbiota in cultures of the green microalga Tetraselmis suecica. Eur. J. Phycol. 2019, 54, 497–508. [Google Scholar] [CrossRef]
- Lawson, C.A.; Raina, J.; Kahlke, T.; Seymour, J.R.; Suggett, D.J. Defining the core microbiome of the symbiotic dinoflagellate, Symbiodinium. Environ. Microbiol. Rep. 2017, 10, 7–11. [Google Scholar] [CrossRef]
- Camp, E.F.; Kahlke, T.; Nitschke, M.R.; Varkey, D.; Fisher, N.L.; Fujise, L.; Goyen, S.; Hughes, D.J.; Lawson, C.; Ros, M.; et al. Revealing changes in the microbiome of Symbiodiniaceae under thermal stress. Environ. Microbiol. 2020, 22, 1294–1309. [Google Scholar] [CrossRef] [PubMed]
- Maire, J.; Girvan, S.K.; Barkla, S.E.; Perez-Gonzalez, A.; Suggett, D.J.; Blackall, L.L.; van Oppen, M.J.H. Intracellular bacteria are common and taxonomically diverse in cultured and in hospite algal endosymbionts of coral reefs. ISME J. 2021, 15, 2028–2042. [Google Scholar] [CrossRef]
- Aires, T.; Moalic, Y.; Serrao, E.A.; Arnaud-Haond, S. Hologenome theory supported by cooccurrence networks of species-specific bacterial communities in siphonous algae (Caulerpa). FEMS Microbiol. Ecol. 2015, 91, fiv067. [Google Scholar] [CrossRef]
- Kimes, N.E.; Johnson, W.R.; Torralba, M.; Nelson, K.E.; Weil, E.; Morris, P.J. The Montastraea faveolate microbiome: Ecological and temporal influences on a Caribbean reef-building coral in decline. Environ. Microbiol. 2013, 15, 2082–2094. [Google Scholar] [CrossRef]
- Sauvage, T.; Schmidt, W.E.; Yoon, H.S.; Paul, V.J.; Fredericq, S. Promising prospects of nanopore sequencing for algal hologenomics and structural variation discovery. BMC Genom. 2019, 20, 850. [Google Scholar] [CrossRef]
- Ricci, F.; Tandon, K.; Black, J.R.; Cao, K.-A.L.; Blackall, L.L.; Verbruggen, H. Host Traits and Phylogeny Contribute to Shaping Coral-Bacterial Symbioses. Msystems 2022, 7, e00044-22. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.-S.; Pinnell, L.J.; Turner, J.W.; Abdulla, H.; Boyd, L.; Linton, E.W.; Zimba, P.V. Preliminary Assessment of Microbial Community Structure of Wind-Tidal Flats in the Laguna Madre, Texas, USA. Biology 2020, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Liu, J.-H.; Zhang, X.-X.; Chen, Y.-G.; Wang, Z.-G.; Li, Q.-Y.; Peng, Q.; Cui, X.-L.; Chen, Y.-G.; Chen, Y. Fodinicurvata sediminis gen. nov., sp. nov. and Fodinicurvata fenggangensis sp. nov., poly-hydroxybutyrate-producing bacteria in the family Rhodospirillaceae. Int. J. Syst. Evol. Microbiol. 2009, 59, 2575–2581. [Google Scholar] [CrossRef] [PubMed]
- Infante-Dominguez, C.; Lawson, P.A.; Johnson, C.N.; Sánchez-Porro, C.; Ventosa, A. Fodinicurvata halophila sp. nov., a moderately halophilic bacterium from a marine saltern. Int. J. Syst. Evol. Microbiol. 2015, 65, 766–771. [Google Scholar] [CrossRef]
- Abraham, W.R.; Rohde, M. The Family Hyphomonadaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 8, pp. 283–299. [Google Scholar]
- Weigel, B.L.; Pfister, C.A. Oxygen metabolism shapes microbial settlement on photosynthetic kelp blades compared to artificial kelp substrates. Environ. Microbiol. Rep. 2020, 13, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Weidner, S.; Arnold, W.; Stackebrandt, E.; Pühler, A. Phylogenetic Analysis of Bacterial Communities Associated with Leaves of the Seagrass Halophila stipulacea by a Culture-Independent Small-Subunit rRNA Gene Approach. Microb. Ecol. 2000, 39, 22–31. [Google Scholar] [CrossRef]
- Fukui, Y.; Abe, M.; Kobayashi, M.; Saito, H.; Oikawa, H.; Yano, Y.; Satomi, M. Algimonas porphyrae gen. nov., sp. nov., a member of the family Hyphomonadaceae, isolated from the red alga Porphyra yezoensis. Int. J. Syst. Evol. Microbiol. 2013, 63, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Hollants, J.; Leroux, O.; Leliaert, F.; Decleyre, H.; De Clerck, O.; Willems, A. Who Is in There? Exploration of Endophytic Bacteria within the Siphonous Green Seaweed Bryopsis (Bryopsidales, Chlorophyta). PLoS ONE 2011, 6, e26458. [Google Scholar] [CrossRef]
- Hollants, J.; Leliaert, F.; Verbruggen, H.; Willems, A.; De Clerck, O. Permanent residents or temporary lodgers: Characterizing intracellular bacterial communities in the siphonous green alga Bryopsis. Proc. R. Soc. B Boil. Sci. 2013, 280, 20122659. [Google Scholar] [CrossRef]
- Carvalho, T.L.G.; Balsemao-Pires, E.; Saraiva, R.M.; Ferreira, P.C.G.; Hemerly, A.S. Nitrogen signalling in plant interactions with associative and endophytic diazotrophic bacteria. J. Exp. Bot. 2014, 65, 5631–5642. [Google Scholar] [CrossRef]
- Roth, F.; Karcher, D.B.; Rädecker, N.; Hohn, S.; Carvalho, S.; Thomson, T.; Saalmann, F.; Voolstra, C.R.; Kürten, B.; Struck, U.; et al. High rates of carbon and dinitrogen fixation suggest a critical role of benthic pioneer communities in the energy and nutrient dynamics of coral reefs. Funct. Ecol. 2020, 34, 1991–2004. [Google Scholar] [CrossRef]
- Fujinami, S.; Takarada, H.; Kasai, H.; Sekine, M.; Omata, S.; Harada, T.; Fukai, R.; Hosoyama, A.; Horikawa, H.; Kato, Y.; et al. Complete genome sequence of Ilumatobacter coccineum YM16-304T. Stand. Genom. Sci. 2013, 8, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Boedeker, C.; Schüler, M.; Reintjes, G.; Jeske, O.; van Teeseling, M.C.F.; Jogler, M.; Rast, P.; Borchert, D.; Devos, D.P.; Kucklick, M.; et al. Determining the bacterial cell biology of Planctomycetes. Nat. Commun. 2017, 8, 14853. [Google Scholar] [CrossRef] [PubMed]
- Maire, J.; Blackall, L.L.; van Oppen, M.J.H. Intracellular Bacterial Symbionts in Corals: Challenges and Future Directions. Microorganisms 2021, 9, 2209. [Google Scholar] [CrossRef]
- Schmitz-Esser, S.; Tischler, P.; Arnold, R.; Montanaro, J.; Wagner, M.; Rattei, T.; Horn, M. The Genome of the Amoeba Symbiont “Candidatus Amoebophilus asiaticus” Reveals Common Mechanisms for Host Cell Interaction among Amoeba-Associated Bacteria. J. Bacteriol. 2010, 192, 1045–1057. [Google Scholar] [CrossRef]
- Ainsworth, T.D.; Krause, L.; Bridge, T.; Torda, G.; Raina, J.-B.; Zakrzewski, M.; Gates, R.D.; Padilla-Gamiño, J.L.; Spalding, H.L.; Smith, C.; et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015, 9, 2261–2274. [Google Scholar] [CrossRef]
- Kawafune, K.; Hongoh, Y.; Hamaji, T.; Nozaki, H. Molecular Identification of Rickettsial Endosymbionts in the Non-Phagotrophic Volvocalean Green Algae. PLoS ONE 2012, 7, e31749. [Google Scholar] [CrossRef]
- Klinges, J.G.; Rosales, S.M.; McMinds, R.; Shaver, E.C.; Shantz, A.A.; Peters, E.C.; Eitel, M.; Wörheide, G.; Sharp, K.H.; Burkepile, D.E.; et al. Phylogenetic, genomic, and biogeographic characterization of a novel and ubiquitous marine invertebrate-associated Rickettsiales parasite, Candidatus Aquarickettsia rohweri, gen. nov., sp. nov. ISME J. 2019, 13, 2938–2953. [Google Scholar] [CrossRef]
- Buerger, P.; Vanstone, R.T.; Maire, J.; van Oppen, M.J.H. Long-Term Heat Selection of the Coral Endosymbiont Cladocopium C1acro (Symbiodiniaceae) Stabilizes Associated Bacterial Communities. Int. J. Mol. Sci. 2022, 23, 4913. [Google Scholar] [CrossRef]
- Singh, R.P.; Reddy, C.R.K. Unraveling the Functions of the Macroalgal Microbiome. Front. Microbiol. 2016, 6, 1488. [Google Scholar] [CrossRef]
- Dittami, S.M.; Duboscq-Bidot, L.; Perennou, M.; Gobet, A.; Corre, E.; Boyen, C.; Tonon, T. Host–microbe interactions as a driver of acclimation to salinity gradients in brown algal cultures. ISME J. 2015, 10, 51–63. [Google Scholar] [CrossRef]
- Ghaderiardakani, F.; Quartino, M.L.; Wichard, T. Microbiome-Dependent Adaptation of Seaweeds Under Environmental Stresses: A Perspective. Front. Mar. Sci. 2020, 7, 575228. [Google Scholar] [CrossRef]
- Voolstra, C.R.; Ziegler, M. Adapting with Microbial Help: Microbiome Flexibility Facilitates Rapid Responses to Environmental Change. Bioessays 2020, 42, e2000004. [Google Scholar] [CrossRef] [PubMed]
- Aires, T.; Serrão, E.A.; Kendrick, G.; Duarte, C.M.; Arnaud-Haond, S. Invasion Is a Community Affair: Clandestine Followers in the Bacterial Community Associated to Green Algae, Caulerpa racemosa, Track the Invasion Source. PLoS ONE 2013, 8, e68429. [Google Scholar] [CrossRef]
- Morrissey, K.L.; Çavaş, L.; Willems, A.; De Clerck, O. Disentangling the Influence of Environment, Host Specificity and Thallus Differentiation on Bacterial Communities in Siphonous Green Seaweeds. Front. Microbiol. 2019, 10, 717. [Google Scholar] [CrossRef] [PubMed]
- Marcelino, V.R.; Verbruggen, H. Multi-marker metabarcoding of coral skeletons reveals a rich microbiome and diverse evolutionary origins of endolithic algae. Sci. Rep. 2016, 6, 31508. [Google Scholar] [CrossRef]
- Gutner-Hoch, E.; Fine, M. Genotypic diversity and distribution of Ostreobium quekettii within scleractinian corals. Coral Reefs 2011, 30, 643–650. [Google Scholar] [CrossRef]
- Verbruggen, H.; Ashworth, M.; LoDuca, S.T.; Vlaeminck, C.; Cocquyt, E.; Sauvage, T.; Zechman, F.W.; Littler, D.; Littler, M.M.; Leliaert, F.; et al. A multi-locus time-calibrated phylogeny of the siphonous green algae. Mol. Phylogenet Evol. 2009, 50, 642–653. [Google Scholar] [CrossRef]
- Famà, P.; Wysor, B.; Kooistra, W.H.; Zuccarello, G.C. Molecular phylogeny of the genus Caulerpa (Caulerpales, Chlorophyta) inferred from chloroplast tufA gene. J. Phycol. 2002, 38, 1040–1050. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample infer-ence from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Yang, S.H.; Tandon, K.; Lu, C.Y.; Wada, N.; Shih, C.J.; Hsiao, S.S.Y.; Jane, W.N.; Lee, T.C.; Yang, C.M.; Liu, C.T.; et al. Meta-genomic, phylogenetic, and functional characterization of predominant endolithic green sulfur bacteria in the coral Isopora palifera. Microbiome 2019, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Neave, M.J.; Rachmawati, R.; Xun, L.; Michell, C.T.; Bourne, D.G.; Apprill, A.; Voolstra, C.R. Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 2017, 11, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Pogoreutz, C.; Rädecker, N.; Càrdenas, A.; Gärdes, A.; Wild, C.; Voolstra, C.R. Dominance of Endozoicomonas bacteria throughout coral bleaching and mortality suggests structural structural inflexibility of the Pocillopora verrucosa microbiome. Ecol. Evol. 2018, 8, 2240–2252. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massé, A.; Detang, J.; Duval, C.; Duperron, S.; Woo, A.C.; Domart-Coulon, I. Bacterial Microbiota of Ostreobium, the Coral-Isolated Chlorophyte Ectosymbiont, at Contrasted Salinities. Microorganisms 2023, 11, 1318. https://doi.org/10.3390/microorganisms11051318
Massé A, Detang J, Duval C, Duperron S, Woo AC, Domart-Coulon I. Bacterial Microbiota of Ostreobium, the Coral-Isolated Chlorophyte Ectosymbiont, at Contrasted Salinities. Microorganisms. 2023; 11(5):1318. https://doi.org/10.3390/microorganisms11051318
Chicago/Turabian StyleMassé, Anaïs, Juliette Detang, Charlotte Duval, Sébastien Duperron, Anthony C. Woo, and Isabelle Domart-Coulon. 2023. "Bacterial Microbiota of Ostreobium, the Coral-Isolated Chlorophyte Ectosymbiont, at Contrasted Salinities" Microorganisms 11, no. 5: 1318. https://doi.org/10.3390/microorganisms11051318