
Metabarcoding and Metabolomics Reveal the Effect of the Invasive Alien Tree Miconia calvescens DC. on Soil Diversity on the Tropical Island of Mo’orea (French Polynesia)
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
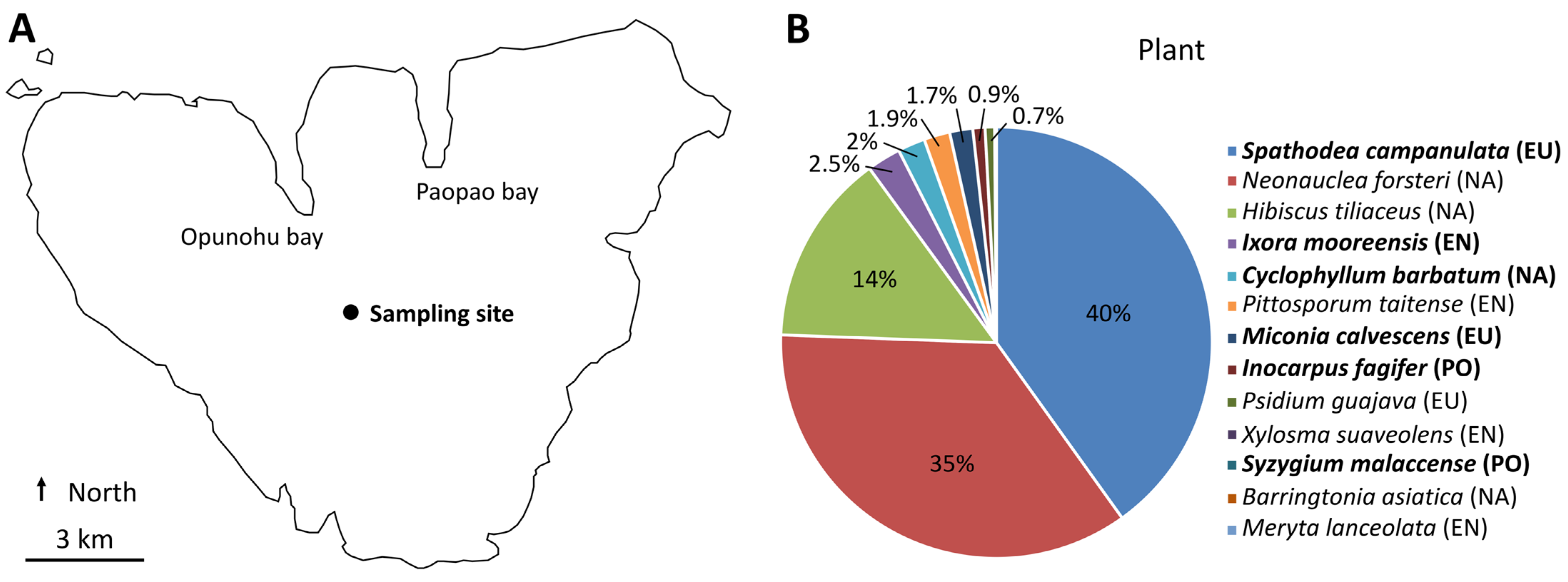
2.1. Sampling Site and Plant Species
2.2. DNA Extraction, PCR, and Sequencing
2.3. Sequence Analyses
2.4. Extraction of Metabolites and UHPLC-HRMS Profiling
2.5. Data Processing for Metabolomics
2.6. Multivariate and Statistical Analyses
2.7. Phylogenetic Analyses
2.8. Metabolite Annotation
3. Results
3.1. Plant Community at the Sampling Site
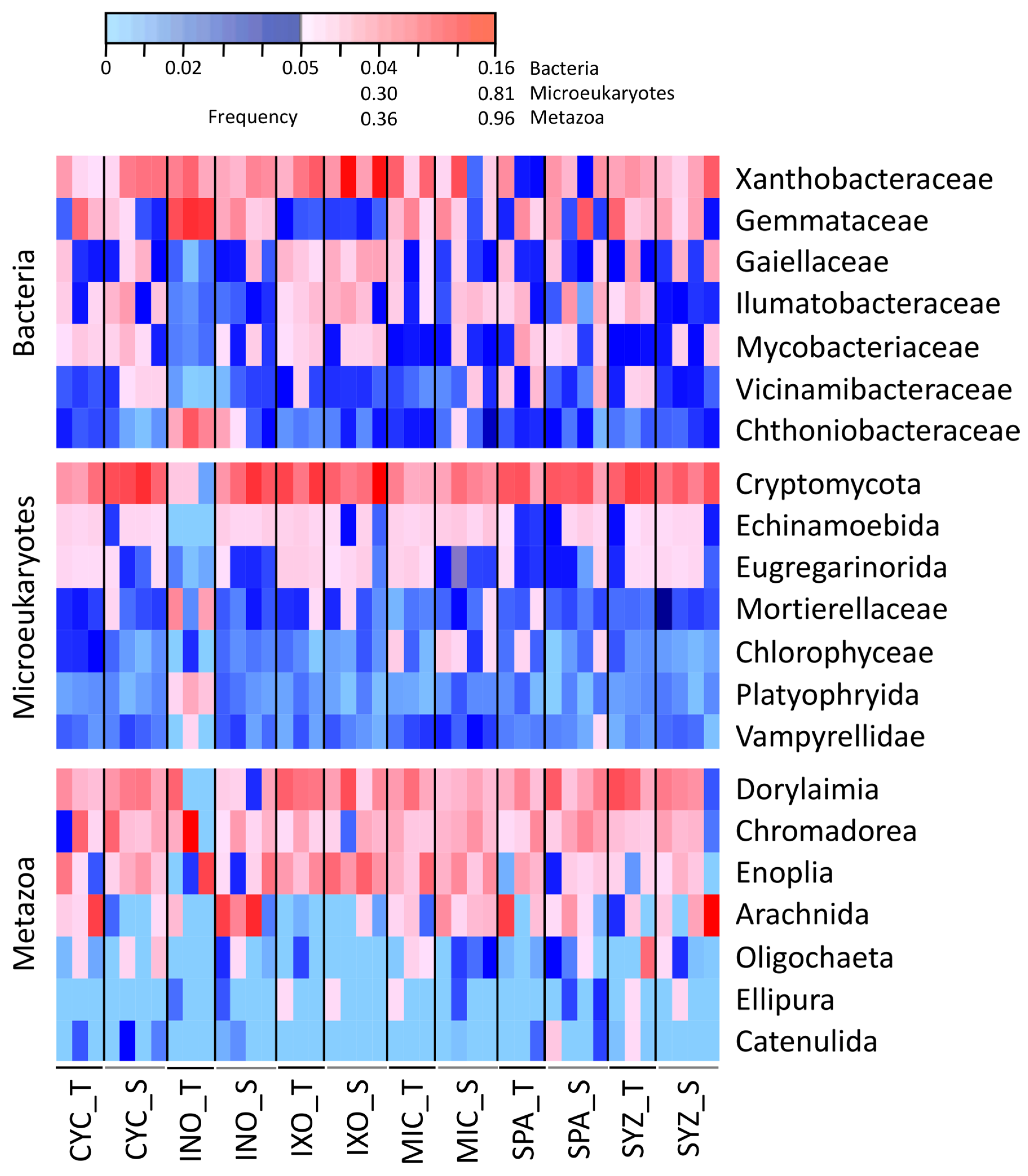
3.2. Dominant Taxa Associated with the Six Plant Species
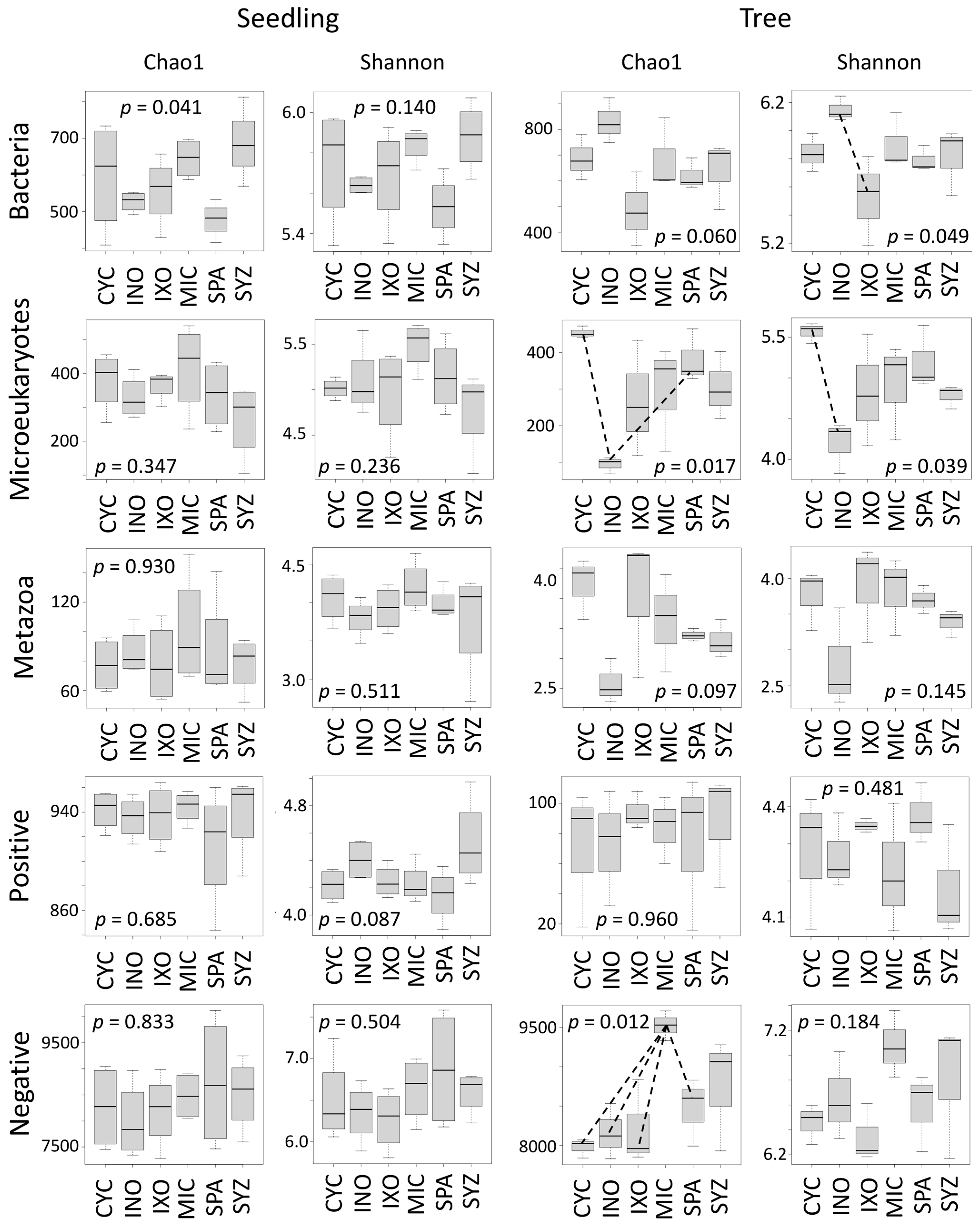
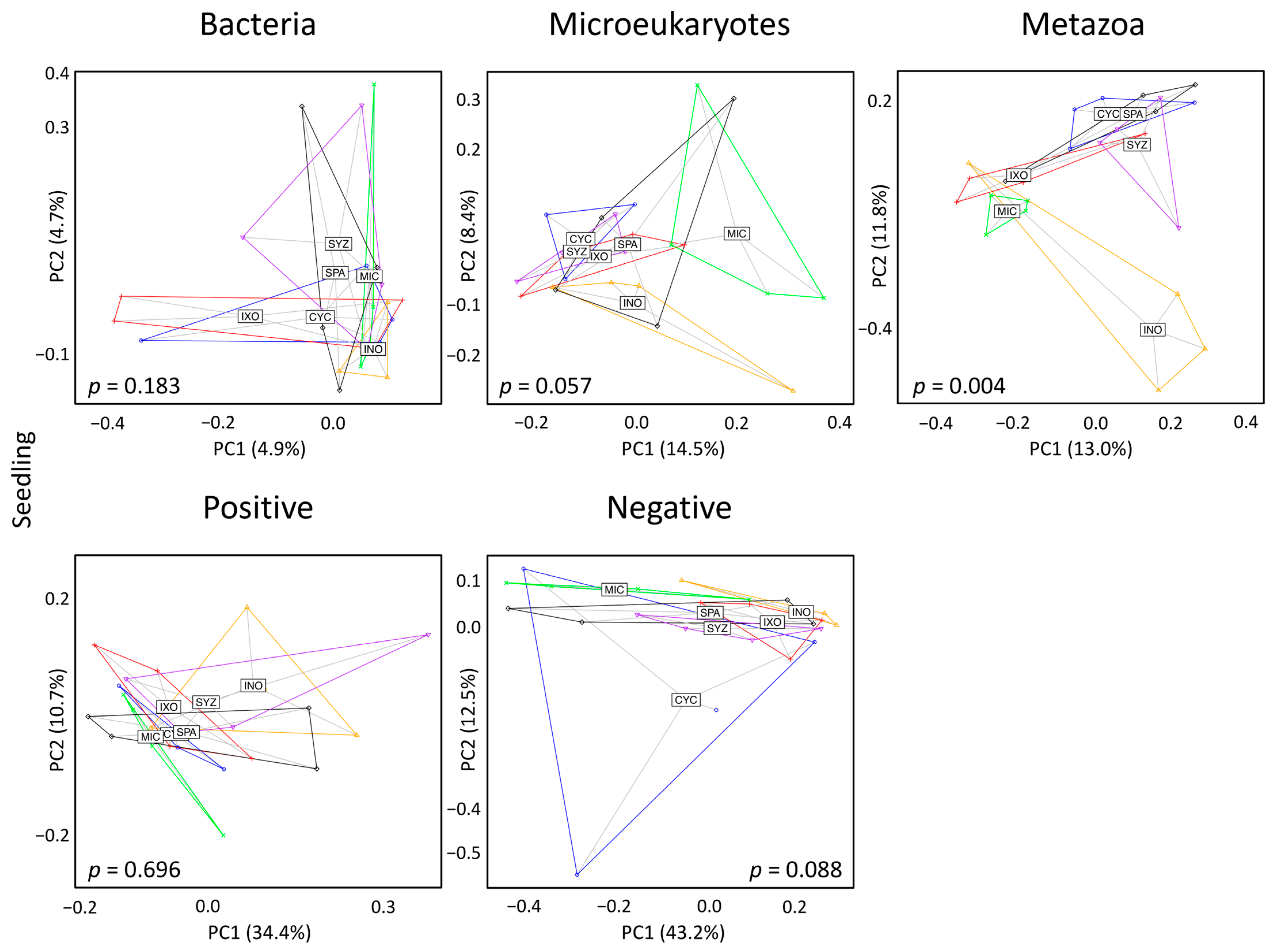
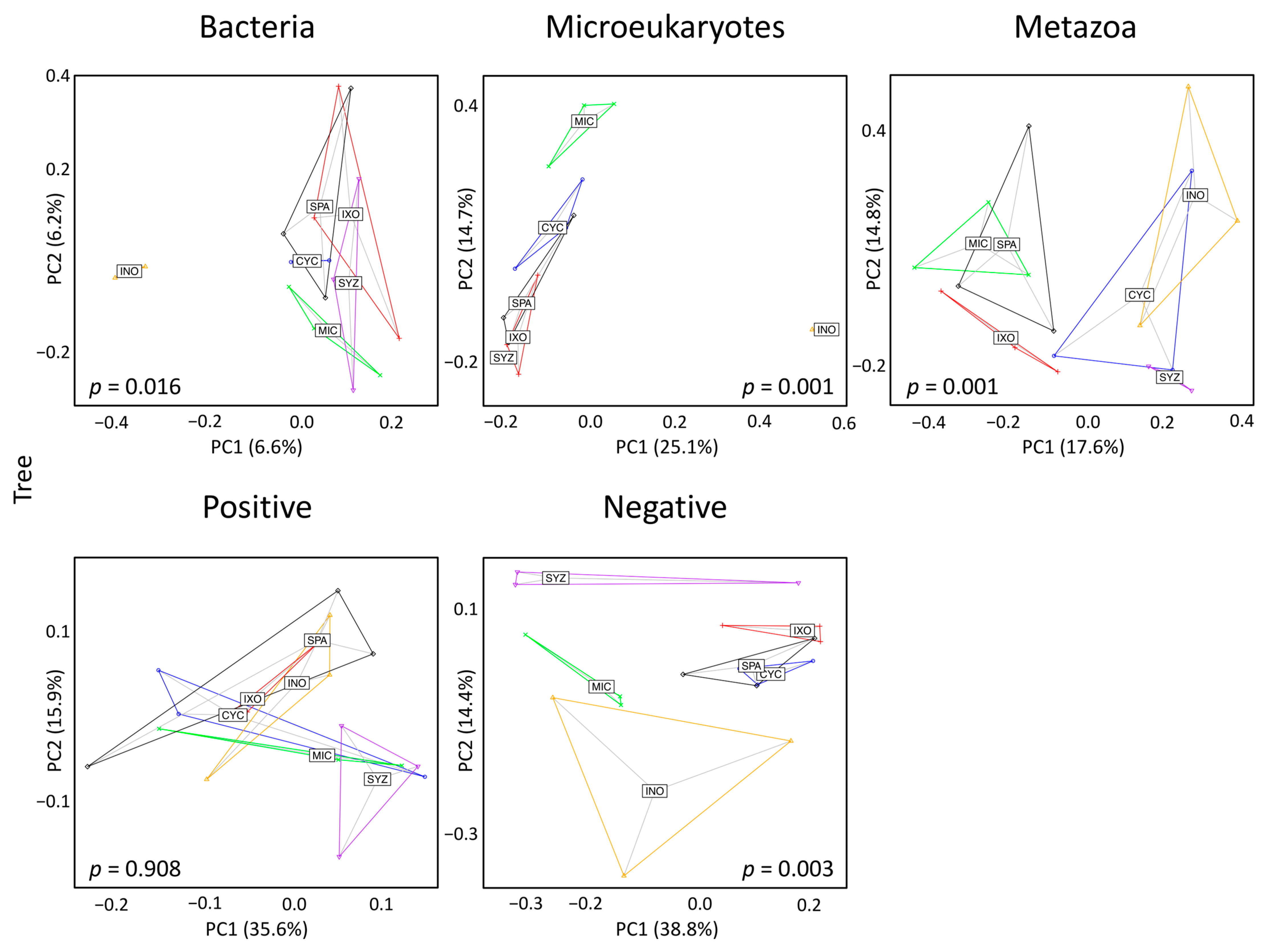
3.3. Seedling Effects on Soil Diversity
3.4. Developmental Effect
3.5. Tree Effects on Soil Diversity
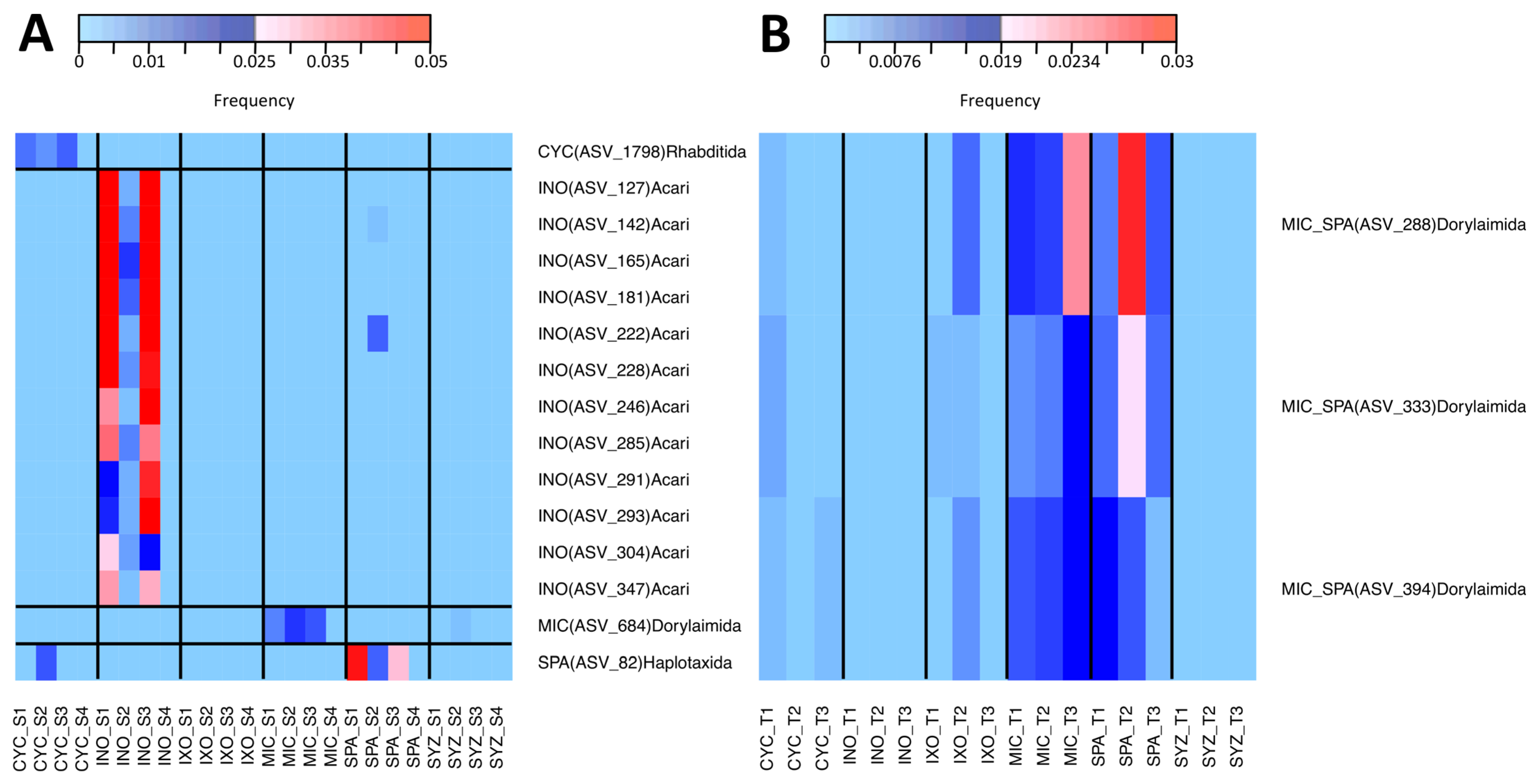
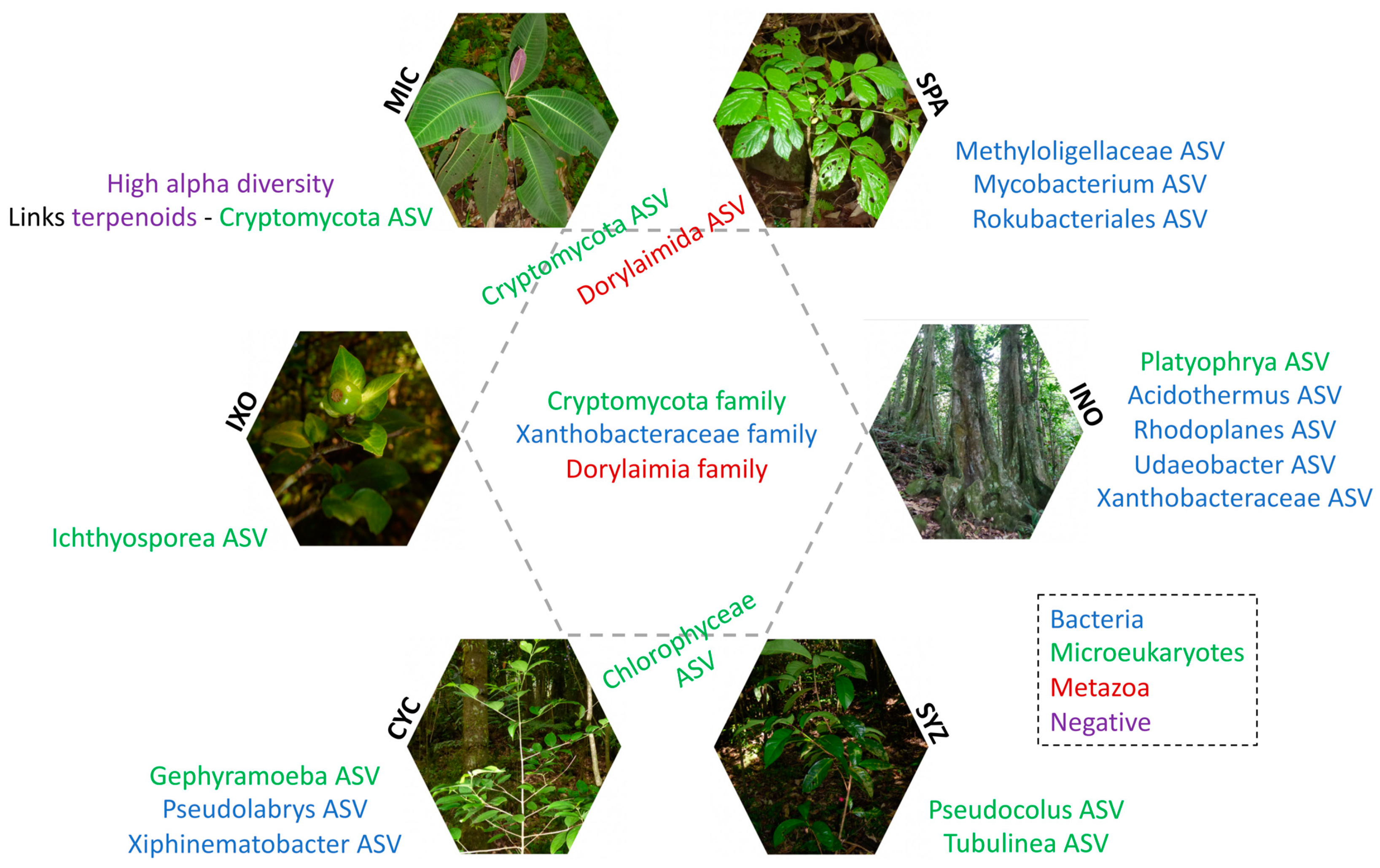
3.6. Specific Taxa of Each Plant Species in the Tree Stage
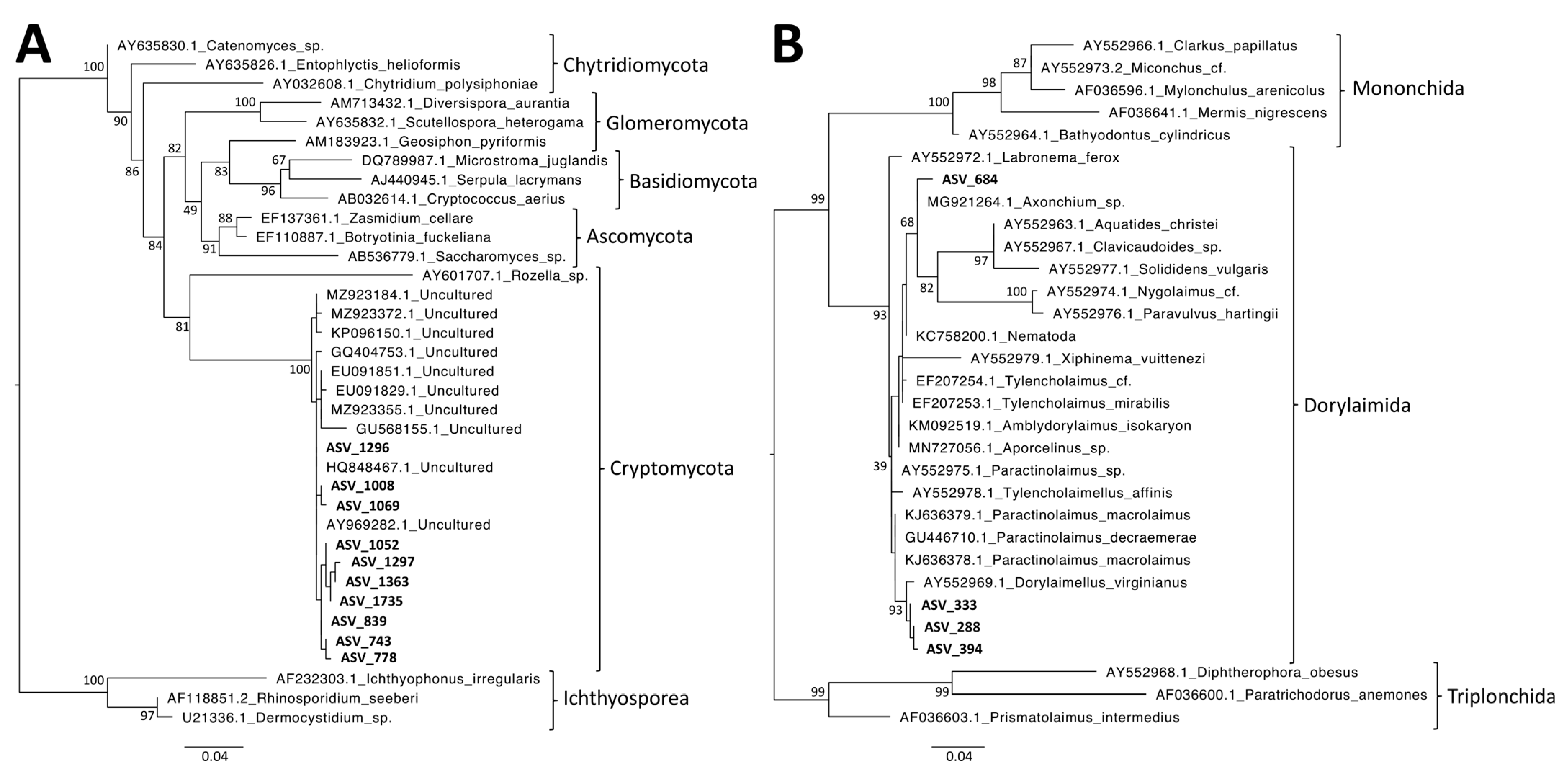
3.7. Phylogenetic Analyses of MIC-Associated Taxa
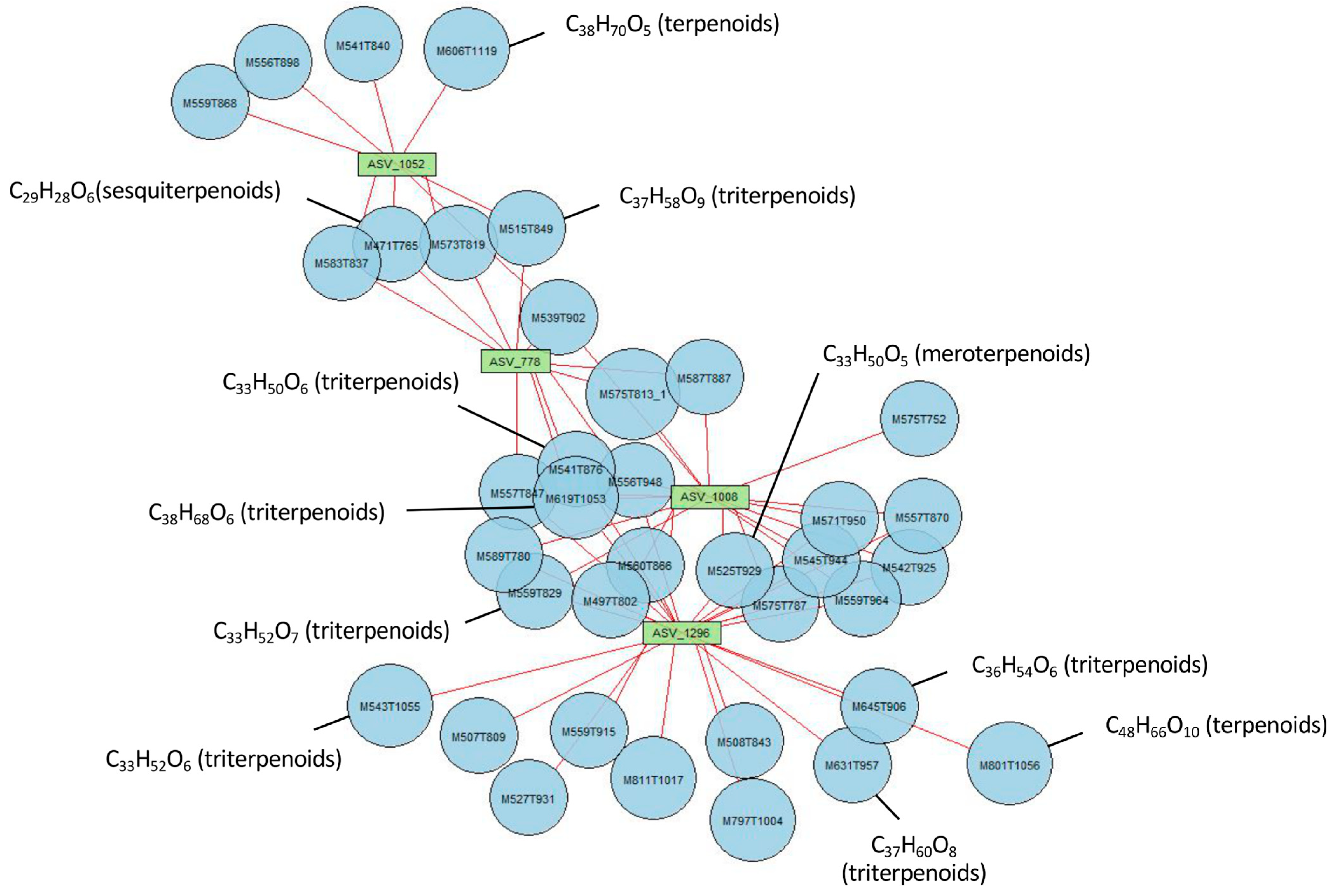
3.8. Specific Metabolites of MIC and Links with Rhizospheric Organisms
4. Discussion
4.1. Trees Had Stronger Effects Than Seedlings on Soil Diversity
4.2. Inocarpus fagifer Strongly Influenced Assemblages of Bacteria, Microeukaryotes, and Metabolites
4.3. Miconia calvescens Strongly Influenced Assemblages of Microeukaryotes and Metabolites
4.4. Improvements Made by This Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vitousek, P.M.; Mooney, H.A.; Lubchenco, J.; Melillo, J.M. Human Domination of Earth’s Ecosystems. Science 1997, 277, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Spatz, D.R.; Zilliacus, K.M.; Holmes, N.D.; Butchart, S.H.M.; Genovesi, P.; Ceballos, G.; Tershy, B.R.; Croll, D.A. Globally Threatened Vertebrates on Islands with Invasive Species. Sci. Adv. 2017, 3, e1603080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney, M.L.; Lockwood, J.L. Biotic Homogenization: A few Winners Replacing Many Losers in the next Mass Extinction. Trends Ecol. Evol. 1999, 14, 450–453. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, C.M.; Dudley, T.L. Biological Invasions as Agents of Change on Islands versus Mainlands. In Islands; Ecological Studies; Vitousek, P.M., Loope, L.L., Adsersen, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1995; Volume 115, pp. 103–121. ISBN 978-3-642-78965-6. [Google Scholar]
- Sax, D.F.; Gaines, S.D. Species Invasions and Extinction: The Future of Native Biodiversity on Islands. Proc. Natl. Acad. Sci. USA 2008, 105, 11490–11497. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Palacios, J.M.; Kreft, H.; Irl, S.D.H.; Norder, S.; Ah-Peng, C.; Borges, P.A.V.; Burns, K.C.; de Nascimento, L.; Meyer, J.-Y.; Montes, E.; et al. Scientists’ Warning—The Outstanding Biodiversity of Islands Is in Peril. Glob. Ecol. Conserv. 2021, 31, e01847. [Google Scholar] [CrossRef]
- Mittermeier, R.A.; Turner, W.R.; Larsen, F.W.; Brooks, T.M.; Gascon, C. Global Biodiversity Conservation: The Critical Role of Hotspots. In Biodiversity Hotspots; Zachos, F.E., Habel, J.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 3–22. ISBN 978-3-642-20991-8. [Google Scholar]
- Lowe, S.; Browne, M.; Boudjelas, S.; De Poorter, M. 100 of the World’s Worst Invasive Alien Species: A Selection from the Global Invasive Species Database. In Encyclopedia of Biological Invasions; University of California Press: Auckland, New Zealand, 2000; pp. 715–716. ISBN 978-0-520-94843-3. [Google Scholar]
- Meyer, J.-Y.; Pouteau, R.; Spotswood, E.; Taputuarai, R.; Fourdrigniez, M. The Importance of Novel and Hybrid Habitats for Plant Conservation on Islands: A Case Study from Moorea (South Pacific). Biodivers. Conserv. 2015, 24, 83–101. [Google Scholar] [CrossRef]
- Meyer, J.-Y.; Florence, J. Tahiti’s Native Flora Endangered by the Invasion of Miconia calvescens DC. (Melastomataceae). J. Biogeogr. 1996, 23, 775–781. [Google Scholar] [CrossRef]
- Chevillotte, H.; Meyer, J.-Y.; Mellado-Forichon, T.; Florence, J.; Emmanuelli, E.; Habert, E.; Galzin, R.; Ferraris, J. Evaluation et Suivi de La Biodiversité Dans l’île de Moorea, Polynésie Française: Approche Méthodologique Appliquée Aux Écosystèmes Terrestres et Marins. Rev. D’écologie (Terre Vie) 2014, 69, 267–284. [Google Scholar] [CrossRef]
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The Role of Root Exudates in Rhizosphere Interactions with Plants and Other Organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef] [Green Version]
- Callaway, R.M.; Hierro, J.L.; Thorpe, A.S. Evolutionary Trajectories in Plant and Soil Microbial Communities: Centaurea Invasions and the Geographic Mosaic of Coevolution. In Species Invasions: Insights into Ecology, Yeast Polarity: Negative Feedback Shifts the Focus; Sinauer: Sunderland, MA, USA, 2005; pp. 341–363. [Google Scholar]
- Dassonville, N.; Guillaumaud, N.; Piola, F.; Meerts, P.; Poly, F. Niche Construction by the Invasive Asian Knotweeds (Species Complex Fallopia): Impact on Activity, Abundance and Community Structure of Denitrifiers and Nitrifiers. Biol. Invasions 2011, 13, 1115–1133. [Google Scholar] [CrossRef]
- Lorenzo, P.; Pereira, C.S.; Rodríguez-Echeverría, S. Differential Impact on Soil Microbes of Allelopathic Compounds Released by the Invasive Acacia dealbata Link. Soil Biol. Biochem. 2013, 57, 156–163. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.M.; Breiner, H.W.; Richards, T.A. Multiple Marker Parallel Tag Environmental DNA Sequencing Reveals a Highly Complex Eukaryotic Community in Marine Anoxic Water. Mol. Ecol. 2010, 19, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Ghosson, H. Development of a Novel Universal Proxy to Assess the Environmental Fate and Impact of Complex (Bio)Pesticides by Mass Spectrometry-Based Metabolomics. Ph.D. Thesis, Université de Perpignan Via Domitia, Perpignan, France, 2020. [Google Scholar]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, F.; Le Corguille, G.; Monsoor, M.; Landi, M.; Pericard, P.; Petera, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A Collaborative Research Infrastructure for Computational Metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Guitton, Y.; Tremblay-Franco, M.; Le Corguillé, G.; Martin, J.-F.; Pétéra, M.; Roger-Mele, P.; Delabrière, A.; Goulitquer, S.; Monsoor, M.; Duperier, C.; et al. Create, Run, Share, Publish, and Reference Your LC–MS, FIA–MS, GC–MS, and NMR Data Analysis Workflows with the Workflow4Metabolomics 3.0 Galaxy Online Infrastructure for Metabolomics. Int. J. Biochem. Cell Biol. 2017, 93, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cáceres, M.D.; Legendre, P. Associations between Species and Groups of Sites: Indices and Statistical Inference. Ecology 2009, 90, 3566–3574. [Google Scholar] [CrossRef]
- Elie, N.; Santerre, C.; Touboul, D. Generation of a Molecular Network from Electron Ionization Mass Spectrometry Data by Combining MZmine2 and MetGem Software. Anal. Chem. 2019, 91, 11489–11492. [Google Scholar] [CrossRef] [Green Version]
- Katajamaa, M.; Miettinen, J.; Orešič, M. MZmine: Toolbox for Processing and Visualization of Mass Spectrometry Based Molecular Profile Data. Bioinformatics 2006, 22, 634–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivon, F.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MZmine 2 Data-Preprocessing to Enhance Molecular Networking Reliability. Anal. Chem. 2017, 89, 7836–7840. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Lê Cao, K.-A. DIABLO: An Integrative Approach for Identifying Key Molecular Drivers from Multi-Omics Assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Chouari, R.; Leonard, M.; Bouali, M.; Guermazi, S.; Rahli, N.; Zrafi, I.; Morin, L.; Sghir, A. Eukaryotic Molecular Diversity at Different Steps of the Wastewater Treatment Plant Process Reveals More Phylogenetic Novel Lineages. World J. Microbiol. Biotechnol. 2017, 33, 44. [Google Scholar] [CrossRef]
- Mullin, P.G.; Harris, T.S.; Powers, T.O. Phylogenetic Relationships of Nygolaimina and Dorylaimina (Nematoda: Dorylaimida) Inferred from Small Subunit Ribosomal DNA Sequences. Nematology 2005, 7, 59–79. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, A.M.; Bandeira, N.; Shen, Z.; Tanner, S.; Briggs, S.P.; Smith, R.D.; Pevzner, P.A. Clustering Millions of Tandem Mass Spectra. J. Proteome Res. 2008, 7, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A Rapid Tool for Turning Tandem Mass Spectra into Metabolite Structure Information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Morelos, V.H.; Soto-Estrada, A.; Pérez-Moreno, J.; Franco-Ramírez, A.; Díaz-Rivera, P. Arbuscular Mycorrhizal Fungi Associated with the Rhizosphere of Seedlings and Mature Trees of Swietenia macrophylla (Magnoliophyta: Meliaceae) in Los Tuxtlas, Veracruz, Mexico. Rev. Chil. Hist. Nat. 2014, 87, 9. [Google Scholar] [CrossRef] [Green Version]
- Huml, L.; Miksatkova, P.; Novy, P.; Drabek, O.; Sabolova, M.; Umar, M.; Tejnecky, V.; Pohorela, B.; Kourimska, L.; Maskova, E.; et al. Fatty Acids, Minerals, Phenolics and Vitamins in the Seeds of Inocarpus Fagifer, a Pacific Island Underutilized Legume. J. Appl. Bot. Food Qual. 2016, 89, 264–269. [Google Scholar] [CrossRef]
- Cooper, J.E. Multiple Responses of Rhizobia to Flavonoids during Legume Root Infection. In Advances in Botanical Research; Elsevier: Amsterdam, The Netherlands, 2004; Volume 41, pp. 1–62. ISBN 978-0-12-005941-6. [Google Scholar]
- Foissner, W.; Wolf, K.W. Morphology and Ontogenesis of Platyophrya bromelicola Nov. Spec., a New Macrostome-Forming Colpodid (Protists, Ciliophora) from Tank Bromeliads of Jamaica. Eur. J. Protistol. 2009, 45, 87–97. [Google Scholar] [CrossRef]
- Hiraishi, A. Characterization of Thermotolerant Phototrophic Bacteria, Rhodoplanes tepidicaeni Sp. Nov. and Rhodoplanes azumiensis Sp. Nov., Isolated from a Geothermal Spring. Int. J. Syst. Evol. Microbiol. 2017, 67, 5038–5045. [Google Scholar] [CrossRef]
- Anders, H.; Power, J.F.; MacKenzie, A.D.; Lagutin, K.; Vyssotski, M.; Hanssen, E.; Moreau, J.W.; Stott, M.B. Limisphaera ngatamarikiensis Gen. Nov., Sp. Nov., a Thermophilic, Pink-Pigmented Coccus Isolated from Subaqueous Mud of a Geothermal Hotspring. Int. J. Syst. Evol. Microbiol. 2015, 65, 1114–1121. [Google Scholar] [CrossRef] [Green Version]
- Thawai, C.; Tanasupawat, S.; Suwanborirux, K.; Itoh, T.; Kudo, T. Micromonospora narathiwatensis Sp. Nov., from Thai Peat Swamp Forest Soils. J. Gen. Appl. Microbiol. 2007, 53, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, X.; Zhang, C.; Li, X.; Zhang, H. Ecological Risk of Long-Term Chlorimuron-Ethyl Application to Soil Microbial Community: An in Situ Investigation in a Continuously Cropped Soybean Field in Northeast China. Environ. Sci. Pollut. Res. 2011, 18, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Wang, C.; Lv, R.; Zhou, J.; Li, X.; Zheng, Y.; Jin, X.; Wang, M.; Ye, Y.; Huang, X.; et al. Community Structure and Succession Regulation of Fungal Consortia in the Lignocellulose-Degrading Process on Natural Biomass. Sci. World J. 2014, 2014, 845721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, H.E.; Parrent, J.L.; Jackson, J.A.; Moncalvo, J.-M.; Vilgalys, R. Fungal Community Analysis by Large-Scale Sequencing of Environmental Samples. Appl. Environ. Microbiol. 2005, 71, 5544–5550. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Li, K.; Nie, M.; Wan, J.-R.; Quan, Z.-X.; Fang, C.-M.; Chen, J.-K.; Gu, J.-D.; Li, B. Responses of Bacterial and Fungal Communities to an Elevation Gradient in a Subtropical Montane Forest of China. Appl. Microbiol. Biotechnol. 2013, 97, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- Brad, T.; Braster, M.; van Breukelen, B.M.; van Straalen, N.M.; Röling, W.F.M. Eukaryotic Diversity in an Anaerobic Aquifer Polluted with Landfill Leachate. Appl. Environ. Microbiol. 2008, 74, 3959–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, K.L.; James, T.Y. Surveying the Biodiversity of the Cryptomycota Using a Targeted PCR Approach. Fungal Ecol. 2015, 14, 62–70. [Google Scholar] [CrossRef]
- Jairajpuri, M.S.; Ahmad, W. Dorylaimida—Free-Living, Predaceous and Plant-Parasitic Nematodes; Oxford and IBH Publishing Co.: New Delhi, India, 1992; ISBN 978-90-04-09229-7. [Google Scholar]
- Yeates, G.W.; Bongers, T. Feeding Habits in Soil Nematode Families and Genera-an Outline for Soil Ecologists. J. Nematol. 1993, 25, 315–331. [Google Scholar] [PubMed]
- Meyer, J.Y.; Taputuarai, R.; Killgore, E. Dissemination and Impacts of the Fungal Pathogen, Colletotrichum gloeosporioides f. Sp. Miconiae, on the Invasive Alien Tree, Miconia calvescens, in Tahiti (South Pacific). In Proceedings of the XII International Symposium on Biological Control of Weeds, La Grande Motte, France, 22–27 April 2007; Julien, M.H., Sforza, R., Bon, M.C., Evans, H.C., Hatcher, P.E., Hinz, H.L., Rector, B.G., Eds.; CAB International: Wallingford, UK, 2008; pp. 594–600. ISBN 978-1-84593-502-3. [Google Scholar]
- Reichert, E.; Johnson, M.T.; Chacón, E.; Anderson, R.S.; Wheeler, T.A. Biology and Host Preferences of Cryptorhynchus melastomae (Coleoptera: Curculionidae), a Possible Biocontrol Agent for Miconia Calvescens (Melastomataceae) in Hawaii. Environ. Entomol. 2010, 39, 1848–1857. [Google Scholar] [CrossRef] [Green Version]
- Seixas, C.D.S.; Barreto, R.W.; Freitas, L.G.; Maffia, L.A.; Monteiro, F.T. Ditylenchus drepanocercus (Nematoda), a Potential Biological Control Agent for Miconia calvescens (Melastomataceae): Host-Specificity and Epidemiology. Biol. Control 2004, 31, 29–37. [Google Scholar] [CrossRef]
- Harrewijn, P.; Oosten, A.M.; Piron, P.G.M. Natural Terpenoids as Messengers: A Multidisciplinary Study of Their Production, Biological Functions and Practical Applications; Springer: Dordrecht, The Netherlands, 2012; ISBN 978-0-7923-6891-5. [Google Scholar]
- de Souza, J.J.; Vieira, I.J.C.; Rodrigues-Filho, E.; Braz-Filho, R. Terpenoids from Endophytic Fungi. Molecules 2011, 16, 10604–10618. [Google Scholar] [CrossRef]
- Matsuda, Y.; Abe, I. Biosynthesis of Fungal Meroterpenoids. Nat. Prod. Rep. 2016, 33, 26–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorissen, H.; Galand, P.E.; Bonnard, I.; Meiling, S.; Raviglione, D.; Meistertzheim, A.-L.; Hédouin, L.; Banaigs, B.; Payri, C.E.; Nugues, M.M. Coral Larval Settlement Preferences Linked to Crustose Coralline Algae with Distinct Chemical and Microbial Signatures. Sci. Rep. 2021, 11, 14610. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Lu, M.; Lu, J.; Yang, W.; Li, B.; Ma, L.; Wang, L. Soil Chemical Properties, Metabolome, and Metabarcoding Give the New Insights into the Soil Transforming Process of Fairy Ring Fungi Leucocalocybe mongolica. J. Fungi 2022, 8, 680. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Lu, J.; Yang, W.; Lu, M.; Wang, J.; Li, S.; Chen, Y.; Hu, L.; Wang, L. Metabarcoding and Metabolome Analyses Reveal Mechanisms of Leymus chinensis Growth Promotion by Fairy Ring of Leucocalocybe mongolica. J. Fungi 2022, 8, 944. [Google Scholar] [CrossRef]
- Mangeot-Peter, L.; Tschaplinski, T.J.; Engle, N.L.; Veneault-Fourrey, C.; Martin, F.; Deveau, A. Impacts of Soil Microbiome Variations on Root Colonization by Fungi and Bacteria and on the Metabolome of Populus tremula × alba. Phytobiomes J. 2020, 4, 142–155. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Wang, X.; Wei, S.; Zhu, Q.; He, S.; Zhou, L. Dynamic Analysis of the Microbial Communities and Metabolome of Healthy Banana Rhizosphere Soil during One Growth Cycle. PeerJ 2022, 10, e14404. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Bacteria | Microeukaryotes | Metazoa | Positive | Negative |
|---|---|---|---|---|---|
| Cyclophyllum barbatum | 0.973 | 0.030 | 0.342 | 0.691 | 0.190 |
| Inocarpus fagifer | 0.030 | 0.029 | 0.050 | 0.528 | 0.087 |
| Ixora mooreensis | 0.461 | 0.358 | 0.554 | 0.557 | 0.831 |
| Miconia calvescens | 0.808 | 0.029 | 0.049 | 0.706 | 0.219 |
| Spathodea campanulata | 0.278 | 0.504 | 0.100 | 0.638 | 0.402 |
| Syzygium malaccense | 0.757 | 0.034 | 0.046 | 0.490 | 0.153 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clerissi, C.; Chaïb, S.; Raviglione, D.; Espiau, B.; Bertrand, C.; Meyer, J.-Y. Metabarcoding and Metabolomics Reveal the Effect of the Invasive Alien Tree Miconia calvescens DC. on Soil Diversity on the Tropical Island of Mo’orea (French Polynesia). Microorganisms 2023, 11, 832. https://doi.org/10.3390/microorganisms11040832
Clerissi C, Chaïb S, Raviglione D, Espiau B, Bertrand C, Meyer J-Y. Metabarcoding and Metabolomics Reveal the Effect of the Invasive Alien Tree Miconia calvescens DC. on Soil Diversity on the Tropical Island of Mo’orea (French Polynesia). Microorganisms. 2023; 11(4):832. https://doi.org/10.3390/microorganisms11040832
Chicago/Turabian StyleClerissi, Camille, Slimane Chaïb, Delphine Raviglione, Benoit Espiau, Cédric Bertrand, and Jean-Yves Meyer. 2023. "Metabarcoding and Metabolomics Reveal the Effect of the Invasive Alien Tree Miconia calvescens DC. on Soil Diversity on the Tropical Island of Mo’orea (French Polynesia)" Microorganisms 11, no. 4: 832. https://doi.org/10.3390/microorganisms11040832