
Adaptation Strategies to High Hydrostatic Pressures in Pseudothermotoga species Revealed by Transcriptional Analyses

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Comparative Genomic Analysis
2.2. Growth under High Hydrostatic Pressure
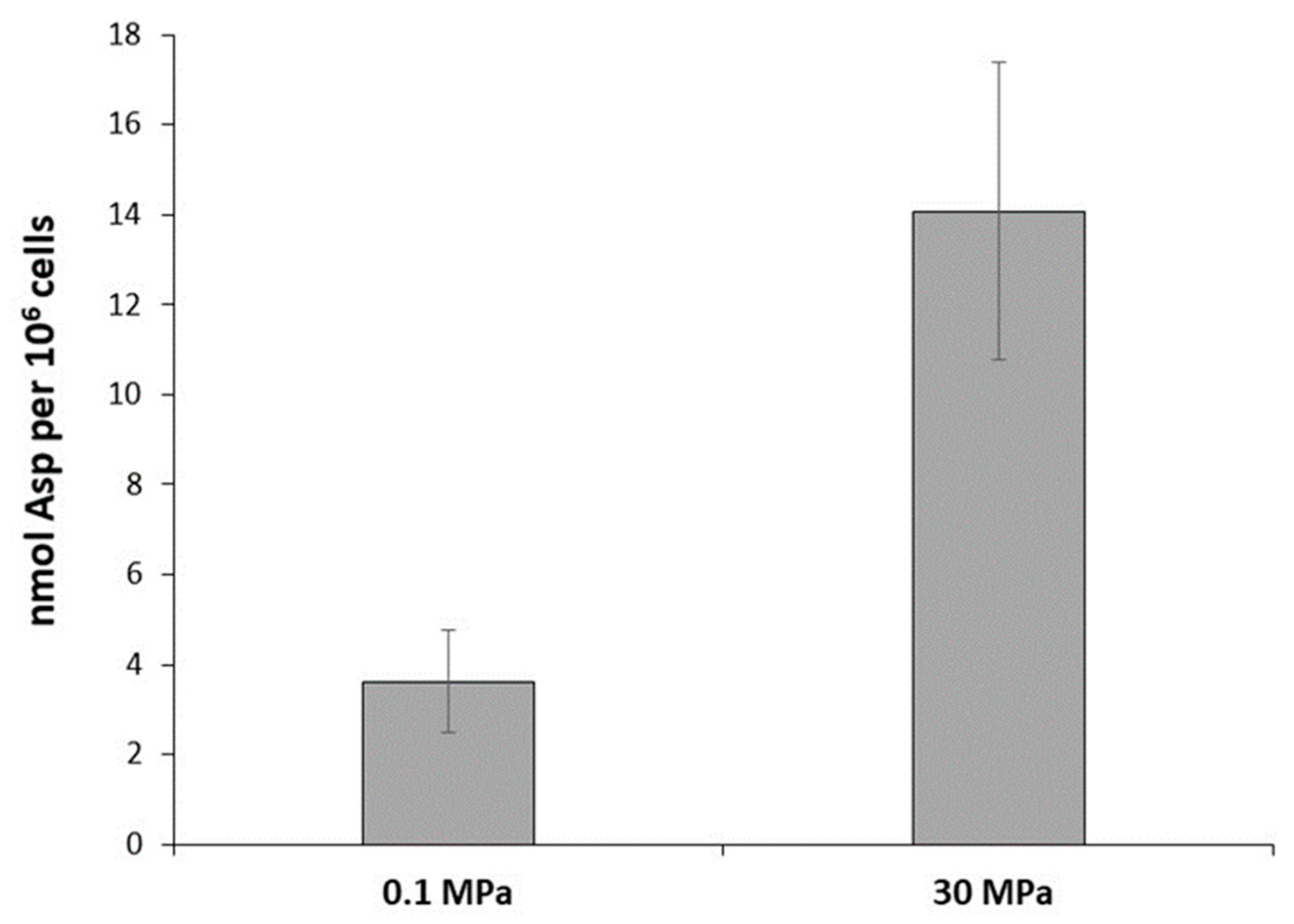
2.3. Aspartate Measurements
2.4. RNA-Seq Experiments
2.5. Bioinformatics Analysis of RNA-Seq Data (Integrated Workflow for RNA-Seq Processing)
3. Results and Discussion
3.1. Comparative Genomics of the Deep and Surface P. elfii Strains
3.2. Overview of the Modifications Induced by Hydrostatic Pressure at the Transcriptome Level of P. elfii DSM9442 and P. elfii subsp. lettingae
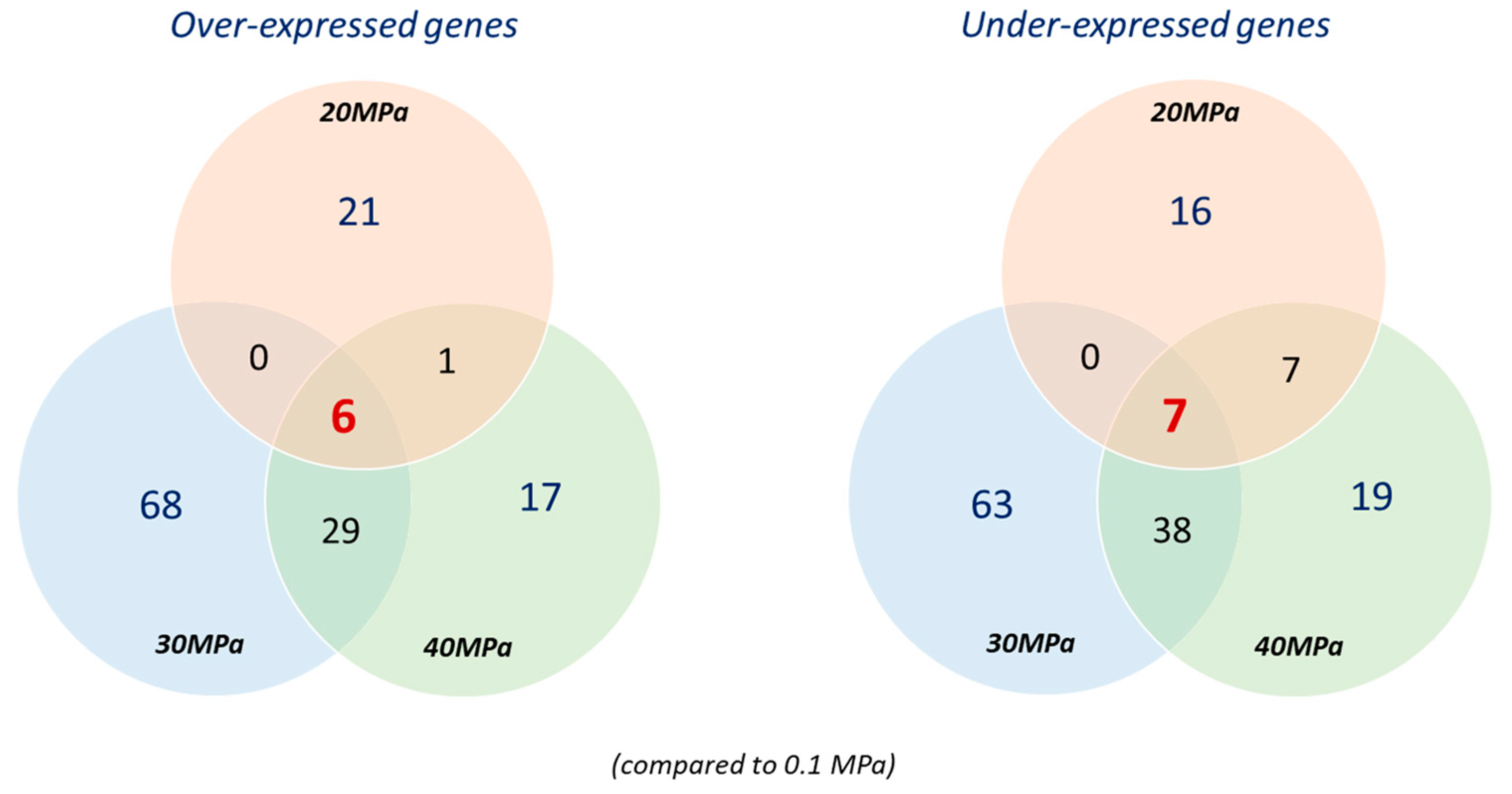
3.3. Differentially Expressed Genes in P. elfii DSM9442 and P. elfii subsp. lettingae in Response to Hydrostatic Pressure
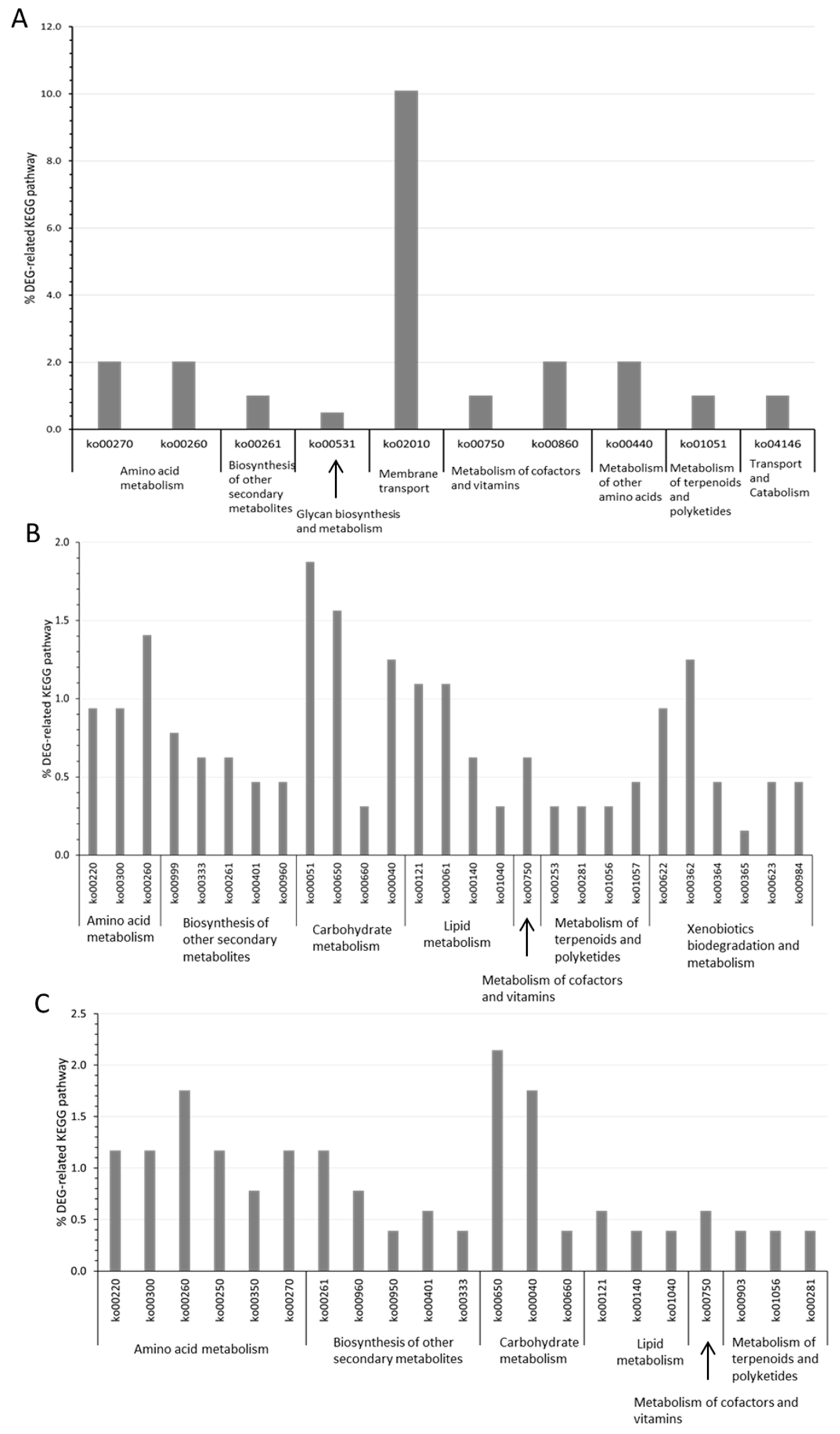
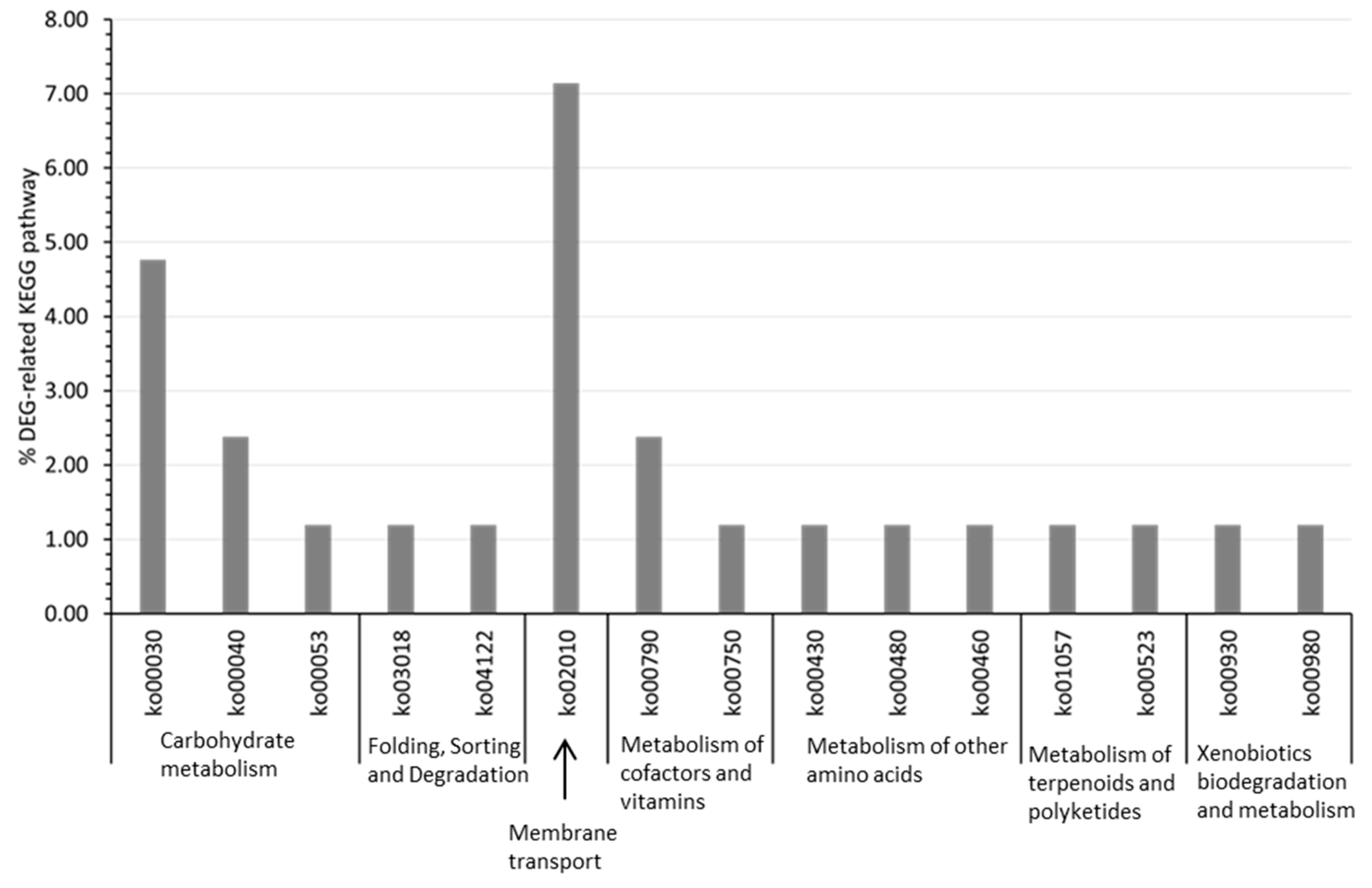
3.4. DEG-Related KEGG Pathway Distribution in P. elfii DSM9442 and P. elfii subsp. lettingae
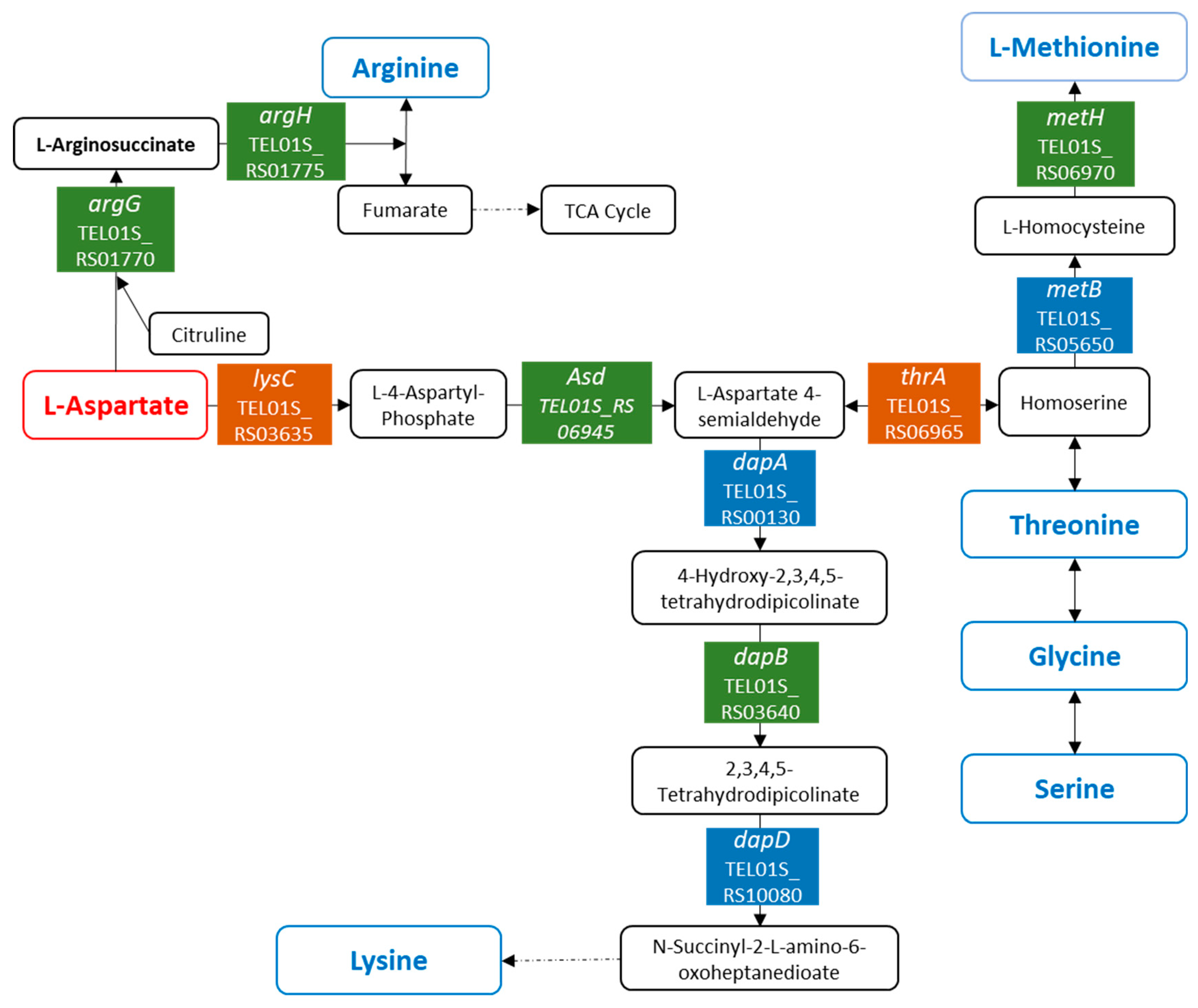
3.5. Variations in the Amino Acid Metabolism in P. elfii in Response to Pressure Changes
3.6. Adaptation Involving Modifications in Membrane Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bartlett, D.H. Pressure effects on in vivo microbial processes. Biochim. Biophys. Acta 2002, 1595, 367–381. [Google Scholar] [CrossRef]
- Simonato, F.; Campanaro, S.; Lauro, F.M.; Vezzi, A.; D’Angelo, M.; Vitulo, N.; Valle, G.; Bartlett, D.H. Piezophilic adaptation: A genomic point of view. J. Biotechnol. 2006, 126, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Lauro, F.; Bartlett, D. Prokaryotic lifestyles in deep sea habitats. Extrem. Life Under Extrem. Cond. 2008, 12, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Lopes, R.; Delgadillo, I.; Saraiva, J. Microorganisms under high pressure--adaptation, growth and biotechnological potential. Biotechnol. Adv. 2013, 31, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Bartlett, D.; Roberts, M. Solute accumulation in the deep-sea bacterium Photobacterium profundum. Extremophiles 2002, 6, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.; Siebenaller, J. Co-evolution of proteins and solutions: Protein adaptation versus cytoprotective micromolecules and their roles in marine organisms. J. Exp. Biol. 2015, 218 Pt 12, 1880–1896. [Google Scholar] [CrossRef] [Green Version]
- Scoma, A.; Barbato, M.; Borin, S.; Daffonchio, D.; Boon, N. An impaired metabolic response to hydrostatic pressure explains Alcanivorax borkumensis recorded distribution in the deep marine water column. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cario, A.; Jebbar, M.; Thiel, A.; Kervarec, N.; Oger, P.M. Molecular chaperone accumulation as a function of stress evidences adaptation to high hydrostatic pressure in the piezophilic archaeon Thermococcus barophilus. Sci. Rep. 2016, 6, 29483. [Google Scholar] [CrossRef] [Green Version]
- Amrani, A.; Bergon, A.; Holota, H.; Tamburini, C.; Garel, M.; Ollivier, B.; Imbert, J.; Dolla, A.; Pradel, N. Transcriptomics reveal several gene expression patterns in the piezophile Desulfovibrio hydrothermalis in response to hydrostatic pressure. PLoS ONE 2014, 9, e106831. [Google Scholar] [CrossRef]
- Amrani, A.; van Helden, J.; Bergon, A.; Aouane, A.; Hania, W.B.; Tamburini, C.; Loriod, B.; Imbert, J.; Ollivier, B.; Pradel, N.; et al. Deciphering the adaptation strategies of Desulfovibrio piezophilus to hydrostatic pressure through metabolic and transcriptional analyses. Environ. Microbiol. Rep. 2016, 8, 520–526. [Google Scholar] [CrossRef]
- Gaussier, H.; Nouailler, M.; Champaud, E.; Garcin, E.B.; Sebban-Kreuzer, C.; Bornet, O.; Garel, M.; Tamburini, C.; Pieulle, L.; Dolla, A.; et al. Glutamate optimizes enzymatic activity under high hydrostatic pressure in Desulfovibrio species: Effects on the ubiquitous thioredoxin system. Extrem. Life Under Extrem. Cond. 2021, 25, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Pradel, N.; Ji, B.; Gimenez, G.; Talla, E.; Lenoble, P.; Garel, M.; Tamburini, C.; Fourquet, P.; Lebrun, R.; Bertin, P.; et al. The first genomic and proteomic characterization of a deep-sea sulfate reducer: Insights into the piezophilic lifestyle of Desulfovibrio piezophilus. PLoS ONE 2013, 8, e55130. [Google Scholar] [CrossRef] [Green Version]
- Moalic, Y.; Hartunians, J.; Dalmasso, C.; Courtine, D.; Georges, M.; Oger, P.; Shao, Z.; Jebbar, M.; Alain, K. The Piezo-Hyperthermophilic Archaeon Thermococcus piezophilus Regulates Its Energy Efficiency System to Cope with Large Hydrostatic Pressure Variations. Front. Microbiol. 2021, 12, 730231. [Google Scholar] [CrossRef]
- Michoud, G.; Jebbar, M. High hydrostatic pressure adaptive strategies in an obligate piezophile Pyrococcus yayanosii. Sci. Rep. 2016, 6, 27289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, F. Dynamic structural changes in microbial membranes in response to high hydrostatic pressure analyzed using time-resolved fluorescence anisotropy measurement. Biophys. Chem. 2013, 183, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xiao, X.; Ou, H.; Gai, Y.; Wang, F. Role and regulation of fatty acid biosynthesis in the response of Shewanella piezotolerans WP3 to different temperatures and pressures. J. Bacteriol. 2009, 191, 2574–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, Y.; Nakayama, A.; Ishihara, K.; Saito, H. Adaptive changes in membrane lipids of barophilic bacteria in response to changes in growth pressure. Appl. Environ. Microbiol. 1998, 64, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Nie, Y.; Chi, C.-Q.; Tang, Y.-Q.; Li, Y.; Wang, X.-B.; Liu, Z.-S.; Yang, Y.; Zhou, J.; Wu, X.-L. Crude oil as a microbial seed bank with unexpected functional potentials. Sci. Rep. 2015, 5, 16057. [Google Scholar] [CrossRef] [Green Version]
- Ollivier, B.; Cayol, J.-L. Fermentative, Iron-Reducing, and Nitrate-Reducing Microorganisms. In Petroleum Microbiology; Magot, B., Ed.; ASM Press: Washington, DC, USA, 2005; pp. 71–88. [Google Scholar]
- Bhandari, V.; Gupta, R. Molecular signatures for the phylum (class) Thermotogae and a proposal for its division into three orders (Thermotogales, Kosmotogales ord. nov. and Petrotogales ord. nov.) containing four families (Thermotogaceae, Fervidobacteriaceae fam. nov., Kosmotogaceae fam. nov. and Petrotogaceae fam. nov.) and a new genus Pseudothermotoga gen. nov. with five new combinations. Antonie Van Leeuwenhoek 2014, 105, 143–168. [Google Scholar]
- Belahbib, H.; Summers, Z.M.; Fardeau, M.-L.; Joseph, M.; Tamburini, C.; Dolla, A.; Ollivier, B.; Armougom, F. Towards a congruent reclassification and nomenclature of the thermophilic species of the genus Pseudothermotoga within the order Thermotogales. Syst. Appl. Microbiol. 2018, 41, 555–563. [Google Scholar] [CrossRef]
- Roumagnac, M.; Pradel, N.; Bartoli, M.; Garel, M.; Jones, A.A.; Armougom, F.; Fenouil, R.; Tamburini, C.; Ollivier, B.; Summers, Z.M.; et al. Responses to the Hydrostatic Pressure of Surface and Subsurface Strains of Pseudothermotoga elfii Revealing the Piezophilic Nature of the Strain Originating from an Oil-Producing Well. Front. Microbiol. 2020, 11, 588771. [Google Scholar] [CrossRef] [PubMed]
- Ravot, G.; Magot, M.; Fardeau, M.-L.; Patel, B.; Prensier, G.; Egan, A.; Garcia, J.-L.; Ollivier, B. Thermotoga elfii sp. nov., a novel thermophilic bacterium from an African oil-producing well. Int. J. Syst. Bacteriol. 1995, 45, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Balk, M.; Weijma, J.; Stams, A. Thermotoga lettingae sp. nov., a novel thermophilic, methanol-degrading bacterium isolated from a thermophilic anaerobic reactor. Int. J. Syst. Evol. Microbiol. 2002, 52 Pt 4, 1361–1368. [Google Scholar] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [Green Version]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.; Mau, B.; Blattner, F.; Perna, N. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Garel, M.; Bonin, P.; Martini, S.; Guasco, S.; Roumagnac, M.; Bhairy, N.; Armougom, F.; Tamburini, C. Pressure-Retaining Sampler and High-Pressure Systems to Study Deep-Sea Microbes under. Front. Microbiol. 2019, 10, 453. [Google Scholar] [CrossRef] [Green Version]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. Lett. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, T.; Smith, A. Predicting the molecular complexity of sequencing libraries. Nat. Methods 2013, 10, 325–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayols, S.; Scherzinger, D.; Klein, H. dupRadar: A Bioconductor package for the assessment of PCR artifacts in RNA-Seq data. BMC Bioinform. 2016, 17, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; McCarthy, D.; Smyth, G. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. A Publ. Protein Soc. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Herrou, J.; Willett, J.W.; Czyż, D.M.; Babnigg, G.; Kim, Y.; Crosson, S. Conserved ABC Transport System Regulated by the General Stress Response Pathways of Alpha- and Gammaproteobacteria. J. Bacteriol. 2017, 199, e00746-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.D.; Wagner, L.M.; Graddis, T.J.; Landick, R.; Antonucci, T.K.; Gibson, A.L.; Oxender, D.L. Nucleotide sequence and genetic characterization reveal six essential genes for the LIV-I and LS transport systems of Escherichia coli. J. Biol. Chem. 1990, 265, 11436–11443. [Google Scholar] [CrossRef]
- Kelly, D.; Thomas, G. The tripartite ATP-independent periplasmic (TRAP) transporters of bacteria and archaea. FEMS Microbiol. Rev. 2001, 25, 323–329. [Google Scholar] [CrossRef]
- Miroshnichenko, M.; Kostrikina, N.; Chernyh, N.; Pimenov, N.; Tourova, T.; Antipov, A.; Spring, S.; Stackebrandt, E.; Bonch-Osmolovskaya, E. Caldithrix abyssi gen. nov., sp. nov., a nitrate-reducing, thermophilic, anaerobic bacterium isolated from a Mid-Atlantic Ridge hydrothermal vent, represents a novel bacterial lineage. Int. J. Syst. Evol. Microbiol. 2003, 53 Pt 1, 11436–11443. [Google Scholar] [CrossRef]
- Hale, C.R.; Majumdar, S.; Elmore, J.; Pfister, N.; Compton, M.; Olson, S.; Resch, A.M.; Glover, C.V.; Graveley, B.R.; Terns, R.M.; et al. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol. Cell 2012, 45, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Liu, M.; Reeves, P. Progress in Our Understanding of Wzx Flippase for Translocation of Bacterial Membrane Lipid-Linked Oligosaccharide. J. Bacteriol. 2017, 200, e00154-17. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Guo, Y.; Liu, J.; Qiu, H.; Zhao, M.; Zou, W.; Li, S. Microbial synthesis of poly-γ-glutamic acid: Current progress, challenges, and future perspectives. Biotechnol. Biofuels 2016, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Redei, G. Glucogenic Amino Acids. In Encyclopedia of Genetics, Genomics, Proteomics and Informatics; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Barreteau, H.; Kovač, A.; Boniface, A.; Sova, M.; Gobec, S.; Blanot, D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 168–207. [Google Scholar] [CrossRef] [Green Version]
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, R.A.; McCubbin, T.; Navone, L.; Stowers, C.; Nielsen, L.K.; Marcellin, E. Microbial Propionic Acid Production. Fermentation 2017, 3, 21. [Google Scholar] [CrossRef]
- Shanbhagm, A. FabG: From a core to circumstantial catalyst. Biotechnol. Lett. 2019, 41, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Z.; Sung, B.H.; Nawab, J.; Siddiqui, M.F.; Ali, A.; Geraldi, A.; Kim, S.C. Enhanced Production of Fatty Acid Ethyl Ester with Engineered fabHDG Operon in Escherichia coli. Microorganisms 2019, 7, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, Y.; Yao, C.; Cao, X.; Tian, J.; Xue, S. FabG can function as PhaB for poly-3-hydroxybutyrate biosynthesis in photosynthetic cyanobacteria Synechocystis sp. PCC 6803. Bioengineered 2017, 8, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Eloe, E.; Lauro, F.; Vogel, R.; Bartlett, D. The deep-sea bacterium Photobacterium profundum SS9 utilizes separate flagellar systems for swimming and swarming under high-pressure conditions. Appl. Environ. Microbiol. 2008, 74, 6298–6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus_Tag | KEGG Name | KEGG Pathway | DEG at 20 MPa * | DEG at 30 MPa * | DEG at 40 MPa * |
|---|---|---|---|---|---|
| TEL01S_RS01780 | argC | ko00220:Arginine biosynthesis | 0 | −1 | −1 |
| TEL01S_RS01785 | argJ | ko00220:Arginine biosynthesis | 0 | −1 | −1 |
| TEL01S_RS01770 | argG | ko00220:Arginine biosynthesis|ko00250:Alanine, aspartate, and glutamate metabolism | 0 | −1 | −1 |
| TEL01S_RS01775 | argH | ko00220:Arginine biosynthesis|ko00250:Alanine, aspartate, and glutamate metabolism | 0 | −1 | −1 |
| TEL01S_RS04500 | glnA | ko00220:Arginine biosynthesis|ko00250:Alanine, aspartate, and glutamate metabolism | 0 | −1 | −1 |
| TEL01S_RS09130 | ko00220:Arginine biosynthesis|ko00250:Alanine, aspartate, and glutamate metabolism|ko00270:Cysteine and methionine metabolism|ko00330:Arginine and proline metabolism|ko00350:Tyrosine metabolism|ko00360:Phenylalanine metabolism|ko00400:Phenylalanine, tyrosine, and tryptophan biosynthesis | 0 | 1 | 1 | |
| TEL01S_RS03835 | glmS | ko00250:Alanine, aspartate, and glutamate metabolism | 0 | −1 | 0 |
| TEL01S_RS09135 | ko00250:Alanine, aspartate, and glutamate metabolism|ko00260:Glycine, serine, and threonine metabolism | 1 | 1 | 1 | |
| TEL01S_RS05590 | soxA | ko00260:Glycine, serine, and threonine metabolism | 0 | −1 | −1 |
| TEL01S_RS10380 | kbl | ko00260:Glycine, serine, and threonine metabolism | 0 | 1 | 1 |
| TEL01S_RS03635 | lysC | ko00260:Glycine, serine, and threonine metabolism|ko00270:Cysteine and methionine metabolism|ko00300:Lysine biosynthesis | 0 | −1 | −1 |
| TEL01S_RS06945 | asd | ko00260:Glycine, serine, and threonine metabolism|ko00270:Cysteine and methionine metabolism|ko00300:Lysine biosynthesis | 0 | −1 | −1 |
| TEL01S_RS06965 | lysC_&_thrA | ko00260:Glycine, serine, and threonine metabolism|ko00270:Cysteine and methionine metabolism|ko00300:Lysine biosynthesis | −1 | −1 | −1 |
| TEL01S_RS09745 | trpB | ko00260:Glycine, serine, and threonine metabolism|ko00400:Phenylalanine, tyrosine, and tryptophan biosynthesis | 0 | −1 | 0 |
| TEL01S_RS09140 | serA | ko00260:Glycine, serine, and threonine metabolism | 0 | 1 | 1 |
| TEL01S_RS08685 | mtnK | ko00270:Cysteine and methionine metabolism | −1 | 0 | 0 |
| TEL01S_RS08690 | mtnA | ko00270:Cysteine and methionine metabolism | −1 | 0 | 0 |
| TEL01S_RS05650 | ko00270:Cysteine and methionine metabolism|ko00260:Glycine, serine, and threonine metabolism | 0 | −1 | 0 | |
| TEL01S_RS06970 | metH | ko00270:Cysteine and methionine metabolism | 0 | −1 | −1 |
| TEL01S_RS03640 | dapB | ko00300:Lysine biosynthesis | 0 | −1 | −1 |
| TEL01S_RS00130 | dapA | ko00300:Lysine biosynthesis | 0 | −1 | 0 |
| TEL01S_RS09220 | murF | ko00300:Lysine biosynthesis | 0 | 1 | 0 |
| TEL01S_RS10080 | dapD | ko00300:Lysine biosynthesis|ko01230:Biosynthesis of amino acids | 0 | −1 | 0 |
| TEL01S_RS08600 | kdd | ko00310:Lysine degradation | 0 | 1 | 0 |
| TEL01S_RS05490 | amiE | ko00330:Arginine and proline metabolism|ko00360:Phenylalanine metabolism|ko00380:Tryptophan metabolism | 0 | 0 | 1 |
| TEL01S_RS06380 | hisC | ko00340:Histidine metabolism|ko00350:Tyrosine metabolism|ko00360:Phenylalanine metabolism|ko00400:Phenylalanine, tyrosine, and tryptophan biosynthesis | 0 | 1 | 1 |
| TEL01S_RS03685 | iaaM | ko00380:Tryptophan metabolism | 0 | −1 | 0 |
| TEL01S_RS05225 | bglX | ko00460:Cyanoamino acid metabolism | 0 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fenouil, R.; Pradel, N.; Belahbib, H.; Roumagnac, M.; Bartoli, M.; Ben Hania, W.; Denis, Y.; Garel, M.; Tamburini, C.; Ollivier, B.; et al. Adaptation Strategies to High Hydrostatic Pressures in Pseudothermotoga species Revealed by Transcriptional Analyses. Microorganisms 2023, 11, 773. https://doi.org/10.3390/microorganisms11030773
Fenouil R, Pradel N, Belahbib H, Roumagnac M, Bartoli M, Ben Hania W, Denis Y, Garel M, Tamburini C, Ollivier B, et al. Adaptation Strategies to High Hydrostatic Pressures in Pseudothermotoga species Revealed by Transcriptional Analyses. Microorganisms. 2023; 11(3):773. https://doi.org/10.3390/microorganisms11030773
Chicago/Turabian StyleFenouil, Romain, Nathalie Pradel, Hassiba Belahbib, Marie Roumagnac, Manon Bartoli, Wajdi Ben Hania, Yann Denis, Marc Garel, Christian Tamburini, Bernard Ollivier, and et al. 2023. "Adaptation Strategies to High Hydrostatic Pressures in Pseudothermotoga species Revealed by Transcriptional Analyses" Microorganisms 11, no. 3: 773. https://doi.org/10.3390/microorganisms11030773